

Abstract
One of the most common complications of childhood obesity is the non-alcoholic fatty liver disease (NAFLD), which is the most common form of liver disease in children. NAFLD is defined by hepatic fat infiltration > 5% hepatocytes, as assessed by liver biopsy, in the absence of excessive alcohol intake, viral, autoimmune and drug-induced liver disease. It encompasses a wide spectrum of liver diseases ranging from simple steatosis to non-alcoholic steatohepatitis, which, in turn, can evolve into cirrhosis and end stage liver disease. Obesity and insulin resistance are the main risk factors for pediatric NAFLD. In fact, NAFLD is strongly associated with the clinical features of insulin resistance especially the metabolic syndrome, prediabetes and type 2 diabetes mellitus (T2D). In particular, it has been clearly shown in obese youth that the prevalence of metabolic syndrome, pre-diabetes and type 2 diabetes increases with NAFLD severity progression. Evidence that not all of the obese patients develop NAFLD suggests that the disease progression is likely to depend on complex interplay between environmental factors and genetic predisposition. Recently, a non-synonymous SNP (rs738409), characterized by a C to G substitution encoding an isoleucine to methionine substitution at the amino acid position 148 in the patatin like phospholipase containing domain 3 gene (PNPLA3), has been associated with hepatic steatosis in a multiethnic cohort of adults as well as in children. Another important polymorphisms that acts with PNPLA3 to convey susceptibility to fatty liver in obese youths is the rs1260326 polymorphism in the glucokinase regulatory protein. The pharmacological approach in NAFLD children poorly adherent to or being unresponsive/partially responsive to lifestyle changes, is aimed at acting upon specific targets involved in the pathogenesis. There are some therapeutic approaches that are being studied in children. This article reviews the current knowledge regarding the pediatric fatty liver disease, the new insights and the future directions.
Keywords: Non alcoholic fatty liver disease, PNPLA3, Obesity, Insulin resistance, Glucokinase regulatory protein, Fructose
Core tip: The prevalence of hepatic steatosis is increased in the last three decades concomitantly with the increased prevalence of pediatric obesity. Non-alcoholic fatty liver disease (NAFLD) is the most common form of liver disease in children. The PNPLA3 rs738409 and the glucokinase regulatory protein rs1260326 are the strongest variants associated with fatty liver in paediatrics. Important risk factors are obesity, insulin resistence, gender, ethnicity and excessive dietetic intake of n-6 polyunsatured fatty acids and fructose. New pharmacological approaches are object of study, in NAFLD children poorly adherent to or being unresponsive/partially responsive to lifestyle changes.
INTRODUCTION
In the last three decades with the increased prevalence of childhood obesity, there has been an increase also of the obesity complications in paediatrics. One of the most common complications of childhood obesity is the non-alcoholic fatty liver disease (NAFLD), which is the most common form of liver disease in children[1].
NAFLD is defined by hepatic fat infiltration > 5% hepatocytes, as assessed by liver biopsy, in the absence of excessive alcohol intake, viral, autoimmune and drug-induced liver disease[2,3]. It encompasses a wide spectrum of liver diseases ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which, in turn, can evolve into cirrhosis and end stage liver disease[3,4].
The prevalence of NAFLD has more than doubled over the past 20 years. According to a landmark study by Schwimmer et al[1] based on autoptic data obtained in 1138 children and adolescents of the San Diego county (CA), its prevalence in the general pediatric population is estimated to be nearly 13%, while among obese and overweight children and, particularly, adolescents it rises up to 46%[1]. Nevertheless, other studies report quite a wide range of steatosis prevalence, likely due to the different diagnostic methods used. In fact, although liver histology is important for NAFLD evaluation, performing biopsies is not always indispensable from a clinical point of view; therefore, surrogate markers are often used in epidemiological and clinical studies. One of the marker most commonly used is liver aminotransferase [aspartate aminotransferase, and alanine aminotransferase (ALT)] evaluation. Children with NAFLD typically have elevated liver enzymes values[5], which is why elevated serum levels of liver enzymes, even though may misrepresent the entity of intrahepatic damage, are used as a non-invasive test to screen for pediatric NAFLD[6] .
RISK FACTORS FOR THE DEVELOPMENT OF PEDIATRIC NAFLD
Obesity and insulin resistance are the main risk factors for pediatric NAFLD[1,7,8]. In fact, NAFLD is strongly associated with the clinical features of insulin resistance especially the metabolic syndrome (MS), prediabetes and type 2 diabetes mellitus (T2D)[9-11]. In particular, it has been clearly shown in obese youth that the prevalence of metabolic syndrome, pre-diabetes and type 2 diabetes increases with progression of NAFLD severity[12].
This picture is strongly contributed by pubertal insulin resistance, a physiologic state characterized by an increased insulin resistance during the adolescence and resolving at the end of the pubertal development and probably consequent to the increase in growth hormone action during this stage of life[8,13]. In fact, although obesity is the most important cause of NAFLD among obese and adolescents, it is important to note that a transient insulin resistant state occurs during puberty[14], and that this state worsens the insulin resistance present in obese children in turn accelerating the progression to MS and type 2 diabetes. In healthy individuals this phenomenon is balanced by an increased insulin secretion by the beta cell, but in obese individuals the co-occurrence of obesity and puberty represents the perfect storm causing such a high degree of insulin resistance that the beta cell is not always able to produce enough insulin to maintain the glycemic control[15-17].
