

Abstract
Vitamin D deficiency is commonly diagnosed among patients with inflammatory bowel disease (IBD). Patients with IBD are at risk of low bone density and increased fractures due to low vitamin D levels, long standing disease, and frequent steroid exposures; as a result, it is well established that vitamin D supplementation in this population is important. There is increasing support for the role of vitamin D in strengthening the innate immune system by acting as an immunomodulator and reducing inflammation in experimental and human IBD. The active form of vitamin D, 1,25(OH)D3, acts on T cells to promote T helper (Th)2/regulatory T responses over Th1/Th17 responses; suppresses dendritic cell inflammatory activity; induces antibacterial activity; and regulates cytokine production in favor of an anti-inflammatory response. Murine and human IBD studies support a therapeutic role of vitamin D in IBD. Risk factors for vitamin D deficiency in this population include decreased sunlight exposure, disease duration, smoking, and genetics. Vitamin D normalization is associated with reduced risk of relapse, reduced risk of IBD-related surgeries, and improvement in quality of life. Vitamin D is an inexpensive supplement which has been shown to improve IBD outcomes. However, further research is required to determine optimal serum vitamin D levels which will achieve beneficial immune effects, and stronger evidence is needed to support the role of vitamin D in inducing disease response and remission, as well as maintaining this improvement in patients’ disease states.
Keywords: Vitamin D, Inflammatory bowel disease, Immune response, Inflammation, Cytokines, Supplementation
Core tip: There is support for the importance of maintaining normal vitamin D levels in inflammatory bowel disease (IBD) patients, demonstrated by its anti-inflammatory actions in the gut. A randomized controlled trial examined the impact of vitamin D supplementation on IBD outcomes and demonstrated a reduced risk of relapse in vitamin D-treated Crohn’s disease patients. Furthermore, vitamin D3 and active vitamin D have been shown to reduce clinical disease activity and improve quality of life in IBD patients. Normalization of vitamin D levels is also associated with a decreased risk for IBD-related surgery. This vitamin has therapeutic benefit in IBD patients.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the intestine that causes abdominal pain, diarrhea, and weight loss and includes two forms, Crohn’s disease and ulcerative colitis[1]. IBD reduces quality of life and may cause financial stress by increasing disability and decreasing the capacity for work[1,2]. The disease is complex and etiology is not completely understood; however, IBD is associated with abnormal immune responses to the body’s natural intestinal bacteria[1,2], which activate the gastrointestinal immune system. It is expected that genetic and environmental interactions play a role in the susceptibility of IBD[1,2]. As there is currently no cure for IBD, medical therapy remains the mainstay treatment for achieving and maintaining remission[2].
It is well-established that vitamin D plays a critical role in improving bone health and is specifically important for patients who are at risk of low bone density. Low bone mineral density is more prevalent among patients with Crohn’s disease and ulcerative colitis compared to healthy controls[3]. Long standing disease, multiple steroid exposures, and low serum 25-hydroxycholecalciferol [25(OH)D3] levels are contributing factors to decreased bone mineral density and increased fractures in patients with IBD[3-5]. Therefore, vitamin D supplementation is essential in this high-risk group. There is, however, growing support for non-traditional actions of vitamin D including anti-inflammatory, anti-proliferative, cell differentiation, and apoptotic effects[6]. These effects have led to examination of vitamin D in the pathogenesis of autoimmune diseases such as IBD[6]. This review will address the regulatory role of vitamin D on immune responses and describe its relevance in regards to inflammatory bowel disease.
VITAMIN D PHYSIOLOGY
Vitamin D metabolism
Vitamin D is present in two major forms. Vitamin D2 (ergocalciferol) is present in plants, yeast, and fungi[6,7], while vitamin D3 (cholecalciferol) can be obtained from animal sources such as oily fish and egg yolk[6-8]. Vitamin D3 is also synthesized endogenously in the skin upon ultraviolet light exposure. Sun light exposure to the skin results in a photochemical conversion of 7-dehydrocholesterol to pre-vitamin D, which then rapidly converts to cholecalciferol. This process is self-limiting to prevent toxicity[6,8].
Vitamin D is fat soluble and absorbed in the small intestine along with dietary fat. After incorporation into chylomicrons, it is rapidly delivered into the venous circulation[3,4]. It is then transported by vitamin D binding proteins (DBPs) to the liver where it is converted to 25(OH)D3 by hepatic 25-hydroxylase[6,7,9]. 25(OH)D3 is inactive, however, it is the main circulating form of vitamin D and the best indicator of vitamin D status[6,9]. Vitamin D is primarily stored in the liver as well as in adipose tissue. Once saturation of these tissues occur, 25(OH)D3 is released to circulate in the blood[4], where it is predominantly bound by DBPs and albumin, leaving little in the free form[3,7]. DBPs transport 25(OH)D3 to the kidney, where it is converted to its active hormonal form, 1α,25-dihydroxycholecalciferol [1,25(OH)2D3], by the enzyme 25-hydroxyvitamin D3-1α-hydroxylase[6-9]. It can now act on its receptor, the vitamin D receptor, in many target tissues, including the intestine, kidney, and bone, thereby altering transcription of target genes[10,11]. In the target tissue, 24-hydroxylase catabolizes 1,25(OH)2D3 and 25(OH)D3 into their inactive metabolites, which are then excreted as calcitroic acid in the urine[3,8,10].
The rate limiting enzyme in the metabolism of vitamin D is 1α-hydroxylase. This enzyme is tightly regulated by plasma parathyroid hormone (PTH) and 1,25(OH)2D3. Active vitamin D production in the kidneys is directed by PTH[4,6,8,9], which upregulates transcription of CYP27B1, the gene encoding for 1α-hydroxylase[9]. This results in an increased production of 1,25(OH)2D3 in the kidney. In turn, 1,25(OH)2D3 takes part in a negative feedback loop to suppress the transcription of PTH and CYP27B1, thereby decreasing production of 1,25(OH)2D3[9]. Simultaneously, 1,25(OH)2D3 induces 24-hydroxylase production[8,9]. This is an autoregulatory mechanism to suppress the actions of 1,25(OH)2D3[9]. An overview of vitamin D sources and metabolism is outlined in Figure 1.
Figure 1.
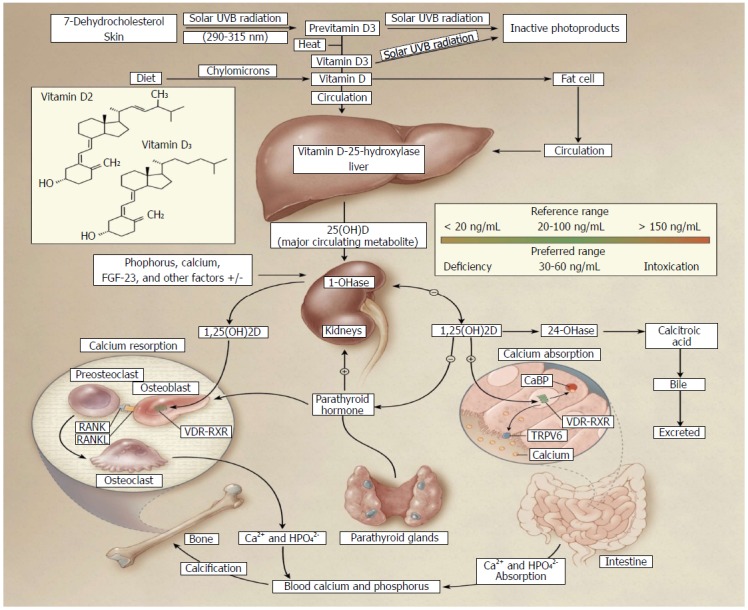
Vitamin D sources and metabolism[8]. Vitamin D can be obtained either from the diet or synthesized in the skin. Under solar ultraviolet (UVB) radiation, 7-dehydrocholesterol in the skin is converted into cholecalciferol (vitamin D3). Vitamin D from the diet enters chylomicrons, which transport it into circulation. Vitamin D is stored in adipose tissue, but when released into circulation, vitamin D binding proteins direct it to the liver where it is converted into its major circulating form, 25-hydroxyvitamin D3 [25(OH)D3] by 25-hydroxylase. In the kidneys, 25(OH)D3 is converted into 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active form, by 1α-hydroxylase. It can now exert its biological effects including calcium absorption, resorption, and bone development. Parathyroid hormone released from the parathyroid glands upregulates hepatic conversion of 1,25(OH)2D3 by stimulating 1α-hydroxylase production; however, autoregulatory mechanisms suppress these actions through negative feedback loops. 1,25(OH)2D3 suppresses parathyroid hormone and 1α-hydroxylase production. Vitamin D is then catabolized by 24-hydroxylase and excreted as calcitroic acid. The recommended optimal range for vitamin D levels is 30-60 ng/mL (75-150 nmol/L). Copyright© 2007 Massachusetts Medical Society. All rights reserved; VDR: Vitamin D receptor; RXR: Retinoid X receptor.
Vitamin D receptor
The vitamin D receptor (VDR) plays an important role in how vitamin D exerts its biological effects. It belongs to a superfamily of nuclear hormone receptors and is specifically activated by 1,25(OH)2D3[11,12]. In response to 1,25(OH)2D3 binding, VDRs regulate gene transcription, thereby producing specific proteins to carry out vitamin D3 biological activity. The DNA binding domain of the zinc finger recognizes vitamin D response elements (VDREs), which are specific DNA sequences on cell-targeted genes[11]. 1,25(OH)2D3 binding results in the formation of the VDR and retinoid X receptor (RXR) heterodimer[12]. This complex binds to VDREs and recruits large coregulatory complexes to these specific genes through the VDR transactivation domain[11,12]. The activities of coregulatory complexes may include nucleosomal remodeling, selective chromatic histone modification, or RNA polymerase II recruitment and initiation. All of these activities work to enhance or suppress gene expression[11].
