

Abstract
AIM: To clarify whether histone deacetylase inhibitors histone deacetylase inhibitors (HDACIs) can sensitize hepatocellular carcinoma (HCC) cells to sorafenib treatment.
METHODS: Bax, Bcl-2, ATG5-ATG12, p21, and p27 protein levels in Hep3B, HepG2, and PLC/PRF/5 cells were examined by Western blot. CCK8 and a fluorometric caspase-3 assay were used to examine cellular viability and apoptosis levels. The effect of Beclin-1 on sensitization of HCC cells to sorafenib was examined by transfecting Beclin-1 siRNA into Hep3B, HepG2, and PLC/PRF/5 cells.
RESULTS: Autophagy inhibition enhances the inhibitory effects of vorinostat and sorafenib alone or in combination on HCC cell growth. Vorinostat and sorafenib synergistically induced apoptosis and cell cycle alterations. Western blot data indicated that HDACIs and Beclin-1 knockdown increased the p53 acetylation level. The knockdown of Beclin-1 enhanced the synergistic effect of the combination of vorinostat with sorafenib.
CONCLUSION: HDACIs can sensitize HCC cells to sorafenib treatment by regulating the acetylation level of Beclin-1.
Keywords: Hepatocellular carcinoma, Histone deacetylase inhibitors, Autophagy, Sorafenib, Chemoresistance
Core tip: In this study, we investigated the antiproliferative effect of sorafenib in combination with the histone deacetylase inhibitor vorinostatin in human hepatoma cell lines (Hep3B, HepG2, and PLC/PRF/5). We also examined whether the combination therapy was enhanced by the inhibition of autophagy. Our results showed that the combination of vorinostat with sorafenib synergistically reduced cell proliferation in hepatocellular carcinoma cells by inducing apoptosis and cell cycle arrest. Synergistic changes in cell cycle and cell survival regulators were also observed.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common cause of cancer-associated mortality in China and one of the leading causes of death in the world[1]. Due to the lack of effective biomarkers for early detection, most patients diagnosed with HCC die within one year because radical resection of the tumors is performed late in the disease process, meaning that the options for these patients are chemotherapy, radiotherapy, or interventional treatment[2]. Importantly, previous clinical trials have shown that patients with HCC do not benefit from traditional systemic chemotherapy[3].
One area of cancer research interest concentrates on epigenetic changes caused by modifications of histone proteins. Acetylation of histones reduces the affinity of histones for DNA, producing an open DNA structure that facilitates gene expression[4]. Histone deacetylases (HDACs) are overexpressed in many types of tumor cells, including human hepatoma cells, and suppress the expression of genes involved in tumor suppression and differentiation[5]. HDACs, along with histone acetyltransferases, reciprocally regulate the acetylation status of the positively charged NH2-terminal histone tails of nucleosomes[6]. HDAC inhibitors (HDACIs) are interesting as cancer therapeutics due to their ability to induce cell differentiation, growth arrest and apoptosis[7]. HDACIs represent a variety of agents that block histone deacetylation genes, thereby modifying chromatin structure and gene transcription[8].
Sorafenib, a multi-target biological agent that targets cancer cells and that was jointly developed by Bayer and Onyx, has shown significant inhibitory effects on tumor cell proliferation and angiogenesis and has become the first clinical drug for HCC approved by the US FDA[9]. By inhibiting the activity of the Raf serine and threonine kinase in the ERK 1/2 signaling pathway, sorafenib effectively controls tumor cell proliferation. In addition, sorafenib also inhibits VEGFR and PDGFR, thus blocking tumor angiogenesis[10]. Recent studies also have found that sorafenib can activate tumor cell autophagy[11]. Numerous drug studies have found that the stimulation of the autophagy pathway can inhibit apoptosis signals; on the contrary, inhibited autophagy can promote apoptosis signals. For example, increased p53 expression can induce rapid apoptosis in lymphoma cells in a Myc-induced lymphoma mouse model, but tumor recurrence occurs soon after. However, inhibition of the autophagy lysosomal pathway by clioquinol (CQ) or Atg5 siRNA can enhance tumor cell apoptosis and reduce recurrence[12]. Klappan et al[13] found that the genetic interference of Beclin-l, Atg5, Atg10, and Atg12 by siRNAs could lead to the inhibition of autophagy, which enhances nutritional deficiencies and causes HeLa cell death. These studies show that autophagy and apoptosis signaling share common signaling pathways (such as PI3K/Akt/mTOR, NF-κB, and ERK) and effector proteins (such as Bcl-2, Bcl-xL, Mcl-1, Atg5, and p53), suggesting that regulating the autophagy pathway should improve HCC chemotherapy[14].
To overcome drug resistance, the combination of HDACIs with existing chemotherapeutic agents has been identified as a potential approach, due to the effect of reducing the dose of other anti-neoplastic drugs. However, whether HDACIs can sensitize HCC cells to sorafenib treatment remains largely unexplored, and very few studies have investigated the activation of the autophagy signaling pathway and its related effects during the combined treatment with sorafenib and HDACIs.