Two other critical risk factors for NAFLD development are represented by the gender and the ethnic background. In fact, NAFLD is more common in boys than in girls[15] with a male to female ratio of 2:1. This has been explained by the liver-protective role of estrogens, as well as by the potentially negative role of androgens in aggravating NASH[18,19]. The beneficial effects of estrogens on liver could be mediated by the beneficial effect on insulin action. Studies showed that insulin sensitivity is greater in premenopausal women compared with age-matched men, and metabolic-related cardiovascular diseases and type 2 diabetes are less frequent in premenopausal women[20,21]. Also, estrogens deficiency leads to increased fat mass and body weight in postmenopausal women, which has been associated with increased intraabdominal fat[22]. Moreover, Camporez et al[23] showed that, in mice, endogenous estrogens are important to protect against high-fat diet induced skeletal muscle insulin resistance, whereas E2 treatment in estrogen-deprived mice increased insulin sensitivity in both liver and skeletal muscle. Also, the estrogens effect is important in turn preventing diet-induced ectopic lipid deposition and hepatic and muscle insulin resistance.
The risk linked to the ethnic background has been investigated in large multiethnic populations. A cornerstone article by Browning et al[15] described for the first time that the prevalence of NAFLD is the highest in the American Hispanic population (45%) and the lowest among African Americans (24%), with the Caucasians showing an intermediate prevalence (33%). Ethnic differences could possibly be due to different degree of insulin resistance, and of visceral adiposity at equivalent body mass index, but may also be a result of genetics as well as socio-economic factors, including type of diet, exercise choice and living location[24].
The accumulation of fat, as triacylglycerol (TAG), in the hepatocyte is the fingerprint of fatty liver. The TAG accumulated in the liver mostly derive from adipose tissue lipolysis (60%) and hepatic de novo lipogenesis (26%) whereas only a small amount directly derives from the diet as chylomicron remnants (14%)[25]. A large body of evidence suggests that not only the amount, but also the quality of dietary fat plays a role in NAFLD development[26]. In particular, recently published literature provides clues that the dietary imbalance between omega-6 (n-6) and omega-3 (n-3) polyunsatured fatty acids (PUFAs) leads to development of an adverse cardiovascular and metabolic profile, thus contributing to the pathogenesis of NAFLD[27]. N-6 and n-3 are essential fatty acids; this means that they are not synthesized by human body. N-6 species are mainly represented by linoleic acid while n-3 are represented by alpha-linolenic acid, mainly found in plants and limited sets of seeds and nuts[28]. N-6 is readily converted by the body into other species such as omega-9 and so incorporated into triglycerides, or converted into arachidonic acid, which is the parent molecule of the main regulators of the inflammatory response including prostaglandins (ciclooxigenase pathway), leukotrienes (lipoxygenase pathway) and tromboxane[28]. It has been demonstrated that individuals with NAFLD have a lower dietary intake of n-3 PUFAs than healthy controls[29] and an increased n-6/n-3 PUFA ratio consumed in the diet[29,30]. Consistent with these data, lipidomic studies have shown that the intrahepatic fat in subjects with steatohepatitis is composed by an excess n-6 PUFA[31]. In particular, studying the three groups of subjects-NAFLD, NASH and healthy controls- it has been observed a progressive increase in the n-6/n-3 ratio from controls to NASH subjects[31].
Another dietary risk factor contributing to the development of NAFLD is the fructose. Nowadays, the majority of fructose consumption comes from the added sugars in the beverages more than from the fruit[32]. Strong evidence exists that high in fructose intake results in increased de novo lipogenesis (DNL), dyslipidemia, insulin resistance, and obesity in humans[33]. Stanhope et al[33], studying the effect of consumption of glucose- or fructose-sweetened beverages providing 25% of energy requirements for 10 wk in overweight and obese subjects, provided the evidence that the consumption of fructose, instead of glucose, specifically increases DNL, promotes dyslipidemia, decreases insulin sensitivity, and increases visceral adiposity in overweight/obese adults.
Progression from NAFLD to NASH
A recent study demonstrated that NAFLD in children is a progressive disease[34]. In that study the authors showed that 6% of subjects with early onset NAFLD develop cirrhosis and end-stage liver disease with the consequent need of liver transplantation.
The oxidative stress seems to explain the progression to NASH and liver fibrosis. Reactive oxygen species (ROS) can induce hepatocellular injury by the inhibition of the mitochondrial respiratory chain enzymes, the inactivation of glyceraldehyde-3-phosphate dehydrogenase and the inactivation of membrane sodium channels. ROS further cause lipid peroxidation, cytokine production, and induce Fas ligand, contributing to hepatocellular injury and fibrosis[12]. The risk of progression varies by ethnicity, in fact, as recently demonstrated, the African American obese children and adolescents show a lower degree of liver damage than Caucasians and Hispanics, independent of the degree of hepatic fat accumulation and insulin resistance. These data suggest that African Americans are protected from hepatic damage even in presence of high degree of hepatic fat accumulation and insulin resistance[35].