Multiple tissues and immune cells express VDRs and the enzymes needed to produce local 1,25(OH)2D3[6,13-15]. This enzyme activity is regulated in a different manner compared to the enzymatic renal production of 1,25(OH)2D3; it is no longer under an endocrine feedback mechanism, but is induced by other factors[6]. These findings have led to the examination of multiple roles of vitamin D in the pathogenesis of autoimmune diseases such as IBD[6].
EFFECTS OF VITAMIN D ON THE IMMUNE SYSTEM
Vitamin D is important in both the innate and adaptive immune systems[13]. Immune cells express VDRs and the enzymes necessary to convert vitamin D3 and 25(OH)D3 into 1,25(OH)2D3, wherein locally produced 1,25(OH)2D3 can exert specific autocrine and paracrine effects without producing unnecessary systemic effects[16,17]. 1,25(OH)2D3 can modulate the adaptive immune responses by altering the actions of activated T and B cells, and it can modulate the innate immune responses by regulating macrophages and dendritic cells[16].
Vitamin D and T-cell differentiation
It has been established that vitamin D is an immune system regulator through its role in targeting CD4+ cells of T lymphocytes to suppress T helper type 1 (Th1) cell driven immune responses[17-19]. Th1 cells produce pro-inflammatory cytokines including IFN-γ, interleukin (IL)-2, and tumour necrosis factor-alpha (TNF-α), which are important for reducing intracellular infections. 1,25(OH)2D3 works to inhibit the over production of these pro-inflammatory cytokines[19].
Overall, the main action of 1,25(OH)2D3 on T-cells is mediating T helper type 1 (Th1)/T helper type 2 (Th2) development and differentiation[17,18]. Vitamin D3 affects the Th1-Th2 balance in favour of Th2 cell development. The development into either Th1 or Th2 from CD4+ T cells is directed by cytokines[18]. Cytokine IL-12 induces Th1 cell development whereas IL-4 induces Th2 cell development. The effects these cytokines have on the Th1-Th2 balance determines which cytokines will be produced and therefore determine the type of immune response. Th1 cells produce pro-inflammatory IFN-γ and lymphotoxin, and Th2 produces anti-inflammatory IL-4, IL-5, and IL-13[17,18]. The increased development of Th2 is a result of direct action of vitamin D3 on CD4+ cells[18], whereas the reduction in Th1 cell development is due to the effects of vitamin D3 on dendritic cells[17,18]. Using mice Ag-specific T cells, Boonstra et al[18] demonstrate that vitamin D3 increases the frequency of IL-4 producing cells in vitro from 8.0%-55.8%, thereby supporting a vitamin D induced Th2 profile. This study also shows that vitamin D3 increases IL-10 and IL-5 producing cells and decreases IFN-γ producing cells[18]. The role of vitamin D in regulating cytokine production has been suggested to limit inflammatory tissue damage, but it also reduces bacterial killing. IFN-γ augments toll-like receptor (TLR) 2/1 induced expression of CYP27B1 and bacterial killing, and IL-4 depresses TLR 2/1 activation of bacterial killing[13]. Therefore, a balanced response is important.
At the transcription level, c-maf and GATA-3 are the transcription factors relating to Th2 development[18]. Vitamin D3 can directly target CD4+ cells to promote Th2 development at the level of transcription[17,18]. In vitro studies show a correlation between increased expression of GATA-3 and c-maf and Th2 cytokine levels, IL-5, IL-10, and IL-4, after vitamin D3 treatment. In addition, there is a reduction in IFN-γ[18]. Boonstra et al[18] conclude that vitamin D likely functions to diminish cell-mediated immune responses by skewing toward a Th2 phenotype, which fights extracellular infections.
Th17 cells are another vitamin D target because of their ability to produce IL-17, a pro-inflammatory cytokine. Vitamin D reduces inflammatory tissue damage by suppressing Th17 development and in doing so, reduces IL-17 production[13]. Furthermore, 1,25(OH)2D3 increases the development of T regulatory cells by acting on naïve CD4+ cells. Regulatory T cells are a group of CD4+ T cells that have immunosuppressive properties by depressing the proliferation of other CD4+ T cells[13]. Vitamin D in combination with another immunosuppressive drug, dexamethasone, has been shown to increase IL-10 producing regulatory T cells. This supports the idea that regulatory T cells located at sites of inflammation downregulate the immune response[19].
Research supports the immunosuppressive effect of vitamin D due to its involvement in T-cell differentiation. The evidence shows that vitamin D plays a role in maintaining a balance between the inflammatory response of Th1/Th17 cells and the immunosuppressive response from Th2/Treg cells.
Vitamin D and dendritic cells
Dendritic cells (DCs) are antigen presenting cells (APCs) and are important in initiating CD4+ T cells responses[20]. Vitamin D inhibits differentiation and maturation of human DCs in vitro[20] by suppressing the IL-12 production from DCs and increasing IL-10 production[17,20]. This is an important immunosuppressive activity as IL-12 is an important cytokine in inducing Th1 development[17,18]. 1,25(OH)2D3 inhibits the differentiation, maturation, and immunostimulatory capacity of DCs[21]. Canning et al[21] confirm that 1,25(OH)2D3 suppresses monocyte differentiation into DCs, thereby generating immature DCs. This suppresses DC ability to stimulate T-cell proliferation.
Lipopolysaccharide (LPS) is a component of the Gram-negative bacterial wall, which induces monocytes/macrophages to produce cytokines. After LPS stimulation, DCs and monocyte-derived macrophages (MACs) have a high local 1-α-hydroxylase production of 1,25(OH)2D3, which positively modulates MAC differentiation and depresses actions of DCs and lymphocytes[14]. This mechanism facilitates a normal immune response as some DCs still mature; vitamin D prevents over stress of this immune response that may lead to pathological effects[13]. Vitamin D skews the development of the immune response towards a non-specific innate immune response and away from an antigen-specific immune response.
Antibacterial activity of vitamin D
As a fundamental part of the innate immune response, human monocytes have been shown to locally produce 1,25(OH)2D3, which triggers increased autophagy, an important mechanism for eliminating pathogens by antibacterial proteins[15]. TLR expressed on monocytes recognize pathogens, and under TLR 2/1 stimulation, CYP27B1, the gene encoding for 1α-hydroxylase, and VDR expression is upregulated[13,15]. 1,25(OH)2D3 acts on monocytes to induce expression of the cathelicidin antimicrobial peptide (CAMP) gene (LL-37), producing a protein that enhances intracellular killing of bacteria[13,15,22]. The CAMP gene is a direct target of the vitamin D receptor[22]. Furthermore, nucleotide-binding oligomerization domain-containing protein 2 (NOD2), a pattern recognition receptor, has an important role in inducing antibacterial activity. NOD2 activation by muramyl dipeptide, a product of Gram-negative and Gram-positive bacteria, stimulates transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which induces gene expression for antimicrobial peptide defensin β2 (DEFB2)[13,23]. Interestingly, 1,25(OH)2D3 induces NOD2 expression in many cell types, thereby increasing cell sensitivity to these bacteria products; as a result 1,25(OH)2D3 enhances NOD2 mediated DEFB2 transcription[13,23].
Anti-inflammatory role of vitamin D
As a part of the innate immune system, macrophages release proinflammatory cytokines and chemokines upon stimulation. This leads to an inflammatory response to protect the body from pathogenic microorganisms[24]. TNF-α is a proinflammatory cytokine that is produced early in the course of inflammation by macrophages and lymphocytes. It is involved in autoimmune diseases, including IBD[25]. Proinflammatory cytokines are positive for host defense, but overproduction leads to unresolved inflammation[26]. Muller et al[27] examine the effect of 1,25(OH)2D3 on LPS-stimulated human blood monocytes, and find it inhibits the release of IL-1α, IL-6, and TNF-α. LPS binds to TLR4 on monocytes to mediate activation of mitogen-activated protein kinase (MAPK)[26]. MAPKs are critical regulators of proinflammatory cytokine production, including IL-6 and TNF-α. 25(OH)D3 treatment has been shown to inhibit LPS-induced IL-6 and TNF-α production in peripheral blood mononuclear cells (PBMC) of healthy donors via upregulation of MKP-1 (MAPK phosphatase-1). MKP-1 switches off cytokine production in monocytes/macrophages after inflammatory stimuli. Interestingly, doses of 25(OH)D3 that relate to vitamin D sufficiency (> 30 ng/mL or 75 nmol/L) significantly inhibited mRNA production of these cytokines[26].
Vitamin D analogues have also been proven to have immunomodulatory effects. Stio et al[28] demonstrate the vitamin D analogue KH 1060 and a monoclonal anti-TNF-α antibody to work synergistically in significantly inhibiting PBMC proliferation and TNF-α production in healthy subjects. In vitro experiments using PBMCs from healthy volunteers were also able to demonstrate the effect of paricalcitol, a vitamin D analogue, in reducing both basal TNF-α and LPS-induced TNF-α[25].
