

Abstract
The somatic mutation burden in healthy white blood cells (WBCs) is not well known. Based on deep whole-genome sequencing, we estimate that approximately 450 somatic mutations accumulated in the nonrepetitive genome within the healthy blood compartment of a 115-yr-old woman. The detected mutations appear to have been harmless passenger mutations: They were enriched in noncoding, AT-rich regions that are not evolutionarily conserved, and they were depleted for genomic elements where mutations might have favorable or adverse effects on cellular fitness, such as regions with actively transcribed genes. The distribution of variant allele frequencies of these mutations suggests that the majority of the peripheral white blood cells were offspring of two related hematopoietic stem cell (HSC) clones. Moreover, telomere lengths of the WBCs were significantly shorter than telomere lengths from other tissues. Together, this suggests that the finite lifespan of HSCs, rather than somatic mutation effects, may lead to hematopoietic clonal evolution at extreme ages.
Mutations are called somatic if they were acquired in a tissue cell during organismal development or later in life, rather than being inherited from a germ cell. As such, somatic mutations lead to genotypic and possibly phenotypic heterogeneity within and between tissues, and they may compromise growth or lead to a growth advantage (Frank 2010). Because somatic mutations often occur during cell division, frequently dividing cell types are more prone to acquire somatic mutations than tissues that rarely divide (Youssoufian and Pyeritz 2002). Consequently, frequently dividing cell types, i.e., epithelial cells, hematopoietic cells, and male germ cells are vulnerable to somatic mutations that may lead to tumor development or other diseases and disorders. Therefore, most studies regarding somatic mutations have been attempts to discover mechanisms leading to cancer and disease (Youssoufian and Pyeritz 2002; Erickson 2010; Hanahan and Weinberg 2011).
It has been estimated that the adult human blood compartment is populated by the offspring of approximately 10,000–20,000 hematopoietic stem cells (HSCs) (Abkowitz et al. 2002). HSCs self-renew about once every 25–50 wk to create two daughter cells equivalent to their parent, and they differentiate to create offspring clones with multipotent progenitor cells that generate the much larger number of diverse blood cells via hematopoiesis (Catlin et al. 2011). Over time, somatic mutations will gradually accumulate within the HSCs, and the genotypes of the HSCs along with their offspring clones will diverge and lead to new clones of varying sizes.
Recent publications show that the genomes of patients with acute myeloid leukemia (AML) contain hundreds of somatic mutations that accumulate with age (Ley et al. 2008; Mardis et al. 2009; Ding et al. 2012), and that most of these mutations occur as random events in HSCs before one of them acquires a specific pathogenic mutation leading to AML (Welch et al. 2012). Similar patterns of clonal evolution have also been shown for the development of chronic lymphocytic leukemia (CLL) (Landau et al. 2013). However, it is currently unknown to what extent healthy HSCs acquire somatic mutations and which types of mutations can be tolerated in the genome during a lifetime without causing disease.
We set out to determine the prevalence and types of single nucleotide and small insertion/deletion mutations that are somatic within the healthy blood genome. Since the occurrence of somatic copy number changes has been shown to increase with age in several tissues in mice (Dolle and Vijg 2002) and also in peripheral blood in cancer-free humans (Forsberg et al. 2012; Jacobs et al. 2012; Laurie et al. 2012), we assumed that single nucleotide somatic mutations might also increase with age. Therefore, we chose a healthy person of extreme age as our subject, anticipating that during a long lifetime, mutations leading to the fittest HSCs might lead to clonal selection and thus the detectability of somatic mutations (Naylor et al. 2005; Gibson et al. 2009). Together, the large number of cellular divisions during a long lifetime and the expected age-dependent clonality could provide better statistical representation of the mutation rate and spectrum. To detect somatic mutations in peripheral blood, we compared its DNA sequence with that from the brain tissue from the same individual. Since cells in occipital brain tissue rarely divide after birth (Spalding et al. 2005), it is expected that these cells do not acquire many somatic mutations, so that DNA isolated from occipital brain tissue may serve as a candid representation of the germline control genome.
Such an analysis of somatic mutations in the healthy white blood cell (WBC) population allowed us to determine the number of (detectable) mutations acquired during a lifetime and to what extent the healthy blood compartment is subject to clonal evolution. Furthermore, we investigated where such somatic mutations occurred in the genome and to what extent the spectrum of somatic mutations compares with the spectrum of germline mutations sustained in offspring populations and with the spectrum of mutations implicated in heritable disease.