GENETIC PREDISPOSITION
Evidence that not all of the obese patients develop NAFLD suggests that disease progression is likely to depend on complex interplay between environmental factors and genetic predisposition (Figure 1).
Figure 1.
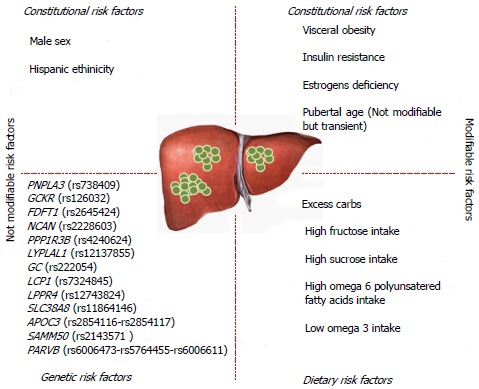
Risk factors for non-alcoholic fatty liver disease development. In this figure, all risk factors for non-alcoholic fatty liver disease (NAFLD) are summarized. We divided the risk factors in modifiable and not modifiable. Among not modifiable risk factors we listed PNPLA3 rs738409, but as underlined in the text, the weight loss can modify the capacity of PNPLA3 polymorphism to lead to hepatic steatosis. PNPLA3: Patatin like phospholipase 3 gene; GCKR: Glucokinase regulatory protein; FDFT1: Farnesyl-diphosphate farnesyltransferase 1; NCAN: Neurocan; PPP1R3B: Protein phosphatase 1 regulatory subunit 3B; LYPLAL1: Lysophospholipase-like 1; GC: Group-specific component; LCP1: Lymphocyte cytosolic protein-1; LPPR4: Lipid phosphate phosphatise-related protein type 4; SLC38A8: Solute carrier family 38 member 8; APOC3: Apolipoprotein C3 gene; SAMM50: Sorting and assembly machinery component; PARVB: Parvin beta.
Recently, a non-synonymous SNP (rs738409), characterized by a C to G substitution encoding an isoleucine to methionine substitution at the amino acid position 148 in the patatin like phospholipase 3 gene (PNPLA3), has been associated with hepatic steatosis in a multiethnic cohort of adults[36] as well as in children[37,38]. PNPLA3 encodes for a triglyceride hydrolase expressed in the liver and adipose tissue[39]. Metabolic studies in transgenic mice revealed that high level expression of PNPLA3I148M in the liver, but not in adipose tissue, affected both hepatic triacylglycerol (TAG) synthesis and catabolism. A surprising finding was that the PNPLA3I148M transgenic mice have significantly increased fatty acid synthesis and an altered spectrum of TAG-fatty acids in the liver, with no evidence of insulin resistance[40]. It is interesting that PNPLA3I148M transgenic mice develop steatosis on a sucrose diet but not on a high-fat diet. Ingestion of sucrose stimulates de novo synthesis of fatty acids[41], whereas most of the hepatic fatty acids in livers of fat-fed mice are derived from circulating non esterified fatty acids (NEFAs). Perhaps PNPLA3 in hepatocytes is exposed preferentially to newly synthesized TAG and is shielded from fatty acids that enter the liver in lipoproteins or are synthesized from circulating NEFAs[40]. Alternatively, PNPLA3 may function specifically under conditions of insulin-stimulated lipid anabolism. The finding that PNPLA3 is virtually absent from livers of fasting animals and is strongly upregulated both transcriptionally[42-45] and post-translationally[39] by carbohydrate refeeding is consistent with the latter hypothesis.
Moreover, purified recombinant PNPLA3 has been found to have 5 enzymatic activities: triacylglycerol, diacylglycerol, and monoacylglycerol hydrolysis[42,46] as well as acyl-CoA thioesterase[42] and lysophosphatidic acid acyltransferase activity[47]. These different activities are not equally affected by the I148M substitution. In vitro assays, the I148M substitution results in a substantial loss of triacylglycerol, monoacylglycerols, and diacylglycerols hydrolytic activity[33]; a modest reduction in acyl-CoA thioesterase activity[33]; and an increase in lysophosphatidic acid acyltransferase activity[47]. None of these activities alone can explain all of the changes in TAG metabolism observed in the PNPLA3 I148M transgenic mice. Therefore, some of the metabolic changes observed in these animals are likely to be secondary rather than direct consequences of altered PNPLA3 activity.
The effect of this polymorphism on liver damage seems to be driven by the size of abdominal fat, expressed as waist to height ratio (W/Hr)[38]. More recently it has been demonstrated that weight loss reduce the effect of this polymorphism in obese children[48].
Other findings suggest also that the influence of PNPLA3 on hepatic fat in obese children and adolescents might be modulated by dietary factors such as n-6/n-3 polynsatured fatty acids (PUFA) intake[49]. Finally, the rs738409 PNPLA3 polymorphysm is considered, regardless of metabolic profile, a risk factor for liver disease. In fact, in a recent study, in a hepatitis C-infected population, the PNPLA3 polymorphism influenced the development of liver steatosis[50]. Moreover, it was also demonstrated as a novel genetic marker associated with progressive ALD (alcoholic liver disease)[51].