The importance of VDRs in the inflammatory response has also been demonstrated. Chen et al[24] report a more robust and prolonged production of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β in bone marrow derived macrophages (BMDMs) from VDR-/- mice compared to wild type (WT) mice after LPS exposure. This suggests a dysregulated and over sustained innate immune response in macrophages under attenuated VDR signalling. Interestingly, 1,25(OH)2D3 and its analogue, paricalcitol, have been shown to reduce LPS-induced TNF-α and IL-6 cytokines in WT BMDMs[24].
Chen et al[24] have also examined novel anti-inflammatory effects of vitamin D by investigating its microRNA-155-SOCS1 target. MicroRNAs (miRNAs) are small noncoding RNAs that control gene expression. Recently, miRNA-155 has been shown to regulate innate immune responses and TLR signaling. It targets the “suppressor of cytokine signalling” (SOCS) family of proteins, specifically SOCS1. miRNA profiling in mice cells was examined after the treatment with LPS, with or without 1,25(OH)2D3. As a result, miR-155 increased the most with LPS and was suppressed the most by vitamin D; miR-155 were elevated in VDR -/- bone marrow derived macrophages (BMDMs) after exposure to LPS compared to the WT BMDMs; and 1,25(OH)2D3 suppressed the induction of miR-155 by LPS in these cells. Additionally, 1,25(OH)2D3 blocked TNF-α, IL-6, and miR-155 induction in human PBMCs. Overall, 1,25(OH)2D3 was found to upregulate SOCS1 through its suppression of miR-155. SOCS1 is important in the negative feedback regulation of LPS-induced inflammation and inhibits TNF-α, IL-6, and IFN-γ pathways. In the absence of VDRs, the negative feedback loop is dysregulated[24].
Seasonal variations in serum vitamin D levels have an effect on the innate immune response. The increase in serum vitamin D levels during the summer months is associated with a significant drop in LPS-induced TNF-α (64%), IL-6 (33%), IL-1β (59%), and IFN-γ (46%) from PBMCs in healthy individuals in vivo compared to LPS-induced levels during the winter months[29]. Caution does need to be taken when considering the difference in physiological up-regulation of vitamin D levels by solar radiation and the doses of vitamin D employed in vitro and in vivo; however, this data does support how the innate immune response can be regulated by the physiological variation of serum vitamin D3 levels during the four seasons of the year[29].
Role of vitamin D in gastrointestinal inflammation
There is support for the role of vitamin D in the immune system, particularly in reducing inflammatory responses. Recently, more studies have demonstrated the role for vitamin D specifically in gastrointestinal inflammation and suggest deficiency is associated with IBD.
The importance of vitamin D in reducing the proinflammatory profile of IBD patients is demonstrated by the impact of cytokine-induced apoptosis and cytokine disruption of epithelial barrier function. Epithelial apoptosis occurs as a normal physiological event in the gastrointestinal tract and the mucosal barrier function is still maintained; however, cytokine-induced apoptosis may disrupt the barrier, leading to abnormal mucosal permeability, a common occurrence among IBD patients[30]. Bruewer et al[30] address the mechanisms by which proinflammatory cytokines disrupt the barrier through non-apoptotic mechanisms. IFN-γ was demonstrated to reduce epithelial gate function, and the effects were increased when combined with TNF-α, resulting in intestinal epithelium paracellular permeability. The authors suggest this allows for the barrier function to quickly normalize when inflammatory cytokines are reduced[30], stressing the importance of medical therapy in maintaining disease remission.
There is evidence to suggest that there are distinct cytokine profiles in Crohn’s disease and ulcerative colitis. Papadakis et al[31] report Crohn’s disease to show a Th1 type of immune response with elevated IL-12, TNF-α, and IFN-γ cytokines, whereas ulcerative colitis presents with increased IL-5 secretion. Specific pro-inflammatory cytokines have been identified in the inflamed mucosa of Crohn’s disease and ulcerative colitis patients such as IL-1, IL-6, IL-8, and TNF-α[31]. Each of these cytokines upregulate the inflammatory cascade leading to more inflammation and tissue damage in the inflamed mucosa[31]. Vitamin D has been shown to target these inflammatory pathways.
TNF-α is a central cytokine in the pathogenesis of inflammatory bowel disease[31]. In the review by Papadakis et al[31], they report three lines of evidence to support the importance of TNF-α in IBD. They explain that anti-TNF therapy has been very successful in treatment of IBD in humans and in animal models, and that a “Crohn’s like” phenotype is expressed in mice that over express TNF-α[31]. Interestingly, a vitamin D analogue has recently been shown to work synergistically with infliximab, an anti-TNF therapy used in the treatment of IBD, to reduce the cytokine TNF-α in human peripheral blood monocytes[28]. Treatment with 1,25(OH)2D3 in the colonic tissue of IL-10 knock-out (KO) mice has also been shown to down-regulate TNF-α-associated genes in these mice[32]. Furthermore, in vitro studies of CD4+ T-cells of healthy controls and patients with Crohn’s disease have shown that 1,25(OH)2D3 increases the production of anti-inflammatory cytokine IL-10 and decreases the production of pro-inflammatory IFN-γ, supporting a therapeutic role of vitamin D in IBD[33].
Stio et al[34] examine the effects of vitamin D on PBMCs from patients with active Crohn’s disease on infliximab, an anti-TNF therapy. After vitamin D treatment, PMBC proliferation decreased in both responders and non-responders, but to a greater extent in responders[34]. Interestingly, vitamin D analogue treatment increased VDR expression in unresponsive patients and VDR levels did not change in responsive patients. The authors suggest infliximab induces VDR expression in the presence of vitamin D in unresponsive patients; as a result, there are differences in sensitivity to vitamin D between responders and non-responders. This suggests that VDR expression and PMBC proliferation may be useful indicators to predict response of Crohn’s disease patients to infliximab therapy[34].
Studies have gone on to suggest that vitamin D deficiency and deficiency in its signaling pathways are contributing factors in the pathogenesis of IBD. Wu et al[35] propose a mechanism by which vitamin D deficiency may cause IBD through the changes in vitamin D receptor signaling in autophagy homeostasis, including increases in TNF-α-induced autophagy. Another study by Wang et al[23] examine the role of vitamin D in NOD2 expression, as NOD2 deficiency, due to mutations in its gene, has been linked to the pathogenesis of Crohn’s disease[22]. These authors show that 1,25(OH)2D3 signaling induces NOD2 expression in human intestinal epithelial cells, thus supporting the idea that vitamin D deficiency plays a causative role in Crohn’s disease.
As discussed previously, local production of vitamin D is an important part of regulating microenvironment inflammatory profiles. DCs from healthy subjects convert 25(OH)D3 to the active form of vitamin D, 1,25(OH)2D3, which could be important in local control of inflammation in Crohn’s disease patients[36]. Bartels et al[36] isolate peripheral monocytes from 20 Crohn’s disease patients who were vitamin D deficient, which were then matured into DCs in vitro. Addition of the active [1,25(OH)2D3] and inactive [25(OH)D3] vitamin D3 metabolites inhibited LPS-induced DC maturation, and supplementation changed the cytokine profile of LPS-matured DC cultures compared to those not supplemented. The production of TNF-α and IL-12 decreased, while unexpectedly, there was a trend towards reduced IL-10 and IL-6 significantly increased[36]. DCs of Crohn’s disease patients could be exposed to LPS in the gut resulting in similar findings[36]. Furthermore, vitamin D supplementation reduced proliferation of the entire lymphocyte population and the CD4+ lymphocytes; therefore, vitamin D could be protective against Crohn’s disease as inflammation in this disease can be characterized as uncontrolled lymphocyte development[36].
While the addition of vitamin D in vitro changes cytokine profiles, there may be differences in the effects of vitamin D in in vivo studies. A study was conducted on Crohn’s disease patients who were treated with 1200 IU of vitamin D3 per day or placebo for 26 wk to assess differences in induced T-cell mediated immune function between the treatment and placebo groups during in vivo[37]. PBMCs were cultured from patients of each group, and there was a significant increase in production of IL-6 in T cells from vitamin D treated patients. IL-6 significantly correlated with serum vitamin D [25(OH)D3] levels (P = 0.02). In the vitamin D treated patients, there was also a trend to increased IL-4 and no effect on TNF-α and IFN-γ production[37]. There was no change in IL-10 production nor in the amount of T regulatory cells CD4+, CD25+, FoxP3+ T cells; however, the amount of proliferating CD4+ cells significantly increased. IL-6 can induce T cell IL-4 production, which may explain the increase in IL-4[37].
There is strong evidence for a protective effect of vitamin D against IBD related inflammatory responses; however, there are mixed findings on the production of IL-6 with the treatment of vitamin D. IL-6 has been shown to increase in certain situations and decrease in others under vitamin D treatment. Interestingly, in vitro and in vivo studies examining vitamin D treatment of LPS-matured DCs and PBMCs from Crohn’s disease patients show increased IL-6 production, whereas these studies examining healthy volunteers demonstrate a decrease in IL-6 after vitamin D treatment. IL-6 has been reported to support Th17 development, which have important anti-microbial roles[36]. Additionally, IL-6 may have anti-inflammatory roles by reducing DC maturation and increasing T cell IL-4 production[36,37]. Therefore, vitamin D does have immunosuppressive properties; however, instead of isolating vitamin D to this effect, it may be better to suggest it is an immunomodulator that strengthens the innate immune response and depresses the adaptive immune reaction[36], which may explain the different actions in healthy volunteers and IBD patients who have intestinal mucosal injury.