The objective of the present study was to determine the synergistic antiproliferative effect of sorafenib in combination with HDACIs and examine the mechanisms underlying the synergistic antiproliferative effects. In particular, we also explored the possibility that the inhibition of autophagy can enhance the synergistic effect of the combination of vorinostat with sorafenib.
MATERIALS AND METHODS
Materials and cell culture
Sorafenib (Nexavar) was purchased from Bayer Pharmaceuticals. Vorinostat and 3-methyladenine (3-MA) were purchased from Sigma. For in vitro studies, various concentrations of sorafenib and vorinostat were dissolved in DMSO. In all experiments, the maximal concentration of DMSO in the medium was 0.02% (v/v), which does not affect cell growth. Hep3B, HepG2, and PLC/PRF/5 human cell lines were obtained from the American Type Culture Collection (ATCC). For cell culture, the following media were used: MEM for Hep3B and HepG2 cells and DMEM for PLC/PRF/5 cells. The ATCC cell bank performed cell line characterizations, and cells were passaged in the laboratory for fewer than 6 mo after thawing. Cells were treated with vorinostat or sorafenib in 5% (v/v) FBS-containing RPMI 1640 medium. For sequential combination treatment with HDACI and sorafenib, the cells were exposed to the former drug for 24 h and then to the next drug for additional 48 h. The single treatment time was consistent with the combined treatment group.
Antibodies for immunoblotting, such as Bax, Bcl-2, ATG5-ATG12, p21, and p27, were obtained from Cell Signaling. Commercially available validated short hairpin RNA molecules to knock down RNA/protein levels were obtained from Qiagen. All other chemicals were purchased from Sigma if not stated otherwise.
Measurement of growth inhibition and cell viability assay
Hep3B, HepG2, or PLC/PRF/5 cells were seeded at a density of 10000 cells/well in 96-well plates and incubated with various concentrations of HDACI, sorafenib, or the combination of the two. The cell number was evaluated by crystal violet staining. For sequential combination treatment with HDACI and sorafenib, the cells were exposed to the former drug for 24 h and then to the next drug for an additional 48 h. Following the method previously described[15], 10 g/L glutaraldehyde was added to the cells in 96-well plates. Then, the cells were stained with 1 g/L crystal violet in phosphate buffered saline (PBS). The excess dye was removed by washing with sterile water. Bound crystal violet was solubilized with 2 mL/L Triton X-100 in PBS. Light extinction, which has a linear dependence on cell number, was read at 570 nm by a microplate reader. The number of cells was determined from the absorbance of each well relative to the average absorbance of the control wells (defined as 100%).
Detection of apoptosis
The activity of caspase-3 was determined using the Fluorometric Caspase 3 Assay Kit (Sigma, United States), and the cell lysates were prepared as described previously[15]. According to the manufacturer’s instructions, the activity of caspase-3 was calculated from the cleavage of the fluorogenic substrate, Ac-DEVD-AMC. The cell lysates were incubated with substrate solution (caspase-3 substrate Ac-DEVD-AMC 20 mg/L, HEPES 20 mmol/L, glycerol 100 mL/L, and DTT 2 mmol/L, pH 7.5) for 1 h at 37 °C, and substrate cleavage was measured with a VersaFluor fluorometer (excitation: 360 nm, emission: 460 nm).
Cell cycle analysis
The cells were fixed with 70% ethanol overnight at 4 °C, and DNA was stained with 60 μg/mL propidium iodide (Sigma, United States) containing 10 U/mL RNaseA for 30 min according to the manufacturer’s instructions. The percentage of cells in the sub-G1 phase (apoptotic cells) was measured by counting 10000 cells using a FACS Calibur flow cytometer (Becton Dickinson, United States) with the ModFit LT 3.0 software.
Western blot analysis
For SDS-PAGE and immunoblotting, the cells were plated at 105 cells/mL in 6-well plates, treated with various types of drugs at the indicated concentrations, and then lysed in whole cell lysis buffer (0.5 mol/L Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, and 0.02% bromophenol blue). The samples were boiled at 100 °C for 5 min. The boiled samples containing 30 μg of protein were subjected to gel electrophoresis. The proteins were then transferred onto PVDF membranes by electroblotting for 90 min. The blots were blocked with 50 g/L non-fat dry milk in a TBS-Tween solution for 1 h at room temperature and then incubated at 4 °C overnight with primary antibodies against different proteins. Anti-β-actin (1:5000) from Sigma served as a loading control. After incubation with horseradish peroxidase-coupled anti-IgG antibodies at room temperature for at least 1 h, the blot was developed using enhanced chemiluminescent detection (GE Healthcare) and subsequently exposed to Hyperfilm ECL film.