Another polymorphism that acts along with the PNPLA3 gene variant to convey susceptibility to fatty liver in obese youths is the rs1260326 polymorphism in the glucokinase regulatory protein (GCKR). This polymorphism is associated with hepatic fat accumulation along with large VLDL and triglyceride levels[52].
Speliotes et al[53], in addition to GCKR, identified variants in novel loci NCAN and LYPLAL1 associated with both increasing computer tomography (CT) hepatic steatosis and histological NAFLD and identified variants in another locus, protein phosphatase 1 regulatory subunit 3B (PPP1R3B), associated with CT steatosis but not histologic NAFLD[53]. Recently Kitamoto et al[54] found that PNPLA3, SAMM50 sorting and assembly machinery component (SAMM50), parvin beta (PARVB) genetic regions was significantly associated with NAFLD in the Japanese population. Adams et al[55] showed that SNPs in two genes expressed in liver were associated with NAFLD in adolescents: group-specific component (GC) and lymphocyte cytosolic protein-1 (LCP1). SNPs in two genes expressed in neurons were also associated with NAFLD: lipid phosphate phosphatise-related protein type 4 (LPPR4) and solute carrier family 38 member 8 (SLC38A8)[55].
TREATMENTS
Diet and lifestyle changes
The goal of lifestyle interventions is a gradual and controlled weight loss achieved by diet and physical exercise (Table 1). This aim is difficult to achieve and only a small percentage of individuals is able to steadily lose weight and exercise regularly[56]. Weight loss in NAFLD patients improves hepatic insulin sensitivity by reducing hepatic NEFAs supply, improves extra-hepatic insulin sensitivity through better glucose utilization and reduces ROS generation and adipose tissue inflammation[56].
Table 1.
Current and future non-alcoholic fatty liver disease treatment strategies
| Present | Under development | |
| Weight loss Physical activity Reduced dietary sucrose intake Reduced dietary fructose intake Reduced dietary omega 6 intake Increased dietary omega 3 intake | Vitamin E Metformin Probiotics Oral treatment with omega 3 | Pentoxifylline Farnesoid X receptor agonists Toll-like receptors modifiers Glucagon-like peptid-1 receptor agonists resistant to DPP-4 mediated degradation DPP-4 inhibitors |
DPP-4: Dipeptidyl peptidase-4.
Currently, there are no evidence-based guidelines establishing the optimal intervention. The only effective interventions are physical activity and dietary changes. In fact, reduction in sugar/sucrose and in soft drinks rich in fructose, most probably not only acts through a reduction in IR and lipogenesis, but also counteracts the recently evidenced hepatic pro-inflammatory/fibrogenetic role of fructose[57]. It should be taken in mind that diet in childhood must be balanced to allow a healthy and harmonic growth, including wellness of bone structures. To intervene on dietary changes does not only mean to reduce the caloric intake, but also the single components of the diet (Table 1). In fact, also diet composition plays an important role in the development of NAFLD, as an increased dietary intake of monounsaturated and polyunsaturated fatty acids (mainly omega 3 PUFA) has been associated with a reduction of hepatic fat content, representing a reasonable intervention especially in the pediatric population[58].
Pharmacological interventions for NAFLD
The pharmacological approach, in NAFLD children poorly adherent to or being unresponsive/partially responsive to lifestyle changes, is aimed at acting upon specific targets involved in etiopathogenesis (Table 1).
Antioxidants, by reducing oxidative stress, protect susceptible components of biological membranes from lipid peroxidation, and may, therefore, prevent the progression of simple steatosis to NASH. The most studied antioxidant in children with NAFLD is alpha tocopherol (vitamin E) and warrants consideration in obesity-related liver dysfunction for children unable to adhere to low-calorie diets[59]. Sanyal et al[60] showed that vitamin E therapy, as compared with placebo, was associated with a significantly higher rate of improvement in NASH (43% vs 19%, P = 0.001) in adults without diabetes. There was no benefit of pioglitazone over placebo for improvement of NASH but serum alanine and aspartate aminotransferase levels were reduced as well as with vitamin E.
For its pathogenic role, insulin resistance appears as an adequate therapeutic target. Metformin is the only insulin-sensitizing agent evaluated in children.
Lavine et al[61] in a more recent large, multicenter, randomised double-bind placebo-controlled trial (TONIC study), evaluated the effect of daily dosing of 800 IU of vitamin E (58 patients), 1000 mg of metformin (57 patients) or placebo (58 patients) for 96 wk of NAFLD course. The patients (aged 8-17 years) with biopsy-confirmed NAFLD and persistently elevated levels of ALT, without diabetes or cirrhosis, were randomly assigned to 1 of 3 groups. At 96 wk neither vitamin E nor metformin was superior to placebo in attaining the primary outcome of sustained reduction in ALT level in pediatric NAFLD; vitamin E and metformin groups, however, showed an improvement in histological hepatocellular ballooning in NAFLD and NASH.