Results
Subject: W115, a supercentenarian
The subject of our study was W115, a woman who lived to the age of 115 and who was regarded as the oldest human being in the world at the time of her death (Holden 2005). At the age of 82, W115 sent a written consent to donate her body to science after death. W115 had no symptoms of hematological illnesses, and autopsy showed that she did not suffer from vascular or dementia-related pathology. She had breast tumor surgery at age 100 and died 15 years later of a gastric tumor that metastasized into her abdomen (den Dunnen et al. 2008). Since W115 never received mutation-inducing chemotherapy, the somatic mutations in the genomes of her tissues are purely a consequence of normal aging.
DNA was isolated from several tissues that were collected during autopsy: whole blood, brain (occipital cortex), artery (media and endothelium), kidney (renal pyramid and minor calyx), heart, liver, lung, spleen, aorta, and the gastric tumor that she died of. DNA was also isolated from the breast tumor that was removed at age 100.
Blood cells had shorter telomeres than cells from other tissues
Telomeres shorten with every cell division (Hastie et al. 1990). To ascertain cellular turnover differences between W115’s whole blood and brain cells, we measured telomere lengths (TLs) in DNA isolated from these and several other W115 tissues (Lin et al. 2010). Telomeres in blood cells were 17× shorter than telomeres in brain cells and the shortest of all tissues tested (Fig. 1). This result supports our expectation that the (precursors of) W115’s blood cells underwent many more divisions than cells isolated from occipital brain. Since the TLs between tissues of a newborn are similar (Okuda et al. 2002) and since occipital brain cells only rarely divide after birth (Spalding et al. 2005), the 17-fold TL reduction of blood cells can be considered relative to birth and thus extremely short (Frenck et al. 1998; Hewakapuge et al. 2008).
Figure 1.

Mean telomere length of W115 tissues. DNA from W115 tissues was isolated using the Qiagen DNA isolation kit and the Promega Wizard kit. Both DNA isolates were measured twice for telomere length (T/S ratio). (*) Blood DNA was isolated only with the Promega Wizard kit.
Detected and confirmed somatic SNVs and indels were mostly novel
To detect somatic point mutations (single nucleotide variants [SNVs]) and short insertions/deletions (indels), we sequenced the DNA isolated from peripheral blood and from brain tissue from W115 to >60× mean read depth for each tissue using SOLiD sequencing (Fig. 2). During subsequent sequence analysis, we identified 612 candidate somatic SNVs in blood that could not be detected in the brain genome. Validation experiments showed that of the candidates with high read depth, almost all novel (i.e., unknown to dbSNP) and only half of the known candidates could be confirmed (Table 1A). Likewise, we identified 107 candidate somatic SNVs in brain that were not detected in the blood genome, but none of these could be confirmed. We also detected 30 candidate indels in the whole blood genome and three in the brain genome (indel detection was genome wide, not only in the nonrepetitive genome). We tested 23 indels in validation experiments and confirmed 22 somatic indels in blood (Table 1B). Together, we conclude that somatic mutations could only be detected in blood and were mostly novel. For a detailed description of somatic variant detection and validation, see Supplemental Material SR1–SR4, the corresponding Supplemental Figures S1–S3, and Supplemental Tables S1–S8.
Figure 2.

Somatic variant detection pipeline SNV and indel detection. Whole-genome sequences of W115 blood and brain tissues were mapped to hg19. Variants were called using both GATK and SAMtools. The SNVs that overlapped between the two genotyping algorithms were considered most trustworthy and were used for further analysis with high stringency (HS-SNV) and low stringency (LS-SNV) filters. Indels were passed through a high stringency (HS-indel) and low stringency (LS-indel) read-depth filter. Candidate somatic SNVs and indels were confirmed by validation with Ion Torrent PGM sequencing and/or Sanger sequencing. The number of confirmed SNVs and indels was extrapolated to account for those not validated. The number of SNVs was also extrapolated to the whole genome, whereas for indel detection the whole genome was assessed.
Table 1.