Transfection of cells with siRNA
Cells were seeded in 60-mm dishes and transfected 24 h after plating. For transfection, 10 nmol/L of the annealed siRNA, the positive sense control double-stranded siRNA targeting GAPDH, or the negative control were used. SiRNA (10 nmol/L; scrambled or experimental for knockdown) was diluted in serum-free medium. Five microliters of HiPerFect Reagent (Qiagen, Valencia, CA) was added to this mixture, and the solution was mixed by pipetting up and down several times, followed by incubation at room temperature for 10 min. The medium in each dish was swirled gently to mix and then incubated at 37 °C for 2 h. Then, 1 mL of 10% (v/v) serum-containing medium was added to each plate, and the cells were incubated at 37 °C for 36 h before treatment with vorinostat or sorafenib. Flow cytometry assays and Western blot analyses were performed at the time points indicated in each figure.
Statistical analysis
All data are presented as mean ± SD of more than three individual experiments. Statistical significance was determined by the Student’s t-test.
RESULTS
Growth inhibitory effects of vorinostat and sorafenib alone or in combination on HCC cells
Using the crystal violet staining test, we first determined the growth inhibitory effects of sorafenib and the HDAC inhibitor, vorinostat (Vor), on human hepatoma cell lines (Hep3B, HepG2, and PLC/PRF/5). As shown in Figure 1, treating different types of hepatoma cells with 0.5-2.5 μmol/L vorinostat or sorafenib for 48 h reduced cell growth in a dose-dependent manner by up to 90%. Moreover, the combination of vorinostat with sorafenib significantly increased growth inhibitory effects in a dose-dependent manner compared to the agents alone. These results showed that the combination of vorinostat with sorafenib synergistically reduces cell proliferation in HCC cells (0.5-2.5 μmol/L vorinostat or sorafenib, P < 0.05, P < 0.01).
Figure 1.
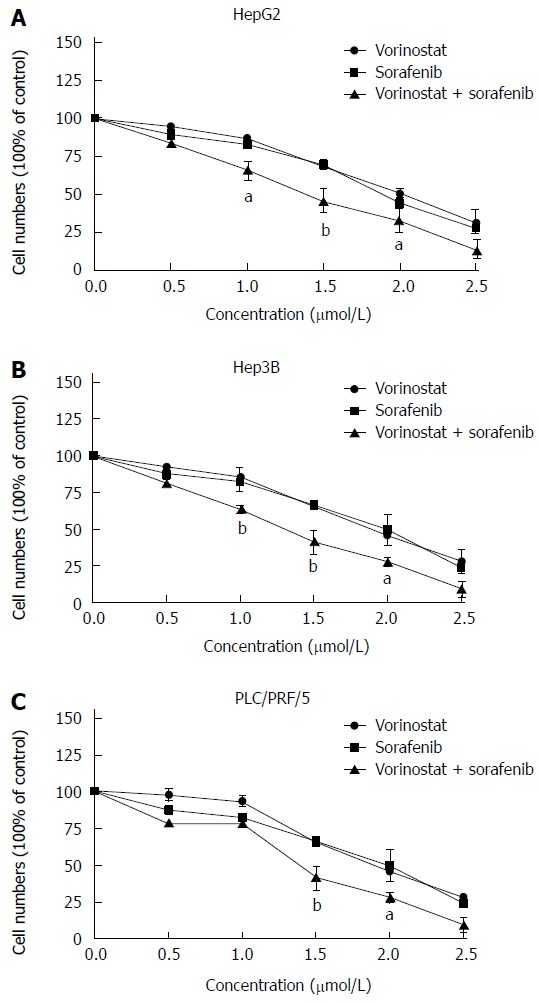
Antiproliferative effects of sorafenib, vorinostat, or the drug combination on human hepatoma cell lines. HepG2 (A), Hep3B (B), and PLC/PRF/5 (C) cells were treated with various concentrations of sorafenib, vorinostat, or the drug combination. The results are expressed as the percentage of viable cells in the control group. The results are expressed as mean ± SD from three independent experiments. aP < 0.05 and bP < 0.01 vs control as evaluated by Student’s t test.
Synergistic induction of apoptosis and cell cycle alterations by vorinostat and sorafenib
To determine the mechanism responsible for the antiproliferative effects of the combination of vorinostat with sorafenib, we next examined the effect of vorinostat and sorafenib individually or in combination on the cell cycle and apoptosis. As shown in Figure 2A, incubating HCC cells with 2.5 μmol/L vorinostat or sorafenib for 24 h resulted in a significant arrest in the G0/G1 phase of the cell cycle, whereas the proportion of cells in the S phase and G2/M phase decreased. Notably, a significant increase in G0/G1 phase arrest and decrease in the S phase and G2/M phase were observed in the vorinostat and sorafenib combination treatment groups for all three HCC cell lines. In addition, the vorinostat and sorafenib combination treatment significantly increased the apoptosis rate, as determined by caspase-3 enzyme activity in HCC cells compared to treatment with either agent alone (Figure 2B).
Figure 2.