Another, single-arm, open-label, small pilot study on metfotmin (500 mg twice daily for 24 wk), conducted in 10 non-diabetic children with biopsy proven NASH and elevated ALT levels showed reduction of hepatic steatosis, as evaluated with Magnetic Resonance Spectroscopy (MRS) and low serum ALT levels[62].
FUTURE DIRECTIONS
A growing body of evidence[63] shows that the gut microbiota controls obesity and visceral fat storage. Specific variations in gut microbiota in early life may determine a major risk factor of obesity and its complications later in life[64]. Small intestinal bacterial overgrowth (SIBO) (a frequent condition in obese individuals, mainly prompted by slowing of the oro-coecal transit time) may promote NAFLD progression to non-alcoholic steatohepatitis by enhancing intestinal permeability and by favouring absorption of endotoxins with pro-inflammatory and pro-fibrogenetic effects on the liver[65].
Probiotics are live microorganisms which when consumed in adequate amounts, confer an healthy benefit to the host[66]. Gut microbiota manipulation with probiotics in rodents with fatty liver reduces intestinal inflammation and improves the epithelial barrier function[67,68]. Therefore, probiotics could represent a new effective treatment also in NAFLD human patients (Table 1). Loguercio and colleagues have shown that probiotics may reduce NAFLD liver injury and may improve liver function tests[69].
Moreover, recent pharmacological studies in NAFLD animal models and in adult humans focusing on the effect of oral treatment with n-3 fatty acids, demonstrate that they have both anti-inflammatory and insulin sensitizing properties, suggesting a potential role in treatment of NAFLD[70]. In NAFLD children n-3-docosahexaenoic acid (DHA) treatment for 6 months improved ultrasonographic fatty liver and insulin sensitivity[71]. Because this treatment is well tolerated in pediatric population, DHA deserve further studies in the management of children with NAFLD.
A series of other interesting approaches, hitherto explored only in NAFLD animal models or in few pilot studies in adults will possibly become in future the object of study in pediatric population (Table 1), as well: (1) tumor necrosis factor-α (TNF-α) and other adipocytokines produced by adipose tissue are involved in NAFLD progression. Pentoxifylline, a phosphodiesterase inhibitor, exerts immunomodulatory functions by antagonizing the TNF-α pathway. In adults with NASH, pentoxifylline treatment showed good tolerability and could decrease serum ALT levels and improve histological features[72]; (2) the nuclear bile acid receptor, Farnesoid X receptor (FXR), strongly expressed in bowel and liver, is probably involved in NAFLD pathogenesis, by mediating control of lipids and glucose homeostasis, and controlling bacterial flora growth. Altogether, these effects may induce reduction of hepatic inflammation and fibrogenesis, through different mechanisms. Therefore, recently developed FXR agonists have a potential role in the pharmacological therapy of NAFLD/NASH[73]; (3) toll-like receptors (TLRs) are receptors sensing microbial components of gut microbiota. A number of recent evidences suggests the role of SIBO and increased intestinal permeability in NAFLD, by exposing via portal vein the liver to an high load of intestinal noxae including lipopolysaccharide and other pathogen-associated molecular patterns[74]. Furthermore, TLRs stimulation causes downstream activation of the inflammatory response. Pro-inflammatory patterns result in production of cytokines and chemokines implicated in progression from simple steatosis to steatohepatitis and fibro-cirrhosis; so therapeutic manipulation of innate immune system through TLRs modifiers, formerly evaluated for autoimmune diseases[75], might be a new potential therapeutic target for pediatric NAFLD, but further studies are necessary; and (4) glucagon-like peptid-1 (GLP-1) is an incretin secreted in response to food intake, allotted to multiple functions, including, the stimulation of glucose-dependent insulin secretion and inhibition of glucagon release. The enzyme dipeptidyl peptidase-4 (DPP-4) rapidly degrades circulating GLP-1 (half-life: 1-2 min). Recent animal model and NAFLD adults studies showed an effective role of GLP-1 receptor agonists resistant to DPP-4 (such as exenatide and liraglutide) or DPP-4 inhibitors (e.g., some gliptins) as a promising new therapy in NAFLD for their ability in modulating fatty acid oxidation, decreasing lipogenesis, and improving hepatic glucose metabolism[76].
CONCLUSION
Non-alcoholic fatty liver disease, because of the rise in the prevalence of childhood obesity, is becoming one of the most important chronic liver disease among children. Evidence that only a sub-group of obese patients develop NAFLD suggests that disease progression is likely to depend on complex interplay between environmental factors and genetic predisposition (Figure 1). Recent researches led us to understand the genetic basis predisposing to NAFLD. Many genes have been identified and many other will be identified and, actually, the most important it appears to be PNPLA3 gene. Probably, all the genetic polymorphisms implicated in NAFLD development could have a summarizing effect. In fact, if more predisposing NAFLD polymorphisms coexist in the same subject, the risk to develop NAFLD and to develop it more severely could increase. Other important findings are related to the diet. For example, strong evidence exists that high in fructose intake, usually present in beverages, results in increased de novo lipogenesis and then in increased risk of NAFLD. Moreover, also high n-6/n-3 PUFA ratio consumed in the diet could predispose the NAFLD development. All these findings must drive the clinical practice: the diet is the first and important approach for the NAFLD prevention and treatment. In fact, there is the striking evidence that the weight loss can reduce the effect of I148M polymorphisms on determining hepatic steatosis. In addition to weight loss, to reduce the fructose intake trough the beverages and increasing the n-3 fatty acids dietary intake could be also useful in contrasting the NAFLD.