Validation of variants detected in blood but not in brain
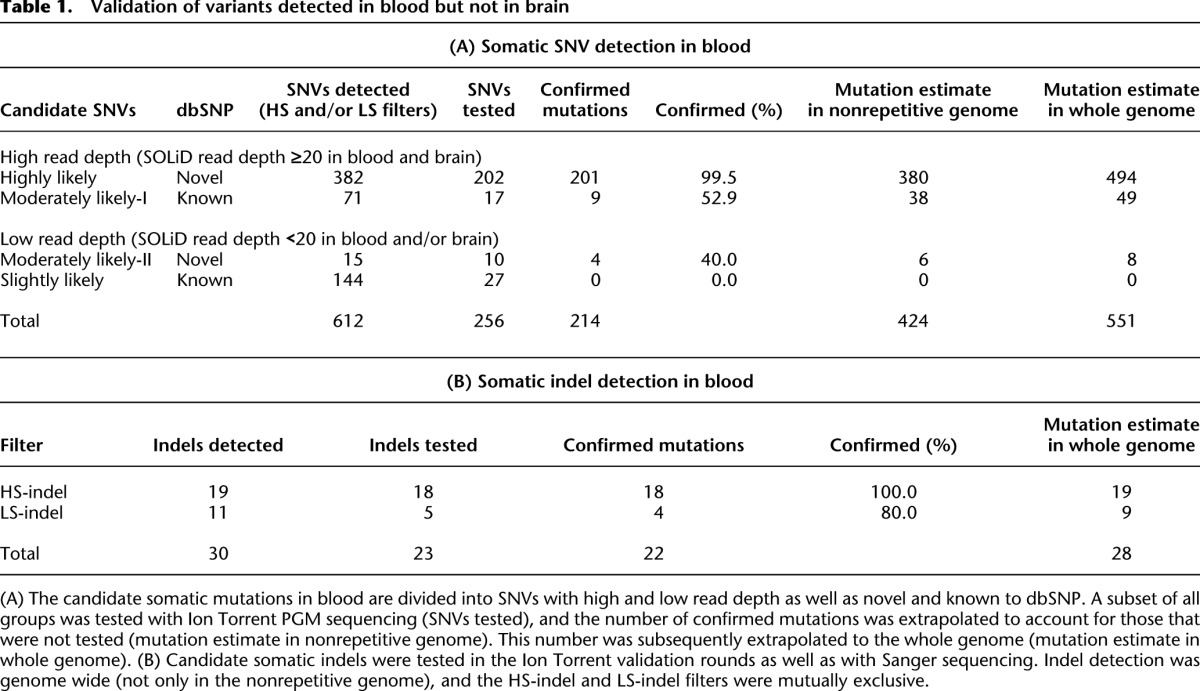
The whole blood genome included roughly 600 somatic mutations
Based on the proportion of tested variants that were confirmed to be somatic mutations, we estimate that there were roughly 424 somatic SNVs in the nonrepetitive genome (Fig. 2; Table 1). Since the nonrepetitive genome comprised 77% of the whole genome (Supplemental Table S1), we estimate that we could have confirmed about 551 somatic SNVs, had we been able to assess the whole genome. Based on the fraction of confirmed somatic indels, we estimate that we could have confirmed 28 somatic indels in the whole genome, of which 22 were in the nonrepetitive genome. Together, we estimate that the nonrepetitive genome included approximately 450 somatic mutations (424 SNVs plus 22 indels) and the whole blood genome included roughly 600 somatic mutations (551 SNVs plus 28 indels). Because our stringent pipeline did not assess the mutation-prone repetitive sequences, and we required the same genotype calling by both the GATK (McKenna et al. 2010; DePristo et al. 2011) and SAMtools variant callers (Li et al. 2009), we consider these numbers to be lower bounds.
Somatic mutations detected in blood were not detected in tumor or other native tissues
The somatic mutations that we detected in blood were not detected in the breast cancer that W115 had at age 100 nor in the gastric tumor she had at age 115. This indicates that the somatic mutations were not derived from tumor cells present in the blood circulation at the time of her death. The validation panel was also used to test samples derived from aorta, artery (endothelium), heart, and kidney (renal pyramid) tissues. None of the confirmed somatic mutations detected in blood were detected in these tissues, and only an occasional mutation detected in blood could be detected in artery (media), kidney (minor calyx), liver, and spleen tissues. In contrast, almost all somatic mutations were detected in DNA derived from lung tissue, but the fraction of reads with the variant allele (the variant allele frequency [VAF]) was much lower in lung tissue than in blood (Fig. 3). Presumably, the DNA isolated from lung tissue was contaminated with blood DNA due to a vast leukocyte presence in the lung tissue. Blood contamination of the brain DNA was kept at a minimum because there were almost no blood cells in the brain blood vessels after the brain was perfused during fixation (Supplemental Material SR5).
Figure 3.

Presence of single nucleotide mutations detected in blood in other W115 tissues. Box plot of the VAF values for the 214 confirmed somatic mutations detected in blood for a variety of other tissues. On each box, the central mark is the median VAF; the edges of the box are the 25th and 75th percentiles, the whiskers extend to the most extreme data points not considered outliers, and outliers are plotted individually as red crosses.