EFFECT OF VITAMIN D IN ANIMAL MODELS OF IBD
VDR KO mice models
Vitamin D and the vitamin D receptor have been shown to have an important role in animal models of colitis. Vitamin D receptor KO mice treated with dextran sodium sulfate (DSS) to induce colitis demonstrate markedly elevated levels of a number of tissue pro-inflammatory cytokines including TNF-α, IL-12p70, and IFN-γ, and have significantly more intestinal injury, fewer intact crypts, more epithelium loss, and more inflammation. This indicates that the absence of VDRs increases the sensitivity of the intestinal mucosa to very low doses of DSS[38], thereby increasing the susceptibility to chemical injury in the gut. This confirms a prominent role of VDR signalling in the regulation of gut inflammation[38].
Oral intake of 1,25(OH)2D3 improves DSS induced colitis in WT mice, and there is an upregulation of anti-inflammatory cytokine IL-10 production, which may help to reduce other cytokine responses[38]. Interestingly, rectal administration of 1,25(OH)2D3 in the WT mice is more effective than oral in regards to decreasing DSS colitis. This is justified by obtaining the same results but only injecting half the dose of vitamin D in the rectum (site of inflammation)[38].
Vitamin D and its receptors play important roles in maintaining gut integrity and protecting the intestine from pathogenic enteric bacterial infection. Wu et al[39] support the role of vitamin D in suppressing bacterial-induced NF-κB activity in the intestine. NF-κB is a regulator of inflammatory cytokines such as IL-6, and after bacterial invasion, VDRs inhibit its actions. Intestinal VDRs protect against bacterial infection, demonstrated by a 6-fold increase in IL-6 and more severe inflammation after a Salmonella in VDR-/- mice compared to VDR+/+ mice[39]. Furthermore, after exposure to Salmonella, mice lacking VDRs had more Salmonella invasion of the intestine compared to WT[39]. Interestingly, VDR expression increased in WT mice as a direct result of an enteric bacterial infection, indicating a role of VDR signalling pathway in responding to specific pathogenic bacteria. VDRs relocated from the surface of colonic epithelial cells to the surface of crypts as well as in the middle and bottom of the crypts after infection[39]. Furthermore, IL-6 was undetectable in VDR+/+ mice and present in VDR-/- mice; as a result, this suggests that VDR-/- mice start with a pre-inflammatory state even before being infected[39].
The intestinal epithelial layer contains a highly specialized immune system and physical barrier to protect against the invasion of pathogens. Intraepithelial lymphocytes (IELs) are found in the intestinal epithelial layer, and they are an important part of maintaining intestinal integrity[40]. Specific IELs, CD8 αα+ T cells, aid in the maintenance of gut health. VDR KO mice have fewer CD8 αα+ T cells in the gut, implying VDRs are important in regulating the growth and maintenance of CD8 αα+ T cells[40]. Moreover, VDR-/- mice have significantly reduced intestinal transepithelial resistance (TER) compared to VDR+/+ mice. TER is reduced when epithelial barrier function is compromised, making it a good indicator of this malfunction. Additionally, tight junctions and desmosomes are severely disrupted in VDR-/- colonic mucosa[41]. The breakdown of the physical barrier that separates the host from its gastrointestinal microorganisms results in intestinal permeability and the development of IBD. These studies demonstrate prominent role of VDR signalling in maintaining the integrity of the epithelial barrier in the intestine and in its protection against IBD[42].
Increased IL-17 production has also been reported in VDR null mice. Th17 cells are pro-inflammatory and have been associated with increased severity of IBD in animal models. VDR KO mice have significantly higher induced Th17 cell and IL-17 production compared to WT mice[43]. Additionally, 1,25(OH)2D3-deficient T cells overproduce IL-17 compared with WT cells[43]. Recombinase-activated gene 2 knockout mice lack mature T and B cells. When WT CD4+ T cells are transferred to VDR/Rag double KO mice, the colonic sections of these recipients show significantly more hyperplasia and inflammation compared to Rag KO recipients[43]. There is an increased expression of IL-17 secreting cells in the gut and periphery of these double KO mice, suggesting VDRs on accessory cells direct Th17 development[43].
Conversely, there are some limitations of vitamin D on host susceptibility to pathogens. While increased production of Th1/Th17 cytokines is related to the pathogenesis of Crohn’s disease, Th1/Th17 responses are critical for the removal of many infectious bacteria[44]. Examining the role of 1,25(OH)2D3 in Citrobacter rodentium (C. rodentium) infected mice, Ryz et al[44] demonstrate that mice treated with 1,25(OH)2D3 have significantly increased scores for edema, goblet cell depletion, hyperplasia and infiltrating inflammatory cells compared to vehicle-treated mice[44]. These mice also have a reduced number of Th17 T cells in their colons and decreased production of Th17 associated antimicrobial peptide TEG3γ, which works as a host defense against infection with C. rodentium[44]. This study demonstrates the potential negative consequences of vitamin D treatment. It can protect against Th17 cell driven damage, but it is important to note these responses are key in host defense against pathogens[44].
IL-10 KO mice models
VDR/IL-10 double KO mice develop severe IBD involving all areas of the small intestine and colon and have the largest change in colon anatomy and inflammation compared to single VDR KO and IL-10 KO mice[45]. WT mice do not express any cytokines and VDR/IL-10 double KO mice express two-three fold higher levels of IL-1β, IL-2, IFN-γ, and TNF-α mRNA in their colons compared to the single KO mice colons[45]. IL-10 KO mice with normal VDR function experience a milder form of disease; thus, VDR deficiency seems to intensify IBD severity. Single VDR KO mice also express a milder form of disease; therefore, IL-10 and VDRs have a collective effect on disease severity[45]. Furthermore, vitamin D deficiency in IL-10 KO mice enhances inflammation in the small intestine in comparison to vitamin D sufficient and vitamin D supplemented IL-10 KO mice[46]. Evidently, vitamin D deficiency augments disease severity; however, dietary intake of vitamin D can be problematic, as few foods are high in vitamin D and weight loss is common in patients with IBD[46]. Therefore, vitamin D supplementation to achieve and maintain sufficient levels is critical.
Trinitrobenze sulfonic acid and DSS induced colitis mice models
Colitis can be induced in mice with treatment of trinitrobenzene sulfonic acid (TNBS). While treatment alone with calcitriol, a vitamin D analogue, significantly reduces colitis in acute TNBS colitis, treatment with both calcitriol and dexamethasone shows the best improvement in health[47]. This is demonstrated by the study by Daniel et al[47], which showed weight gain and improvement in macroscopic, microscopic, and immunological parameters of colitis after treatment of TNBS-induced colitis with the combined therapies. Calcitriol treatment reduced Th1 mediators and increased IL-4, thereby promoting the Th2 subset[47]. Additionally, calcitriol upregulated IL-10 and enhanced regulator T cell function, specifically transforming growth factor beta, and FoxP3 levels. Calcitriol also decreased IL-12p70 and IL-23p19 expression, which are DC mediators. As a result, calcitriol downregulates the pro-inflammatory response of intestinal DCs and counteracts Th1 action[47].
A vitamin D analogue has been shown to provide similar results. ZK156979 is a new low calcemic vitamin D analogue, and its use in treating TNBS-induced colitis in mice results in remarkable disease improvement. Daniel et al[48] show a reduction in colitis-associated hypercalcemia and inflammation as a result of treatment of TNBS-induced colitis with this vitamin D analogue. Infiltration of inflammatory cells, neutrophils and lymphocytes, ulcerations, loss of goblet cells, and fibrosis in the colon were restored after vitamin D treatment[48]. TNF-α and IFN-γ levels decreased, and the Th2 profile was induced with increased production of IL-4 and IL-10[48]. This analogue effectively treats Th1 colitis.
DSS treated vitamin D deficient and sufficient mice both show increased expression of mRNAs for cytokines IL-1, IL-10, IL-17, and IFN-γ[49]. Both types of mice show severe ulceration, granulation, and inflammation; however, symptoms are exacerbated in vitamin D deficient mice[49]. Vitamin D acts to protect DSS treated mice, as there is an increased expression of vitamin D activating enzyme CYP27B1 in these mice[49] and CYP27B1 KO mice are more susceptible to colitis after DSS treatment[50]. Lagishetty et al[49] examine the impact of vitamin D status on antibacterial activity in DSS-induced experimental colitis. The expression of an antimicrobial protein, angiogenin-4 (Ang4), decreased in vitamin D deficient mice[49]. Ang4 has bactericidal activity in the intestine and a decreased expression resulted in a 50-fold increase of bacterial infiltration in vitamin D deficient mice[49]. It is worthy to note dietary restrictions in these mice resulted in 25(OH)D3 concentrations less than 50 nmol/L, which is consistent with human parameters of deficiency. The authors note that the consequences of vitamin D deficiency resulted after treatment with DSS; therefore, vitamin D deficiency may be important after inflammation has occurred[49]. On the other hand, the expression of Ang4 was associated with vitamin D status; therefore, the authors hypothesize impaired vitamin D status may be a predisposing factor for IBD due to the regulation of enteric bacteria[49].
Although these animal results are limited in their application to humans as chemical induced colitis in mice may not fully be representative of human IBD forms, they are important in examining treatment and disease activity outcomes. Investigation into determining whether other mouse models better replicate human IBD will be important[50]. The differences in results between models with impaired vitamin D status as opposed to deletion of VDRs or the CYP27B1 gene expression are also important in determining IBD susceptibility[50].