In conclusion, waiting the new approaches, the dear and old diet is always a fundamental and irreplaceable NAFLD therapy.
Footnotes
P- Reviewers: Borzio M, Celikbilek M, Gatselis NK, Izumi N S- Editor: Zhai HH L- Editor: A E- Editor: Wu HL
Supported by The American Heart Association (AHA), No. 13SDG14640038; 2012 Yale Center for Clinical Investigation (YCCI) scholar award to Santoro N; CTSA Grant Number UL1 RR024139 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH), and NIH roadmap for Medical Research
References
- 1.Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–1393. doi: 10.1542/peds.2006-1212. [DOI] [PubMed] [Google Scholar]
- 2.Aly FZ, Kleiner DE. Update on fatty liver disease and steatohepatitis. Adv Anat Pathol. 2011;18:294–300. doi: 10.1097/PAP.0b013e318220f59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]
- 4.Day CP. Non-alcoholic fatty liver disease: a massive problem. Clin Med. 2011;11:176–178. doi: 10.7861/clinmedicine.11-2-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nobili V, Reale A, Alisi A, Morino G, Trenta I, Pisani M, Marcellini M, Raucci U. Elevated serum ALT in children presenting to the emergency unit: Relationship with NAFLD. Dig Liver Dis. 2009;41:749–752. doi: 10.1016/j.dld.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez G, Gallego S, Breidenassel C, Moreno LA, Gottrand F. Is liver transaminases assessment an appropriate tool for the screening of non-alcoholic fatty liver disease in at risk obese children and adolescents? Nutr Hosp. 2010;25:712–717. [PubMed] [Google Scholar]
- 7.Tominaga K, Kurata JH, Chen YK, Fujimoto E, Miyagawa S, Abe I, Kusano Y. Prevalence of fatty liver in Japanese children and relationship to obesity. An epidemiological ultrasonographic survey. Dig Dis Sci. 1995;40:2002–2009. doi: 10.1007/BF02208670. [DOI] [PubMed] [Google Scholar]
- 8.Pacifico L, Poggiogalle E, Cantisani V, Menichini G, Ricci P, Ferraro F, Chiesa C. Pediatric nonalcoholic fatty liver disease: A clinical and laboratory challenge. World J Hepatol. 2010;2:275–288. doi: 10.4254/wjh.v2.i7.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadeau KJ, Klingensmith G, Zeitler P. Type 2 diabetes in children is frequently associated with elevated alanine aminotransferase. J Pediatr Gastroenterol Nutr. 2005;41:94–98. doi: 10.1097/01.mpg.0000164698.03164.e5. [DOI] [PubMed] [Google Scholar]
- 10.Manco M, Marcellini M, Devito R, Comparcola D, Sartorelli MR, Nobili V. Metabolic syndrome and liver histology in paediatric non-alcoholic steatohepatitis. Int J Obes (Lond) 2008;32:381–387. doi: 10.1038/sj.ijo.0803711. [DOI] [PubMed] [Google Scholar]
- 11.Sundaram SS, Zeitler P, Nadeau K. The metabolic syndrome and nonalcoholic fatty liver disease in children. Curr Opin Pediatr. 2009;21:529–535. doi: 10.1097/MOP.0b013e32832cb16f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giorgio V, Prono F, Graziano F, Nobili V. Pediatric non alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013;13:40. doi: 10.1186/1471-2431-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barshop NJ, Sirlin CB, Schwimmer JB, Lavine JE. Review article: epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;28:13–24. doi: 10.1111/j.1365-2036.2008.03703.x. [DOI] [PubMed] [Google Scholar]
- 14.Spiteller G. Linoleic acid peroxidation--the dominant lipid peroxidation process in low density lipoprotein--and its relationship to chronic diseases. Chem Phys Lipids. 1998;95:105–162. doi: 10.1016/s0009-3084(98)00091-7. [DOI] [PubMed] [Google Scholar]
- 15.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 16.Poustchi H, George J, Esmaili S, Esna-Ashari F, Ardalan G, Sepanlou SG, Alavian SM. Gender differences in healthy ranges for serum alanine aminotransferase levels in adolescence. PLoS One. 2011;6:e21178. doi: 10.1371/journal.pone.0021178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology. 2009;50:1282–1293. doi: 10.1002/hep.23119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobanova YS, Scherbakov AM, Shatskaya VA, Evteev VA, Krasil’nikov MA. NF-kappaB suppression provokes the sensitization of hormone-resistant breast cancer cells to estrogen apoptosis. Mol Cell Biochem. 2009;324:65–71. doi: 10.1007/s11010-008-9985-0. [DOI] [PubMed] [Google Scholar]
- 19.Xu JW, Gong J, Chang XM, Luo JY, Dong L, Jia A, Xu GP. Effects of estradiol on liver estrogen receptor-alpha and its mRNA expression in hepatic fibrosis in rats. World J Gastroenterol. 2004;10:250–254. doi: 10.3748/wjg.v10.i2.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuutila P, Knuuti MJ, Mäki M, Laine H, Ruotsalainen U, Teräs M, Haaparanta M, Solin O, Yki-Järvinen H. Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography. Diabetes. 1995;44:31–36. doi: 10.2337/diab.44.1.31. [DOI] [PubMed] [Google Scholar]