Somatic mutations were not predicted to have a functional selective advantage
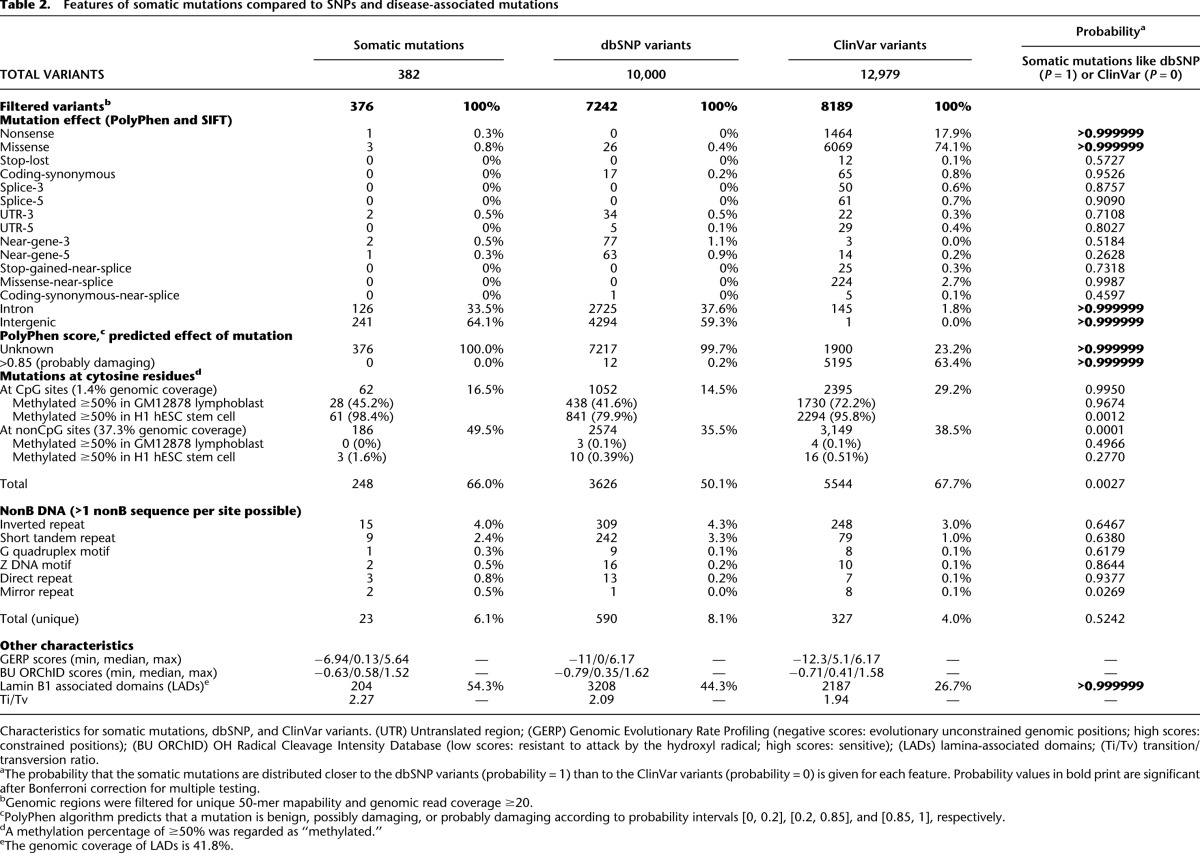
To characterize the somatic mutations acquired in the healthy blood compartment, we used the complete group of 382 “highly likely” somatic mutations because almost all variants in this group that were tested in validation experiments were confirmed to be true mutations (Table 1; Supplemental Material SR2). Of these mutations, 376 passed consistency filters and were used for further analysis (Methods; Supplemental Material SM7). None of the 376 somatic mutations that mapped to coding regions were predicted to have a deleterious effect on protein function by the SIFT and PolyPhen algorithms (Kumar et al. 2009; Ng et al. 2009). For details of functional effect prediction, see Supplemental Material SR6. Furthermore, none of the mutations were previously associated with clinical outcome; they do not appear in the COSMIC catalogue of somatic mutations in cancer (Forbes et al. 2011) or in the Human Gene Mutation Database (HGMD) (http://www.hgmd.org). In particular, none have been implicated in any form of leukemia. For further characterization, we compared the somatic mutations to a random set of mostly nonpathogenic polymorphisms (dbSNP) and with single nucleotide mutations associated with disease (ClinVar) (Table 2; Methods; Supplemental Material SM8). Like the somatic mutations, most of the dbSNP variants mapped to noncoding regions with unknown functional effect, whereas almost all ClinVar variants mapped to coding regions and were predicted to have a “probably damaging effect” on protein function (Table 2; Wei et al. 2011). Concluding, somatic mutations, like dbSNP variants, were not predicted to have a functional selective advantage.