EFFECT OF VITAMIN D IN HUMAN IBD
Geographical distribution of vitamin D deficiency in IBD
Epidemiological evidence for a role of vitamin D in IBD is seen in the geographical distribution of disease, with higher incidences and prevalence in temperate climates and lower risks in persons living near the equator[6,51,52]. Additional environmental, lifestyle, or genetic factors that have similar associations with geographical location may play a part in the association between sun exposure and IBD[51]; however, this “north-south” gradient in the risk of Crohn’s disease and ulcerative colitis is likely explained by the variation in sun exposure, a major determinant of vitamin D levels[51,52], thereby strengthening the role of vitamin D in IBD.
Vitamin D deficiency has been well described in IBD patients from all over the world with varying prevalence. In Ireland, 63% of Crohn’s disease patients had 25-OH vitamin D levels < 50 nmol/L; however, using the higher cut-off to define vitamin D deficiency (< 80 nmol/L), 90% of this Crohn’s disease cohort was vitamin D deficient[53]. The frequency of vitamin D deficiency in Canada is approximately 8% (< 25 nmol/L) with an additional 22% having insufficiency (< 40 nmol/L)[54]. A Japanese study found that 27.3% of Crohn’s patients were vitamin D deficient (< 10 ng/mL or < 25 nmol/L) compared to only 6.7% of controls[55]. In an IBD cohort of children, adolescents, and young adults from Philadelphia and Pennsylvania, 16% of the Crohn’s disease patients enrolled were vitamin D deficient (< 38 nmol/L)[56]. In a cohort study of IBD patients in Wisconsin, 49.8% of patients had levels < 50 nmol/L with 10.9% having levels < 25 nmol/L[57]. Lastly, in London, United Kingdom, 68% of the IBD cohort was vitamin D deficient (< 50 nmol/L)[58].
Higher 25-OH vitamin D plasma levels, predicted on the basis of a validate regression model in the Nurses’ Health Study, have been shown to be associated with a lower incidence of Crohn’s disease and ulcerative colitis[59]; therefore, obtaining and maintaining optimal vitamin D levels are important, and yet, cut-off values used to define vitamin D deficiency among studies are variable. It is, however, generally accepted that vitamin D deficiency is defined as a serum 25-OH vitamin D level < 75 nmol/L[8,60].
Risk factors of vitamin D deficiency in IBD
Many risk factors for vitamin D deficiency in IBD have been reported. Seasonal variation is evident in most studies with lower levels seen in winter months[53,54,56,58]. Lower vitamin D levels have also been associated with longer disease duration and smoking[53,57]. Poor health outcomes such as the need for intestinal resection, a structuring disease phenotype, the need for oral corticosteroids within 3 mo of diagnosis, and a diagnosis of pancolitis in ulcerative colitis are more prevalent in severely vitamin D deficient patients (< 25 nmol/L) compared to those with normal levels (> 50 nmol/L)[58]. Intestinal resection may be a risk factor as the prevalence of at least one intestinal resection is significantly higher in those with vitamin D deficiency than those with adequate levels (P < 0.05)[58].
The role of ethnicity as a risk factor for vitamin D deficiency in IBD has also been examined. A one year prospective study was conducted to examine the association between vitamin D levels, ethnicity, and human IBD. A significantly higher percentage of South Asian IBD patients were vitamin D deficient (< 50 nmol/L) compared to Caucasians[61]. For both Caucasians and South Asians with Crohn’s disease, an inverse relationship was found between clinical disease severity and vitamin D levels. For all IBD patients, CRP levels were inversely related to vitamin D levels; however, none of these results reached a significant association[61]. Chatu et al[58] also examine ethnicity as a predictor of vitamin D deficiency in an IBD cohort. No differences were found in median vitamin D levels among Crohn’s disease and ulcerative colitis patients; however, the median vitamin D level was significantly lower in non-Caucasians (Asian and Black) compared to Caucasians. The multivariate regression analysis showed a history of IBD related surgery and ethnicity to be independently associated with vitamin D deficiency in Crohn’s disease and ethnicity alone to be independently associated with vitamin D deficiency in ulcerative colitis[58]. In an IBD cohort of children, adolescents, and young adults from Philadelphia and Pennsylvania, deficiency was more prevalent among African American subjects, Crohn’s disease patients with upper gastrointestinal tract involvement, and patients with a significantly greater lifetime exposure to glucocorticoid therapy[56]. Other risk factors may include decreased nutrition intake due to Crohn’s associated anorexia[56,62], fear of GI discomfort from dairy due to lactose intolerance, and active disease associated with decreased physical activity resulting in reduced sun exposure[56].
Genetic variants in the VDR and DBP have been shown to be associated with increased risk of IBD[63,64]. Analysis of the frequency of common genetic variants in the DBP have shown the DBP 420 variant Lys to be less common in IBD patients as compared to healthy controls (P = 0.034)[63]. A meta-analysis found that VDR gene polymorphisms are associated with the susceptibility to IBD. The TT genotype of TaqI was associated with Crohn’s disease in Europeans (OR = 1.23; 95%CI: 1.02-1.49)[64]. This polymorphism is a base substitution resulting in two encodings of isoleucine instead of an amino acid change. It has been suggested to result in lower VDR mRNA levels and less vitamin D/VDR inhibition on immune activation[64]. Additionally, this variant was significantly associated with IBD in males. Furthermore, an increased risk of ulcerative colitis in Asians was significantly associated with the ff genotype of FokI on the promoter region of the VDR (OR = 1.65; 95%CI: 1.11-2.45)[64]. This was compared to the FF genotype in Asians. The fourth finding was associated with decreased Crohn’s disease susceptibility if one was a carrier of the “a” allele (Aa + aa genotypes) of ApAI (OR = 0.81; 95%CI: 0.67-0.97)[64]. These studies demonstrate a genetic role explaining the high prevalence of vitamin D deficiency among IBD patients.
Vitamin D and disease activity
There is strong evidence to support a high prevalence of significantly lower serum 25-OH vitamin D levels among the IBD population, which have been shown to correlate with increased disease activity. Vitamin D levels have been shown to correlate negatively with disease activity assessed by the Harvey Bradshaw score[57,61,62,64] or Crohn’s disease activity index (CDAI) score[65]. Joseph et al[66] show that Crohn’s disease patients have significantly lower vitamin D levels than their age- and sex-matched controls who were patients diagnosed with irritable bowel syndrome[66]. Predictors of vitamin D status in this study were disease activity and sun exposure. In regards to severity of disease, patients with mild disease had vitamin D levels similar to the controls, but vitamin D levels were significantly lower in patients with moderate-severe Crohn’s disease[66]. As expected, lower vitamin D levels were observed in patients who had jejunal involvement of their disease[66]. This association has also been reported in patients with ulcerative colitis. Blanck et al[67] conducted a cross-sectional study and reported a larger number of patients with clinically active disease, using the six-point partial Mayo index, in the vitamin D deficient group compared to the vitamin D sufficient group (P = 0.04). Patients were stratified based on their vitamin D levels as either vitamin D sufficient, insufficient, and deficient, and there continued to be a trend towards more active disease as vitamin D levels decreased[67]. Furthermore, in a retrospective review of South Asian patients with IBD, patients with vitamin D deficiency appeared to have a more aggressive disease course with 14% of deficient patients requiring surgical management[68]; therefore, optimizing vitamin D status and assessing the relationship between vitamin D treatment and disease activity is important[68].
Despite a high prevalence of vitamin D deficiency among IBD patients, serum vitamin D levels may not always be associated with disease activity. Hassan et al[69] found no association between low vitamin D levels and increased disease activity in IBD patients. Vitamin D deficiency may also be explained by the increased risk of intestinal malabsorption among the IBD population, particularly if patients have undergone small bowel resections or use cholestyramine for postresectional diarrhea. Cholestryamine reduces bile acids, which are required for vitamin D absorption[6]. It has been demonstrated that Crohn’s disease patients with quiescent disease have on average a 30% decrease in their ability to absorb vitamin D in comparison to normal subjects after supplementation with 50000 IU of vitamin D2[70]. Furthermore, Suibhne et al[53] report vitamin D deficiency to be common among Crohn’s disease patients in clinical remission. Even in the summer, vitamin D deficiency among these patients continued to remain high (50%). About 40% of these patients were supplementing with vitamin D (200-400 IU), but it was not enough to maintain optimal vitamin D levels[53]. The location of disease, disease activity, or prior resection may not be the only factors affecting vitamin D bioavailability[70].
According to the previous studies, vitamin D status has been reported to be inversely correlated with disease activity[57,61,62,64,67]. However, Abreu et al[71] demonstrate a significant positive correlation between modified Harvey Bradshaw indices and 1,25(OH)2D3 levels in Crohn’s disease patients taking corticosteroids. This correlation, however, did not exist in the patients who were not taking this drug[71]. The increase in systemic 1,25(OH)2D3 may be a result of increased inflammation as a result of Crohn’s disease[71]. This can be explained by the expression of 1α-hydroxylase in the intestine and increased expression of 1α-hydroxylase in inflamed biopsies of Crohn’s patients[71]. The authors conclude that elevated 1,25(OH)2D3 is an additional risk factor for osteoporosis in this study population as they determine glucocorticoid use and high 1,25(OH)2D3 levels to be independent risk factors for low bone mineral density. They also suggest that 1,25(OH)2D3 may be a direct cause of bone loss or a surrogate marker for the type of intestinal inflammation leading to osteoporosis[71]. The evidence from this review would support the latter. It should be noted that the previous studies measured 25(OH)D3 to determine vitamin D status, not 1,25(OH)2D3 levels.