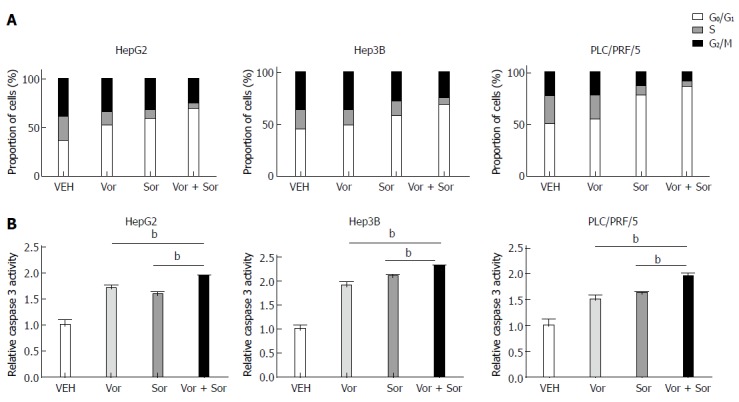
Sorafenib, vorinostat, or the drug combination induces cell cycle arrest and apoptosis in human hepatoma cell lines. HepG2, Hep3B and PLC/PRF/5 cells were treated with various concentrations of sorafenib (Sor), vorinostat (Vor), or the drug combination. A: Cell cycle distributions were analyzed by flow cytometry, and the percentage of cells in the G0/G1, S, or G2/M phases of the cell cycle is indicated; B: The apoptosis-specific caspase-3 activity induced by various concentrations of sorafenib, vorinostat, or the drug combination. The results are expressed as mean ± SD from three independent experiments. bP < 0.01 using Student’s t test.
Synergistic changes in cell cycle and cell survival regulators
To identify the potential mechanisms of the combined action of vorinostat and sorafenib, their effects on the regulatory proteins that govern cell cycle and cell survival were investigated using Western blot. The HCC cells treated for 48 h with vorinostat or sorafenib showed a significant decrease in anti-apoptotic Bcl-2, whereas the expression of pro-apoptotic Bax was increased in all cell lines (Figure 3A). Furthermore, the changes induced by the combination treatment were much more profound. Moreover, upon combination treatment, a more significant increase in the expression of p21 (Waf-1/CIP1) was observed compared with the individual treatments. By contrast, no obvious change in the cyclin-dependent kinase inhibitor p27 was observed in any treatment group.
Figure 3.
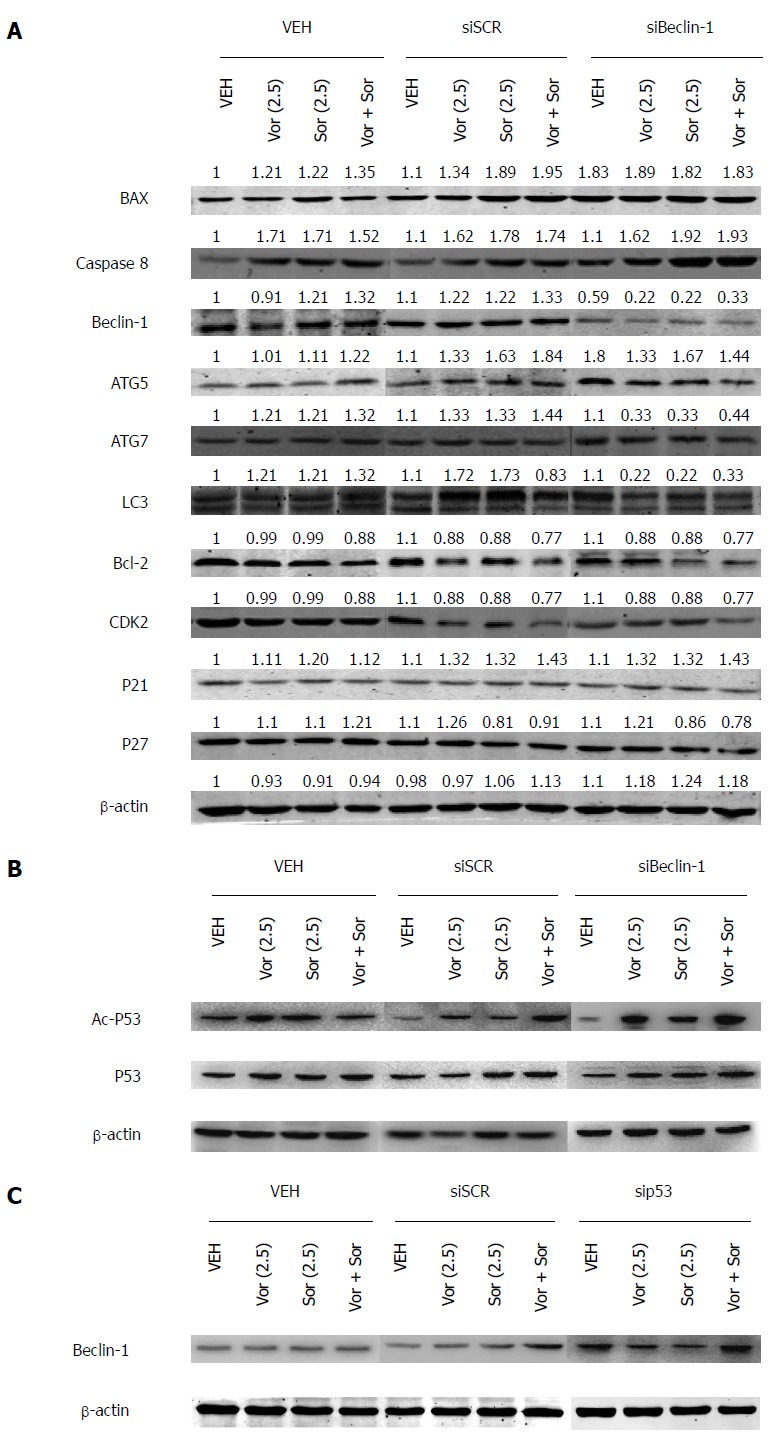
Sorafenib, vorinostat, or the drug combination induces the modulation of apoptosis-, cell cycle- and autophagy-related proteins. A: Representative images of Western blot showing the effect of treatment with siBeclin-1 and vorinostat/sorafenib on apoptosis-, cell cycle-, and autophagy-related proteins in HepG2 cells. siSCR, siRNA scramble; B: Representative images of Western blot showing the acetylated p53 level in HepG2 cells treated with or without siBeclin-1 or vorinostat/sorafenib. siSCR, siRNA scramble; C: Protein level of Beclin-1 in HepG2 cells infected with sip53 combined with or without vorinostat/sorafenib. All experiments were performed independently in triplicate. siSCR, siRNA scramble.