- 21.Donahue RP, Bean JA, Donahue RA, Goldberg RB, Prineas RJ. Insulin response in a triethnic population: effects of sex, ethnic origin, and body fat. Miami Community Health Study. Diabetes Care. 1997;20:1670–1676. doi: 10.2337/diacare.20.11.1670. [DOI] [PubMed] [Google Scholar]
- 22.Lovejoy JC, Champagne CM, de Jonge L, Xie H, Smith SR. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int J Obes (Lond) 2008;32:949–958. doi: 10.1038/ijo.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camporez JP, Jornayvaz FR, Lee HY, Kanda S, Guigni BA, Kahn M, Samuel VT, Carvalho CR, Petersen KF, Jurczak MJ, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. 2013;154:1021–1028. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham RC, Burke A, Stettler N. Ethnic and sex differences in the association between metabolic syndrome and suspected nonalcoholic fatty liver disease in a nationally representative sample of US adolescents. J Pediatr Gastroenterol Nutr. 2009;49:442–449. doi: 10.1097/MPG.0b013e31819f73b4. [DOI] [PubMed] [Google Scholar]
- 25.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortez-Pinto H, Jesus L, Barros H, Lopes C, Moura MC, Camilo ME. How different is the dietary pattern in non-alcoholic steatohepatitis patients? Clin Nutr. 2006;25:816–823. doi: 10.1016/j.clnu.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Toshimitsu K, Matsuura B, Ohkubo I, Niiya T, Furukawa S, Hiasa Y, Kawamura M, Ebihara K, Onji M. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition. 2007;23:46–52. doi: 10.1016/j.nut.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Mozaffarian D, Wu JH. Omega-3 fatty acids and cardiovascular disease: effects on risk factors, molecular pathways, and clinical events. J Am Coll Cardiol. 2011;58:2047–2067. doi: 10.1016/j.jacc.2011.06.063. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Chen D. The optimal dose of omega-3 supplementation for non-alcoholic fatty liver disease. J Hepatol. 2012;57:468–469; author reply 468-469;. doi: 10.1016/j.jhep.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 30.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 31.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 32.Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med. 2008;10:160. [PMC free article] [PubMed] [Google Scholar]
- 33.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. 2009;58:1538–1544. doi: 10.1136/gut.2008.171280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santoro N, Feldstein AE, Enoksson E, Pierpont B, Kursawe R, Kim G, Caprio S. The association between hepatic fat content and liver injury in obese children and adolescents: effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care. 2013;36:1353–1360. doi: 10.2337/dc12-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoro N, Kursawe R, D’Adamo E, Dykas DJ, Zhang CK, Bale AE, Calí AM, Narayan D, Shaw MM, Pierpont B, et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. 2010;52:1281–1290. doi: 10.1002/hep.23832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giudice EM, Grandone A, Cirillo G, Santoro N, Amato A, Brienza C, Savarese P, Marzuillo P, Perrone L. The association of PNPLA3 variants with liver enzymes in childhood obesity is driven by the interaction with abdominal fat. PLoS One. 2011;6:e27933. doi: 10.1371/journal.pone.0027933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, Hobbs HH. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen AM, Teitelbaum A. Effect of glucose, fructose, sucrose and starch on lipgenesis in rats. Life Sci. 1968;7:23–29. doi: 10.1016/0024-3205(68)90343-3. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res. 2005;46:2477–2487. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Polson DA, Thompson MP. Adiponutrin mRNA expression in white adipose tissue is rapidly induced by meal-feeding a high-sucrose diet. Biochem Biophys Res Commun. 2003;301:261–266. doi: 10.1016/s0006-291x(02)03027-9. [DOI] [PubMed] [Google Scholar]
- 45.Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes. 2006;55:148–157. [PMC free article] [PubMed] [Google Scholar]
- 46.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 47.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marzuillo P, Grandone A, Perrone L, del Giudice EM. Weight loss allows the dissection of the interaction between abdominal fat and PNPLA3 (adiponutrin) in the liver damage of obese children. J Hepatol. 2013;59:1143–1144. doi: 10.1016/j.jhep.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 49.Santoro N, Savoye M, Kim G, Marotto K, Shaw MM, Pierpont B, Caprio S. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS One. 2012;7:e37827. doi: 10.1371/journal.pone.0037827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zampino R, Coppola N, Cirillo G, Boemio A, Pisaturo M, Marrone A, Macera M, Sagnelli E, Perrone L, Adinolfi LE, et al. Abdominal fat interacts with PNPLA3 I148M, but not with the APOC3 variant in the pathogenesis of liver steatosis in chronic hepatitis C. J Viral Hepat. 2013;20:517–523. doi: 10.1111/jvh.12053. [DOI] [PubMed] [Google Scholar]