Vitamin D supplementation in IBD
Vitamin D supplementation has traditionally been recommended in patients with IBD for management of bone disease. There is now increasing evidence for the potential immunomodulatory effects of supplementation. To date, the optimal level of 25-OH vitamin D for immunomodulatory effects is not known[6].
Ananthakrishnan et al[72] demonstrate an increased risk for IBD-related surgery in patients who have low plasma 25(OH)D levels. This association was found to be similar in both patients with Crohn’s disease and ulcerative colitis. Furthermore, Crohn’s disease patients who initially had a low level, which then was normalized, were significantly less likely to undergo surgery in comparison to patients who continued to maintain a low vitamin D level[72]. A significantly lower C-reactive protein was also seen in these “normalized” patients. There was no association seen in ulcerative colitis patients, which the authors suggest may be due to a higher plasma 25(OH)D threshold in these patients to obtain any immune effects. Another explanation may be that vitamin D has a stronger interaction in Crohn’s disease vs ulcerative colitis[72]. Furthermore, Zator et al[73] report a significant association between earlier cessation of anti-TNF therapy in IBD patients who had insufficient vitamin D levels prior to initiation of anti-TNF therapy, suggesting vitamin D may be an important adjuvant treatment aiding in the maintenance of response to this therapy. These studies denote the importance of repleting and maintaining sufficient vitamin D levels in patients who have IBD, specifically above 30 ng/mL (75 nmol/L), to reduce the risk of flares and to maintain response to IBD-therapies[72,73].
One randomized placebo-controlled study has assessed the effectiveness of vitamin D supplementation in improving Crohn’s disease activity. In comparison to the placebo, oral vitamin D supplementation of 1200 IU in adult patients with Crohn’s disease in remission was shown to increase the 25-OH vitamin D levels and reduce the risk of relapse from 29% to 13% at 1 year (P = 0.06)[74]. Although this difference in relapse was not statistically significant, the difference is clinically meaningful and does warrant further study. The authors did discuss the risk of type II error as an explanation for not reaching statistical significance[74,75].
A prospective study completed by Miheller et al[76] compares supplementation with active vitamin D (alfacalcidiol) to non-active vitamin D (cholecalciferol) in Crohn’s patients. Looking at the clinical course of Crohn’s disease at 6 wk, disease activity significantly decreased in the active vitamin D group; however, there was no difference by the end of the trial at 12 mo[76]. Active vitamin D treatment resulted in a significant decrease in CDAI scores and CRP levels, as well as improvement in quality of life scores. It has prominent short-term effects and may be due to improved immune responses[76].
A systematic review[75] was completed to examine the efficacy of vitamin D supplementation for treating colitis in humans and animals. With a primary outcome of inducing or maintaining remission of the disease, the above study by Miheller et al[76] was included. The authors conclude that the inability to sustain differences in clinical outcomes with active vitamin D does not undermine the efficacy of vitamin D[75]. It shows the additional improvement of the active form to the plain form. In comparison, Yang et al[77] assessed the improvement in vitamin D status, clinical disease activity, and quality of life scores in 18 mild-moderate Crohn’s disease patients who underwent vitamin D3 supplementation for 24 wk. Patients were started with 1000 IU/d of vitamin D3, and the dose was increased every two wk by 1000 IU until serum 25(OH)D3 levels were above 40 ng/mL (100 nmol/L) or the patients were taking 5000 IU/d. After 24 wk, the maximum dose of 5000 IU/d was required by 78% of patients and effectively raised serum 25(OH)D levels. The assessment of CDAI scores at 24 wk showed that 78% of patients achieved clinical response defined by a decreased CDAI score of 70 points or more. Additionally, 67% of patients were in remission and disease-specific quality of life significantly improved[77]. Table 1 outlines the four studies examining vitamin D supplementation on IBD outcomes.
Table 1.
Vitamin D supplementation improves disease activity and outcomes in human inflammatory bowel disease
| Ref. | n | Methodology | Aims | Intervention | Definition of improvement | Conclusions |
| Miheller et al[76] | 37 | Prospective single cohort study | Compare the effects of active vitamin D and plain vitamin D on bone health and disease status in Crohn’s disease patients | Group A: 2 μg × 0.25 μg alfacalcidiol (active vitamin D) daily Group B: 1000 IU cholecalciferol daily (plain vitamin D) | Assessment: Osteocalcin (OC) and beta-CrossLaps (βCL) concentrations | Significant reduction in βCL and OC concentrations showed decelerated bone resorption and bone turnover in the active vitamin D group compared to the plain vitamin D group at 6 wk and 3 mo (P < 0.05) Significant reduction in disease activity and improved quality of life in the active vitamin D group at 6 wk (P < 0.05) No difference at 12 mo |
| CDAI, CRP, IBD-questionnaire (IBD-Q) | ||||||
| Improvement: Significant decrease in OC and βCL concentrations | ||||||
| Significant decrease in CDAI and IBDQ scores and CRP concentrations | ||||||
| Jørgensen et al[74] | 94 | A multi-centre randomized double blinded placebo controlled trial | Assess the efficacy of vitamin D supplementation in reducing the risk of relapse in Crohn’s disease patients compared to placebo | Treatment group: 1200 IU vitamin D3 + 1200 mg calcium/d Placebo group: placebo + 1200 mg calcium/d | Assessment: CDAI Improvement: Decreased proportion of patients who achieve a CDAI score of 150+ and a 70 point increase in CDAI compared to baseline | Decreased risk of relapse (29% to 13%) at 1 yr with vitamin D treatment, but did not reach significance (P = 0.06) |
| Ananthakrishnan et al[72] | 3217 | Retrospective study | Assess the association between plasma 25(OH)D3 levels and IBD related surgeries and hospitalizations Assess changes in these outcomes after normalization of IBD patients’ vitamin D levels | None | Assessment: Risk of IBD-related surgery or hospitalization (OR); Improvement: Reduction in risk (OR < 1) for surgery or hospitalization | Low levels of vitamin D significantly increased risk for IBD-related surgery and hospital admissions |
| (OR = 2.05; 95%CI: 1.53-2.75 for Crohn’s disease and OR = 1.75; 95%CI: 1.21-2.52 for ulcerative colitis) | ||||||
| Achieving normal vitamin D levels decreased risk of Crohn’s disease-related surgery | ||||||
| (OR = 0.56; 95%CI: 0.32-0.98) | ||||||
| Yang et al[77] | 18 | Prospective clinical pilot study | Establish the oral dose of vitamin D3 required to achieve serum 25(OH)D3 concentrations above 40 ng/mL (100 nmol/L) in mild-moderate Crohn’s disease patients Assess improvement in disease activity and quality of life after vitamin D supplementation in Crohn’s disease patients | Initiated on 1000 IU per day of vitamin D3 for 2 wk. Increased dose every two wk by 1000 IU/d until achievement of serum 25(OH)D level of 40 ng/mL occurred or patients were taking a total of 5000 IU/d | Assessment: Dose of vitamin D3 and serum 25(OH)D3 levels CDAI and IBDQ Improvement: Increase in serum 25(OH)D3 levels Reduction in CDAI scores of > 70 points or achievement of CDAI score < 150, and increase in IBDQ scores | Vitamin D supplementation improved vitamin D status at 24 wk (P < 0.001). 78% of patients required 5000 IU/d of vitamin D3, suggesting this is an effective dose in raising serum 25(OH)D3 levels in mild-moderate Crohn’s patients |
| Vitamin D treatment significantly improved disease activity and quality of life at 24 wk (P < 0.001) |
CDAI: Crohn’s disease activity index; CRP: C-reactive protein; IBD: Inflammatory bowel disease.
In a randomized clinical trial of children and adolescents with IBD, weekly high dose vitamin D2 (50000 IU) or daily vitamin D3 (2000 IU) were superior to daily vitamin D2 (2000 IU) in raising the serum 25-OH vitamin D level at 6 wk (25.4 ± 2.5, 16.4 ± 2.0, 9.3 ± 1.8 ng/mL, respectively)[78]. Adherence may be improved with large doses, and dosing according to individual levels may achieve target levels most effectively[3]. Recent studies have suggested that optimal targets for serum 25-OH vitamin D levels are greater than 30 ng/mL (75-80 nmol/L), with levels between 21-29 ng/mL (51-74 nmol/L) being defined as insufficient and levels < 20 ng/mL (< 50 nmol/L) being defined as deficient[60].
CONCLUSION
Recent advances in the understanding of the effects and mechanism of action of vitamin D on the mucosal and systemic immune system and subsequently on intestinal inflammation suggests it has a role to play in the therapeutic management of IBD. Furthermore, both epidemiologic and emerging retrospective and prospective clinical evidence supports a significant beneficial role of vitamin D supplementation in patients with IBD. While the precise level of 25(OH)D3 that needs to be achieved for these therapeutic effects is unknown, it has been established that levels of 75 nmol/L or higher are generally adequate[3,8,26,60,72]. Given the safety profile and low cost of vitamin D, its addition to the therapeutic armamentarium as a supplement to induction and maintenance therapy should be strongly considered.