Activation of autophagy has been reported to mediate drug resistance and promote the survival of cancer cells. Therefore, we examined autophagy marker proteins, such as Beclin-1, ATG5, ATG7, and LC3, after drug treatment. As shown in Figure 3A, a significant increase in Beclin-1, ATG5, ATG7, and LC3 was observed in the cells treated with vorinostat or sorafenib, suggesting that the combined action of vorinostat and sorafenib generate a Beclin1-dependent protective form of autophagy.
HDACIs induce p53 acetylation
HDACIs induced apoptosis of human hepatoma HepG2 cells in a p53-dependent manner[16]; therefore, Western blot was used to further determine the acetylation and protein levels of p53. The HCC cells treated for 48 h with vorinostat or sorafenib or the combination (2.5 μmol/L) showed a significant increase in acetylated p53, but not in the total p53 level. Moreover, the acetylation of p53 increased noticeably after Beclin-1 knockdown, which indicated that Beclin-1 might negatively regulate the p53 acetylation level (Figure 3B). Furthermore, the Beclin-1 level was also increased in sip53 cells (Figure 3C).
Knockdown of Beclin-1 or the autophagy inhibitor 3-MA enhances the synergistic effect of the combination of vorinostat with sorafenib
To observe whether the inhibition of autophagy can enhance the synergistic effect of the combination of vorinostat with sorafenib, the knockdown of Beclin-1 was performed by transient transfection of small interfering RNA (siRNA) oligos. The knockdown efficiency was confirmed by quantitative RT-PCR (data not shown). As shown in Figures 4 and 5, the knockdown of Beclin-1 increased the growth inhibitory effects of the combination of vorinostat with sorafenib, as well as the cell cycle alterations and induction of apoptosis. Furthermore, the knockdown of Beclin-1 also increased the expression of Bax and p21 compared with the control group (Figure 3A). Consistently, the autophagy inhibitor 3-MA also enhanced the synergistic effect of the combination of vorinostat with sorafenib (Figure 5). These data indicate that the inhibition of autophagy enhanced the synergistic effect of the combination of vorinostat with sorafenib.
Figure 4.
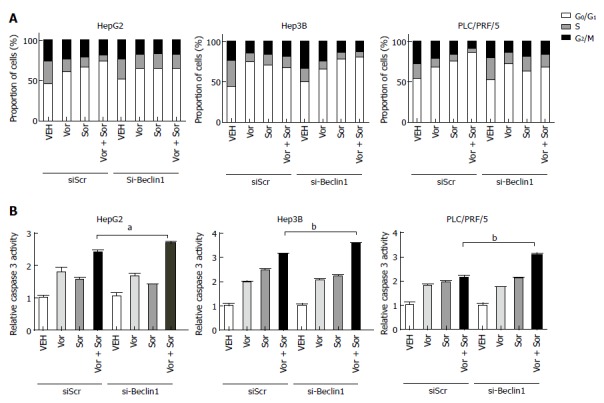
Knockdown of Beclin-1 enhances the synergistic effect of the combination of vorinostat with sorafenib. A: Knockdown of Beclin-1 enhanced the vorinostat/sorafenib drug combination-stimulated cell cycle alterations. siSCR, siRNA scramble; B: Knockdown of Beclin-1 enhanced the vorinostat/sorafenib drug combination-stimulated apoptosis. The results are expressed as mean ± SD from three independent experiments. aP < 0.05 and bP < 0.01 using Student’s t test. siSCR, siRNA scramble.