- 51.Dubuquoy C, Burnol AF, Moldes M. PNPLA3, a genetic marker of progressive liver disease, still hiding its metabolic function? Clin Res Hepatol Gastroenterol. 2013;37:30–35. doi: 10.1016/j.clinre.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 52.Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781–789. doi: 10.1002/hep.24806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, Teranishi H, Mizusawa S, Ueno T, Chayama K, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. 2013;132:783–792. doi: 10.1007/s00439-013-1294-3. [DOI] [PubMed] [Google Scholar]
- 55.Adams LA, White SW, Marsh JA, Lye SJ, Connor KL, Maganga R, Ayonrinde OT, Olynyk JK, Mori TA, Beilin LJ, et al. Association between liver-specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology. 2013;57:590–600. doi: 10.1002/hep.26184. [DOI] [PubMed] [Google Scholar]
- 56.Nobili V, Alisi A, Raponi M. Pediatric non-alcoholic fatty liver disease: preventive and therapeutic value of lifestyle intervention. World J Gastroenterol. 2009;15:6017–6022. doi: 10.3748/wjg.15.6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology. 2013;57:2525–2531. doi: 10.1002/hep.26299. [DOI] [PubMed] [Google Scholar]
- 58.Zelber-Sagi S, Ratziu V, Oren R. Nutrition and physical activity in NAFLD: an overview of the epidemiological evidence. World J Gastroenterol. 2011;17:3377–3389. doi: 10.3748/wjg.v17.i29.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vajro P, Mandato C, Franzese A, Ciccimarra E, Lucariello S, Savoia M, Capuano G, Migliaro F. Vitamin E treatment in pediatric obesity-related liver disease: a randomized study. J Pediatr Gastroenterol Nutr. 2004;38:48–55. doi: 10.1097/00005176-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 60.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, Abrams SH, Scheimann AO, Sanyal AJ, Chalasani N, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305:1659–1668. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwimmer JB, Middleton MS, Deutsch R, Lavine JE. A phase 2 clinical trial of metformin as a treatment for non-diabetic paediatric non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2005;21:871–879. doi: 10.1111/j.1365-2036.2005.02420.x. [DOI] [PubMed] [Google Scholar]
- 63.Musso G, Gambino R, Cassader M. Gut microbiota as a regulator of energy homeostasis and ectopic fat deposition: mechanisms and implications for metabolic disorders. Curr Opin Lipidol. 2010;21:76–83. doi: 10.1097/MOL.0b013e3283347ebb. [DOI] [PubMed] [Google Scholar]
- 64.Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, Groen AK, Hoekstra JB, Stroes ES, Nieuwdorp M. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:112–120. doi: 10.1111/j.1463-1326.2011.01483.x. [DOI] [PubMed] [Google Scholar]
- 65.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schrezenmeir J, de Vrese M. Probiotics, prebiotics, and synbiotics--approaching a definition. Am J Clin Nutr. 2001;73:361S–364S. doi: 10.1093/ajcn/73.2.361s. [DOI] [PubMed] [Google Scholar]
- 67.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 68.Esposito E, Iacono A, Bianco G, Autore G, Cuzzocrea S, Vajro P, Canani RB, Calignano A, Raso GM, Meli R. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J Nutr. 2009;139:905–911. doi: 10.3945/jn.108.101808. [DOI] [PubMed] [Google Scholar]
- 69.Loguercio C, Federico A, Tuccillo C, Terracciano F, D’Auria MV, De Simone C, Del Vecchio Blanco C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39:540–543. doi: 10.1097/01.mcg.0000165671.25272.0f. [DOI] [PubMed] [Google Scholar]
- 70.Masterton GS, Plevris JN, Hayes PC. Review article: omega-3 fatty acids - a promising novel therapy for non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2010;31:679–692. doi: 10.1111/j.1365-2036.2010.04230.x. [DOI] [PubMed] [Google Scholar]
- 71.Huang JS, Barlow SE, Quiros-Tejeira RE, Scheimann A, Skelton J, Suskind D, Tsai P, Uko V, Warolin JP, Xanthakos SA. Childhood obesity for pediatric gastroenterologists. J Pediatr Gastroenterol Nutr. 2013;56:99–109. doi: 10.1097/MPG.0b013e31826d3c62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li W, Zheng L, Sheng C, Cheng X, Qing L, Qu S. Systematic review on the treatment of pentoxifylline in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2011;10:49. doi: 10.1186/1476-511X-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuchs M. Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid x receptor as an emerging treatment target. J Lipids. 2012;2012:934396. doi: 10.1155/2012/934396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 76.Lee J, Hong SW, Rhee EJ, Lee WY. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab J. 2012;36:262–267. doi: 10.4093/dmj.2012.36.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]