Footnotes
Supported by A graduate studentship from the Center of Excellence for Gastrointestinal and Immunity Research (CEGIIR) and the Alberta Innovates-Health Solutions Inflammatory Bowel Disease Consortium to Reich KM
P- Reviewers: Gazouli M, Ingle SB, Nikfar S, Spisni E, Thorne K S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
- 1.Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53 Suppl 5:V1–16. doi: 10.1136/gut.2004.043372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–1794. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 3.Garg M, Lubel JS, Sparrow MP, Holt SG, Gibson PR. Review article: vitamin D and inflammatory bowel disease--established concepts and future directions. Aliment Pharmacol Ther. 2012;36:324–344. doi: 10.1111/j.1365-2036.2012.05181.x. [DOI] [PubMed] [Google Scholar]
- 4.Javorsky BR, Maybee N, Padia SH, Dalkin AC. Vitamin D deficiency in gastrointestinal disease. Pract Gastroenterol. 2006;29:52–72. [Google Scholar]
- 5.Miznerova E, Hlavaty T, Koller T, Toth J, Holociova K, Huorka M, Killinger Z, Payer J. The prevalence and risk factors for osteoporosis in patients with inflammatory bowel disease. Bratisl Lek Listy. 2013;114:439–445. doi: 10.4149/bll_2013_092. [DOI] [PubMed] [Google Scholar]
- 6.Raman M, Milestone AN, Walters JR, Hart AL, Ghosh S. Vitamin D and gastrointestinal diseases: inflammatory bowel disease and colorectal cancer. Therap Adv Gastroenterol. 2011;4:49–62. doi: 10.1177/1756283X10377820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lips P. Vitamin D physiology. Prog Biophys Mol Biol. 2006;92:4–8. doi: 10.1016/j.pbiomolbio.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 9.Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metab Clin North Am. 2010;39:243–253, table of contents. doi: 10.1016/j.ecl.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christakos S, Dhawan P, Liu Y, Peng X, Porta A. New insights into the mechanisms of vitamin D action. J Cell Biochem. 2003;88:695–705. doi: 10.1002/jcb.10423. [DOI] [PubMed] [Google Scholar]
- 11.Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D3. Rheum Dis Clin North Am. 2012;38:13–27. doi: 10.1016/j.rdc.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasmin R, Williams RM, Xu M, Noy N. Nuclear import of the retinoid X receptor, the vitamin D receptor, and their mutual heterodimer. J Biol Chem. 2005;280:40152–40160. doi: 10.1074/jbc.M507708200. [DOI] [PubMed] [Google Scholar]
- 13.Hewison M. Vitamin D and immune function: an overview. Proc Nutr Soc. 2012;71:50–61. doi: 10.1017/S0029665111001650. [DOI] [PubMed] [Google Scholar]
- 14.Fritsche J, Mondal K, Ehrnsperger A, Andreesen R, Kreutz M. Regulation of 25-hydroxyvitamin D3-1 alpha-hydroxylase and production of 1 alpha,25-dihydroxyvitamin D3 by human dendritic cells. Blood. 2003;102:3314–3316. doi: 10.1182/blood-2002-11-3521. [DOI] [PubMed] [Google Scholar]
- 15.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 16.Guillot X, Prati C, Saidenberg-Kermanac’h N, Semerano L, Falgarone G, Boissier MC, Wendling D. Inflammation and vitamin D. Watson RR, editor. Handbook of vitamin D in human health: Prevention, treatment and toxicity. The Netherlands: Wageningen Academic Publishers; 2013. pp. 372–390. [Google Scholar]
- 17.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O’Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001;167:4974–4980. doi: 10.4049/jimmunol.167.9.4974. [DOI] [PubMed] [Google Scholar]
- 19.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, de Waal-Malefyt R, Coffman RL, Hawrylowicz CM, O’Garra A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000;164:2405–2411. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- 21.Canning MO, Grotenhuis K, de Wit H, Ruwhof C, Drexhage HA. 1-alpha,25-Dihydroxyvitamin D3 (1,25(OH)(2)D(3)) hampers the maturation of fully active immature dendritic cells from monocytes. Eur J Endocrinol. 2001;145:351–357. doi: 10.1530/eje.0.1450351. [DOI] [PubMed] [Google Scholar]
- 22.Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005;19:1067–1077. doi: 10.1096/fj.04-3284com. [DOI] [PubMed] [Google Scholar]
- 23.Wang TT, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, Dionne S, Servant MJ, Bitton A, Seidman EG, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem. 2010;285:2227–2231. doi: 10.1074/jbc.C109.071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Liu W, Sun T, Huang Y, Wang Y, Deb DK, Yoon D, Kong J, Thadhani R, Li YC. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting microRNA-155-SOCS1 in macrophages. J Immunol. 2013;190:3687–3695. doi: 10.4049/jimmunol.1203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eleftheriadis T, Antoniadi G, Liakopoulos V, Kartsios C, Stefanidis I, Galaktidou G. Paricalcitol reduces basal and lipopolysaccharide-induced (LPS) TNF-alpha and IL-8 production by human peripheral blood mononuclear cells. Int Urol Nephrol. 2010;42:181–185. doi: 10.1007/s11255-009-9541-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Leung DY, Richers BN, Liu Y, Remigio LK, Riches DW, Goleva E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188:2127–2135. doi: 10.4049/jimmunol.1102412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller K, Haahr PM, Diamant M, Rieneck K, Kharazmi A, Bendtzen K. 1,25-Dihydroxyvitamin D3 inhibits cytokine production by human blood monocytes at the post-transcriptional level. Cytokine. 1992;4:506–512. doi: 10.1016/1043-4666(92)90012-g. [DOI] [PubMed] [Google Scholar]
- 28.Stio M, Treves C, Martinesi M, Bonanomi AG. Biochemical effects of KH 1060 and anti-TNF monoclonal antibody on human peripheral blood mononuclear cells. Int Immunopharmacol. 2005;5:649–659. doi: 10.1016/j.intimp.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Khoo AL, Chai LY, Koenen HJ, Sweep FC, Joosten I, Netea MG, van der Ven AJ. Regulation of cytokine responses by seasonality of vitamin D status in healthy individuals. Clin Exp Immunol. 2011;164:72–79. doi: 10.1111/j.1365-2249.2010.04315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 31.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–298. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Mahon BD, Froicu M, Cantorna MT. Calcium and 1 alpha,25-dihydroxyvitamin D3 target the TNF-alpha pathway to suppress experimental inflammatory bowel disease. Eur J Immunol. 2005;35:217–224. doi: 10.1002/eji.200425491. [DOI] [PubMed] [Google Scholar]
- 33.Bartels LE, Jørgensen SP, Agnholt J, Kelsen J, Hvas CL, Dahlerup JF. 1,25-dihydroxyvitamin D3 and dexamethasone increase interleukin-10 production in CD4+ T cells from patients with Crohn’s disease. Int Immunopharmacol. 2007;7:1755–1764. doi: 10.1016/j.intimp.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Stio M, Treves C, Martinesi M, d’Albasio G, Bagnoli S, Bonanomi AG. Effect of anti-TNF therapy and vitamin D derivatives on the proliferation of peripheral blood mononuclear cells in Crohn’s disease. Dig Dis Sci. 2004;49:328–335. doi: 10.1023/b:ddas.0000017460.90887.11. [DOI] [PubMed] [Google Scholar]
- 35.Wu S, Sun J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov Med. 2011;11:325–335. [PMC free article] [PubMed] [Google Scholar]
- 36.Bartels LE, Jørgensen SP, Bendix M, Hvas CL, Agnholt J, Agger R, Dahlerup JF. 25-Hydroxy vitamin D3 modulates dendritic cell phenotype and function in Crohn’s disease. Inflammopharmacology. 2013;21:177–186. doi: 10.1007/s10787-012-0168-y. [DOI] [PubMed] [Google Scholar]
- 37.Bendix-Struve M, Bartels LE, Agnholt J, Dige A, Jørgensen SP, Dahlerup JF. Vitamin D3 treatment of Crohn’s disease patients increases stimulated T cell IL-6 production and proliferation. Aliment Pharmacol Ther. 2010;32:1364–1372. doi: 10.1111/j.1365-2036.2010.04463.x. [DOI] [PubMed] [Google Scholar]
- 38.Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol. 2007;8:5. doi: 10.1186/1471-2172-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S, Liao AP, Xia Y, Li YC, Li JD, Sartor RB, Sun J. Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. Am J Pathol. 2010;177:686–697. doi: 10.2353/ajpath.2010.090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruce D, Cantorna MT. Intrinsic requirement for the vitamin D receptor in the development of CD8αα-expressing T cells. J Immunol. 2011;186:2819–2825. doi: 10.4049/jimmunol.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, Bissonnette M, Li YC. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–G216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