Figure 5.
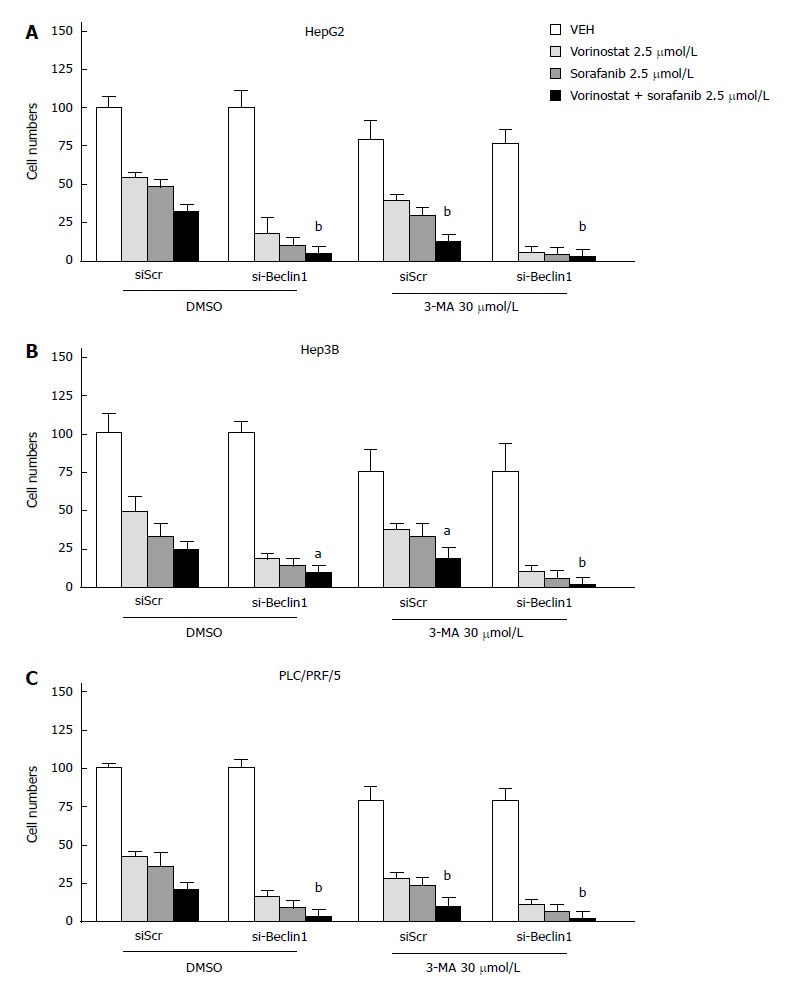
Knockdown of Beclin-1 or the autophagy inhibitor 3-MA enhances the growth inhibitory effects of the combination of vorinostat with sorafenib. A: HepG2; B: Hep3B; C: PLC/PRF/5. The results are expressed as mean ± SD from three independent experiments. aP < 0.05 and bP < 0.01 vs the siSCR DMSO group using Student’s t test.
DISCUSSION
Previous studies have confirmed that the inhibition of HDAC activity stimulates apoptosis in a variety of cancers, including breast and prostate cancer, neuroblastoma, hepatoma, gastrointestinal neuroendocrine tumor cells, and some types of hematologic malignancies[17]. HDACIs have been found to induce apoptosis, reduce tumor growth, and inhibit angiogenesis in hematological malignancies and solid tumors[18]. Defective histone acetylation regulatory enzymes have been identified in malignant cells, and HDAC inhibition may have anticancer properties through the restoration of normal acetylation[19]. Vorinostat has induced histone acetylation, cell cycle arrest, apoptosis, and anti-tumor activity in preclinical cancer models[20].
Sorafenib is the first drug that was approved for the clinical treatment of HCC and exhibits significant inhibitory effects on tumor cell proliferation and angiogenesis[9]. A better understanding of the mechanisms that underlie these effects would allow for an understanding of its efficacy and assist in predicting synergistic effects with other drugs. Recent studies also have found that sorafenib can activate tumor cell autophagy[11], but the precise role of autophagy in survival or death within these studies was not investigated. The present study was designed to explore the effect of combination treatment with sorafenib and HDACI on HCC cells. Meanwhile, we sought to determine the role of autophagy in the response of tumor cells to sorafenib or vorinostat and to understand how the levels of autophagy caused by HDACI could cause the additional effect of cell death by combined treatment with the multi-RTK inhibitor sorafenib.
Our study found that sorafenib or vorinostat potently inhibited the growth of HCC cells HEPG2, HEP3B, and PLC/PRF/5 in a dose-dependent manner, and the combination treatment exhibited higher antiproliferative activity. Submicromolar concentrations of sorafenib or vorinostat were sufficient to significantly inhibit the proliferation of Hep3B, HepG2, and PLC/PRF/5 cells, and the sorafenib or vorinostat concentration of half-maximal anti-neoplastic effects (IC50) was approximately 1.0 and 1.2 μmol/L, respectively, in all cell lines. The drug combination exhibited elevated anti-tumor effects in vitro compared with the individual agents in HCC cells.
The mechanisms involved in the HDACI-induced apoptosis are complex and differ among cell types[21]. HDACIs have been shown to up-regulate pro-apoptotic Fas, a member of the tumor necrosis factor receptor superfamily, and the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors/death receptors DR4 and DR5. Through these factors, HDACIs trigger an extrinsic pathway that is paralleled by the down-regulation of the caspase-8 inhibitor c-FLIP, leading to caspase-8 and subsequently caspase-3 activation[22]. Moreover, HDACIs have been reported to sensitize AML cells to HDACIs via ROS-mediated activation of the extrinsic apoptotic pathway[23]. Up-regulation of pro-apoptotic Bak and induction of the pro-apoptotic protein Bax can stimulate the mitochondrial apoptosis pathway[24]. HDACIs can also inhibit the expression of anti-apoptotic proteins, such as Bcl-2, Bcl-xL, XIAP, Mcl-1, and survivin[25]. Here, we demonstrated that the pro-apoptotic effect of HDAC inhibition by vorinostat or sorafenib or the combination in HCC cells is regulated by the activation of caspase-3 and a shift in the balance of pro-apoptotic Bax over anti-apoptotic Bcl-2. The combination of sorafenib with vorinostat significantly increased the expression of caspase-3 compared with the individual drug treatments. However, the knockdown of Beclin-1 enhanced the efficacy of the three treatment groups, indicating that autophagy may participate in the additional increase in tumor cell inhibition caused by the drugs alone or in combination.
Flow cytometry was performed to analyze the cell cycle to further determine the growth inhibitory activity of sorafenib and vorinostat. After treatment with the drugs, we found that cell cycle progression was blocked in both the G0/G1 and G2/M phases. The induction of cell cycle arrest was connected with an increase in the expression of the cyclin-dependent kinase inhibitor (CDKI) p21 Waf-1/Cip1, which is a key component of the cell cycle checkpoints, such as the G1/S and G2/M checkpoints. Accordingly, sorafenib and vorinostat were found to inhibit both the G1/S and G2/M transition, and the combination exhibited a higher inhibitory effect. With the knockdown of Beclin-1, the percentage of cells in the G0/G1 and G2/M phases increased again, in agreement with the fact that autophagy was involved in the additional levels of tumor cell inhibition caused by the drug combination. However, no cell-specific inhibitory effect was observed during any of the experiments. SiRNA knockdown partially reduced sorafenib HDACI lethality in hepatoma cells. Because sorafenib and vorinostat therapy will soon be explored in a phase I trial in hepatomas, our data suggest that the incorporation of GX15-070 (obatoclax) together with sorafenib-HDACI therapy may provide significant additional value in tumor control, including tumors that lack extrinsic pathway signaling. As previously reported by other groups, autophagy either protects cells from toxic stress or facilitates the toxicity of the stress, all of which seem to be based on the stimulus[26].
In conclusion, our study indicates that HDACIs and sorafenib interact in a highly synergistic manner to enhance the antiproliferative activity in HCC cells in vitro. Autophagy is activated and plays a compensatory role during treatment with sorafenib, vorinostat, or the drug combination. Importantly, the knockdown of Beclin-1 enhanced the synergistic effect of the combination of vorinostat with sorafenib, suggesting that the combination of HDAC inhibitors with sorafenib is a promising approach to reduce the dose of other anti-neoplastic drugs and to overcome drug resistance. Future animal studies will be required to fully verify the importance of autophagy in the treatment with sorafenib and vorinostat (or other HDACIs) as a therapeutic in HCC.
COMMENTS
Background
Hepatocellular carcinoma (HCC) is the most common cause of cancer-associated mortality in China and one of the leading causes of death in the world. Although sorafenib, a multi-target and multi-kinase inhibitor, currently sets the new standard for advanced HCC, the tumor response rates are actually quite low. Therefore, it is important to improve the response to sorafenib in HCC.
Research frontiers
Histone deacetylases (HDACs) are overexpressed in many types of tumor cells, including human hepatoma cells. Histone deacetylase inhibitors (HDACIs) are interesting as cancer therapeutics, and the combination of HDACIs with existing chemotherapeutic agents has been identified as a potential approach to overcome drug resistance.
Innovations and breakthroughs
Whether HDACIs can sensitize HCC cells to sorafenib treatment remains largely unexplored. The study indicates that HDACIs and sorafenib interact in a highly synergistic manner to enhance the antiproliferative activity in HCC cells in vitro. Autophagy is activated and plays a compensatory role during treatment with sorafenib, vorinostat, or the drug combination. Knockdown of Beclin-1 or the autophagy inhibitor 3-MA enhanced the synergistic effect of the combination of vorinostat with sorafenib.