- 42.Palmer MT, Weaver CT. Linking vitamin d deficiency to inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2245–2256. doi: 10.1097/MIB.0b013e31828a3b6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruce D, Yu S, Ooi JH, Cantorna MT. Converging pathways lead to overproduction of IL-17 in the absence of vitamin D signaling. Int Immunol. 2011;23:519–528. doi: 10.1093/intimm/dxr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryz NR, Patterson SJ, Zhang Y, Ma C, Huang T, Bhinder G, Wu X, Chan J, Glesby A, Sham HP, et al. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to Citrobacter rodentium by suppressing mucosal Th17 responses. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1299–G1311. doi: 10.1152/ajpgi.00320.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Froicu M, Zhu Y, Cantorna MT. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006;117:310–318. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 47.Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther. 2008;324:23–33. doi: 10.1124/jpet.107.127209. [DOI] [PubMed] [Google Scholar]
- 48.Daniel C, Radeke HH, Sartory NA, Zahn N, Zuegel U, Steinmeyer A, Stein J. The new low calcemic vitamin D analog 22-ene-25-oxa-vitamin D prominently ameliorates T helper cell type 1-mediated colitis in mice. J Pharmacol Exp Ther. 2006;319:622–631. doi: 10.1124/jpet.106.107599. [DOI] [PubMed] [Google Scholar]
- 49.Lagishetty V, Misharin AV, Liu NQ, Lisse TS, Chun RF, Ouyang Y, McLachlan SM, Adams JS, Hewison M. Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology. 2010;151:2423–2432. doi: 10.1210/en.2010-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu N, Nguyen L, Chun RF, Lagishetty V, Ren S, Wu S, Hollis B, DeLuca HF, Adams JS, Hewison M. Altered endocrine and autocrine metabolism of vitamin D in a mouse model of gastrointestinal inflammation. Endocrinology. 2008;149:4799–4808. doi: 10.1210/en.2008-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nerich V, Jantchou P, Boutron-Ruault MC, Monnet E, Weill A, Vanbockstael V, Auleley GR, Balaire C, Dubost P, Rican S, et al. Low exposure to sunlight is a risk factor for Crohn’s disease. Aliment Pharmacol Ther. 2011;33:940–945. doi: 10.1111/j.1365-2036.2011.04601.x. [DOI] [PubMed] [Google Scholar]
- 52.Khalili H, Huang ES, Ananthakrishnan AN, Higuchi L, Richter JM, Fuchs CS, Chan AT. Geographical variation and incidence of inflammatory bowel disease among US women. Gut. 2012;61:1686–1692. doi: 10.1136/gutjnl-2011-301574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suibhne TN, Cox G, Healy M, O’Morain C, O’Sullivan M. Vitamin D deficiency in Crohn’s disease: prevalence, risk factors and supplement use in an outpatient setting. J Crohns Colitis. 2012;6:182–188. doi: 10.1016/j.crohns.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Siffledeen JS, Siminoski K, Steinhart H, Greenberg G, Fedorak RN. The frequency of vitamin D deficiency in adults with Crohn’s disease. Can J Gastroenterol. 2003;17:473–478. doi: 10.1155/2003/391308. [DOI] [PubMed] [Google Scholar]
- 55.Tajika M, Matsuura A, Nakamura T, Suzuki T, Sawaki A, Kato T, Hara K, Ookubo K, Yamao K, Kato M, et al. Risk factors for vitamin D deficiency in patients with Crohn’s disease. J Gastroenterol. 2004;39:527–533. doi: 10.1007/s00535-003-1338-x. [DOI] [PubMed] [Google Scholar]
- 56.Sentongo TA, Semaeo EJ, Stettler N, Piccoli DA, Stallings VA, Zemel BS. Vitamin D status in children, adolescents, and young adults with Crohn disease. Am J Clin Nutr. 2002;76:1077–1081. doi: 10.1093/ajcn/76.5.1077. [DOI] [PubMed] [Google Scholar]
- 57.Ulitsky A, Ananthakrishnan AN, Naik A, Skaros S, Zadvornova Y, Binion DG, Issa M. Vitamin D deficiency in patients with inflammatory bowel disease: association with disease activity and quality of life. JPEN J Parenter Enteral Nutr. 2011;35:308–316. doi: 10.1177/0148607110381267. [DOI] [PubMed] [Google Scholar]
- 58.Chatu S, Chhaya V, Holmes R, Neild P, Kang J, Pollok RC, Poullis A. Factors associated with vitamin D deficiency in a multicultural inflammatory bowel disease cohort. Frontline Gastroenterol. 2013;4:51–56. doi: 10.1136/flgastro-2012-100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ananthakrishnan AN, Khalili H, Higuchi LM, Bao Y, Korzenik JR, Giovannucci EL, Richter JM, Fuchs CS, Chan AT. Higher predicted vitamin D status is associated with reduced risk of Crohn’s disease. Gastroenterology. 2012;142:482–489. doi: 10.1053/j.gastro.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holick MF. Vitamin D and health: evolution, biologic functions, and recommended dietary intakes for vitamin D. Clin Rev Bone Miner Metab. 2009;8:2–19. [Google Scholar]
- 61.Fu YT, Chatur N, Cheong-Lee C, Salh B. Hypovitaminosis D in adults with inflammatory bowel disease: potential role of ethnicity. Dig Dis Sci. 2012;57:2144–2148. doi: 10.1007/s10620-012-2130-7. [DOI] [PubMed] [Google Scholar]
- 62.Harries AD, Brown R, Heatley RV, Williams LA, Woodhead S, Rhodes J. Vitamin D status in Crohn’s disease: association with nutrition and disease activity. Gut. 1985;26:1197–1203. doi: 10.1136/gut.26.11.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eloranta JJ, Wenger C, Mwinyi J, Hiller C, Gubler C, Vavricka SR, Fried M, Kullak-Ublick GA. Association of a common vitamin D-binding protein polymorphism with inflammatory bowel disease. Pharmacogenet Genomics. 2011;21:559–564. doi: 10.1097/FPC.0b013e328348f70c. [DOI] [PubMed] [Google Scholar]
- 64.Xue LN, Xu KQ, Zhang W, Wang Q, Wu J, Wang XY. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: a meta-analysis. Inflamm Bowel Dis. 2013;19:54–60. doi: 10.1002/ibd.22966. [DOI] [PubMed] [Google Scholar]
- 65.Jørgensen SP, Hvas CL, Agnholt J, Christensen LA, Heickendorff L, Dahlerup JF. Active Crohn’s disease is associated with low vitamin D levels. J Crohns Colitis. 2013;7:e407–e413. doi: 10.1016/j.crohns.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 66.Joseph AJ, George B, Pulimood AB, Seshadri MS, Chacko A. 25 (OH) vitamin D level in Crohn’s disease: association with sun exposure & amp; disease activity. Indian J Med Res. 2009;130:133–137. [PubMed] [Google Scholar]
- 67.Blanck S, Aberra F. Vitamin d deficiency is associated with ulcerative colitis disease activity. Dig Dis Sci. 2013;58:1698–1702. doi: 10.1007/s10620-012-2531-7. [DOI] [PubMed] [Google Scholar]
- 68.Boyd CA, Limdi JK. Vitamin D deficiency and disease outcomes in South Asian patients with IBD. Dig Dis Sci. 2013;58:2124–2125. doi: 10.1007/s10620-013-2654-5. [DOI] [PubMed] [Google Scholar]
- 69.Hassan V, Hassan S, Seyed-Javad P, Ahmad K, Asieh H, Maryam S, Farid F, Siavash A. Association between Serum 25 (OH) Vitamin D Concentrations and Inflammatory Bowel Diseases (IBDs) Activity. Med J Malaysia. 2013;68:34–38. [PubMed] [Google Scholar]
- 70.Farraye FA, Nimitphong H, Stucchi A, Dendrinos K, Boulanger AB, Vijjeswarapu A, Tanennbaum A, Biancuzzo R, Chen TC, Holick MF. Use of a novel vitamin D bioavailability test demonstrates that vitamin D absorption is decreased in patients with quiescent Crohn’s disease. Inflamm Bowel Dis. 2011;17:2116–2121. doi: 10.1002/ibd.21595. [DOI] [PubMed] [Google Scholar]
- 71.Abreu MT, Kantorovich V, Vasiliauskas EA, Gruntmanis U, Matuk R, Daigle K, Chen S, Zehnder D, Lin YC, Yang H, et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut. 2004;53:1129–1136. doi: 10.1136/gut.2003.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ananthakrishnan AN, Cagan A, Gainer VS, Cai T, Cheng SC, Savova G, Chen P, Szolovits P, Xia Z, De Jager PL, et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease. Inflamm Bowel Dis. 2013;19:1921–1927. doi: 10.1097/MIB.0b013e3182902ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zator ZA, Cantu SM, Konijeti GG, Nguyen DD, Sauk J, Yajnik V, Ananthakrishnan AN. Pretreatment 25-Hydroxyvitamin D Levels and Durability of Anti-Tumor Necrosis Factor-α Therapy in Inflammatory Bowel Diseases. JPEN J Parenter Enteral Nutr. 2014;38:385–391. doi: 10.1177/0148607113504002. [DOI] [PubMed] [Google Scholar]
- 74.Jørgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, Bartels LE, Kelsen J, Christensen LA, Dahlerup JF. Clinical trial: vitamin D3 treatment in Crohn’s disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. doi: 10.1111/j.1365-2036.2010.04355.x. [DOI] [PubMed] [Google Scholar]
- 75.Nicholson I, Dalzell AM, El-Matary W. Vitamin D as a therapy for colitis: a systematic review. J Crohns Colitis. 2012;6:405–411. doi: 10.1016/j.crohns.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Miheller P, Muzes G, Hritz I, Lakatos G, Pregun I, Lakatos PL, Herszényi L, Tulassay Z. Comparison of the effects of 1,25 dihydroxyvitamin D and 25 hydroxyvitamin D on bone pathology and disease activity in Crohn’s disease patients. Inflamm Bowel Dis. 2009;15:1656–1662. doi: 10.1002/ibd.20947. [DOI] [PubMed] [Google Scholar]
- 77.Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of vitamin d supplementation in a pilot study of Crohn’s patients. Clin Transl Gastroenterol. 2013;4:e33. doi: 10.1038/ctg.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pappa HM, Mitchell PD, Jiang H, Kassiff S, Filip-Dhima R, DiFabio D, Quinn N, Lawton RC, Varvaris M, Van Straaten S, et al. Treatment of vitamin D insufficiency in children and adolescents with inflammatory bowel disease: a randomized clinical trial comparing three regimens. J Clin Endocrinol Metab. 2012;97:2134–2142. doi: 10.1210/jc.2011-3182. [DOI] [PMC free article] [PubMed] [Google Scholar]