Applications
These findings highlight that the combination of HDAC inhibitors with sorafenib appears to be a promising approach to considerably improve treatment response in HCC patients.
Peer review
The manuscript described the experiments that aimed to clarify the mechanism of enhancing hepatoma cell death by using the combination of two known anticancer agents: sorafenib and vorinostat. They concluded that inhibition of autophagy could enhance the effect of vorinostat/sorafenib.
Footnotes
Supported by Eastern Hepatobiliary Surgery Hospital of the Second Military Medical University
P- Reviewers: Leardkamolkarn V, Lu MY, Fouad YMM S- Editor: Wen LL L- Editor: Wang TQ E- Editor: Liu XM
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye QH, Wang L, Zhou J, Qiu SJ, Li Y, et al. A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2004;130:187–196. doi: 10.1007/s00432-003-0511-1. [DOI] [PubMed] [Google Scholar]
- 3.Verslype C, Van Cutsem E, Dicato M, Arber N, Berlin JD, Cunningham D, De Gramont A, Diaz-Rubio E, Ducreux M, Gruenberger T, et al. The management of hepatocellular carcinoma. Current expert opinion and recommendations derived from the 10th World Congress on Gastrointestinal Cancer, Barcelona, 2008. Ann Oncol. 2009;20 Suppl 7:vii1–vii6. doi: 10.1093/annonc/mdp281. [DOI] [PubMed] [Google Scholar]
- 4.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 5.Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 7.Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gammoh N, Marks PA, Jiang X. Curbing autophagy and histone deacetylases to kill cancer cells. Autophagy. 2012;8:1521–1522. doi: 10.4161/auto.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 10.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 11.Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, Grant S, Dent P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220–2231. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klappan AK, Hones S, Mylonas I, Brüning A. Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochem Cell Biol. 2012;137:25–36. doi: 10.1007/s00418-011-0869-0. [DOI] [PubMed] [Google Scholar]
- 14.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutter AP, Maaser K, Grabowski P, Bradacs G, Vormbrock K, Höpfner M, Krahn A, Heine B, Stein H, Somasundaram R, et al. Peripheral benzodiazepine receptor ligands induce apoptosis and cell cycle arrest in human hepatocellular carcinoma cells and enhance chemosensitivity to paclitaxel, docetaxel, doxorubicin and the Bcl-2 inhibitor HA14-1. J Hepatol. 2004;41:799–807. doi: 10.1016/j.jhep.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG, Schuppan D. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol. 2002;36:233–240. doi: 10.1016/s0168-8278(01)00257-4. [DOI] [PubMed] [Google Scholar]
- 17.Baradari V, Huether A, Höpfner M, Schuppan D, Scherübl H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr Relat Cancer. 2006;13:1237–1250. doi: 10.1677/erc.1.01249. [DOI] [PubMed] [Google Scholar]
- 18.Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69:1911–1934. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol. 2005;125:1045–1052. doi: 10.1111/j.0022-202X.2005.23925.x. [DOI] [PubMed] [Google Scholar]
- 21.Henderson C, Mizzau M, Paroni G, Maestro R, Schneider C, Brancolini C. Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA) J Biol Chem. 2003;278:12579–12589. doi: 10.1074/jbc.M213093200. [DOI] [PubMed] [Google Scholar]
- 22.Natoni F, Diolordi L, Santoni C, Gilardini Montani MS. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and the extrinsic apoptotic pathways. Biochim Biophys Acta. 2005;1745:318–329. doi: 10.1016/j.bbamcr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Yaseen A, Chen S, Hock S, Rosato R, Dent P, Dai Y, Grant S. Resveratrol sensitizes acute myelogenous leukemia cells to histone deacetylase inhibitors through reactive oxygen species-mediated activation of the extrinsic apoptotic pathway. Mol Pharmacol. 2012;82:1030–1041. doi: 10.1124/mol.112.079624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MG, Pak JH, Choi WH, Park JY, Nam JH, Kim JH. The relationship between cisplatin resistance and histone deacetylase isoform overexpression in epithelial ovarian cancer cell lines. J Gynecol Oncol. 2012;23:182–189. doi: 10.3802/jgo.2012.23.3.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider B, Münkel S, Krippner-Heidenreich A, Grunwald I, Wels WS, Wajant H, Pfizenmaier K, Gerspach J. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis. 2010;1:e68. doi: 10.1038/cddis.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin AP, Park MA, Mitchell C, Walker T, Rahmani M, Thorburn A, Häussinger D, Reinehr R, Grant S, Dent P. BCL-2 family inhibitors enhance histone deacetylase inhibitor and sorafenib lethality via autophagy and overcome blockade of the extrinsic pathway to facilitate killing. Mol Pharmacol. 2009;76:327–341. doi: 10.1124/mol.109.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]