Table 2.
Features of somatic mutations compared to SNPs and disease-associated mutations
Spectrum of functional elements of somatic mutations is similar to dbSNP variants and different from disease-associated variants
To determine whether somatic mutations in the healthy blood compartment located to specific functional genomic elements, we intersected mutated loci with functional elements tracked by ENCODE; for tracks, see Supplemental Table S9 (The ENCODE Project Consortium 2012). We then compared the enrichment/depletion spectra with those of the dbSNP and ClinVar variants. Somatic mutations and dbSNP variants did not cluster at confined genomic locations (Supplemental Fig. S5; Supplemental Material SR7, SM9). They were, however, significantly enriched in Lamin B1 associated domains (LADs), in gene-poor Giemsa positive, strongly A/T-rich heterochromatin, in solvent-accessible sites (BU ORChID), and at methylated cytosines (Fig. 4; Supplemental Table S9; Balasubramanian et al. 1998; Greenbaum et al. 2007; Guelen et al. 2008; Meissner et al. 2008). In contrast, they were significantly depleted in regions with histone methylation/acetylation associated with active gene transcription, especially in regions with high H3K36me3 levels, associated with transcriptional activation and elongation (Ram et al. 2011) and at conserved loci (GERP) (Supplemental Fig. S6; Davydov et al. 2010). The ClinVar variants, on the other hand, were especially depleted in regions with high H3K9me3 levels, associated with gene repression and silencing.
Figure 4.
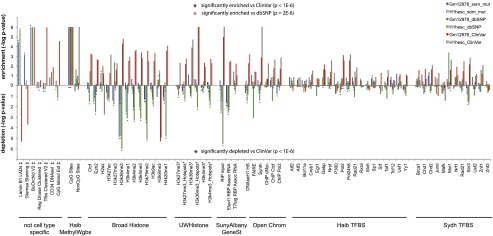
Enrichment and depletion of somatic mutations in functional genomic elements tracked by ENCODE. Enrichment depletion analysis: (y-axis) −log P-values indicate the enrichment or depletion of somatic mutations (blue bar), dbSNP variants (green bar), and ClinVar (red bar) variants for each functional element tracked by ENCODE. The lowest possible P-value = 1 × 10−6, or –log P-value = 6. Comparative analysis: (Black stars) Variant set is significantly enriched (star above bars) or depleted (star below bars) relative to ClinVar variants, P < 1 × 10−6. Pink star (BU ORChID track only) Variant set is significantly enriched relative to dbSNP variants. Comparisons of other variant sets did not yield significant differences. (‡) Tracks are not specific for cell lines. (*) Track not available for H1 hESC, track for H7 human embryonic stem cell line used instead.
In a second comparative analysis, we analyzed whether the genomic functional elements were differentially enriched or depleted with loci of somatic mutations, dbSNP and ClinVar variants (details in Supplemental Material SM9). The somatic mutations were significantly more enriched in solvent accessible sites (BU ORChID track) compared to dbSNP loci, but in all other functional elements, dbSNP variants were similarly enriched/depleted. In contrast, the somatic mutation and dbSNP spectra differed significantly from that of the disease-associated variants (Fig. 4; Supplemental Fig. S6; Supplemental Table S9). In short, somatic mutations overlap with functional elements similar to nonpathogenic dbSNP variants, but not with disease-associated variants, supporting their harmless nature.
Mutations occurred in a cell with a stem-cell-like methylation signature
A subset of the somatic mutations may have resulted from the spontaneous deamination of methylated cytosines, forming a thymine at that location. Indeed, 62 of the 376 somatic mutations mapped in putatively methylated CpG sites, indicating a significantly increased mutation-likelihood at CpG loci (P-value < 1 × 10−6) (Fig. 4; Supplemental Table S9). To determine whether the methylation signature of a stem or a lymphoblastoid cell could explain the loci of the detected somatic mutations, we compared their loci with the methylation status of CpG sites of the H1 hESC stem cell line and GM12878 lymphoblastoid cell line as tracked by ENCODE, HAIB Methyl RRBS (Meissner et al. 2008). In the GM12878 cell line, 50.7% of the CpG sites were methylated, and 28/62 of the somatic mutations coincided with these loci, which could be expected by chance (P-value = 0.8) (Table 2). In contrast, 85.4% of the CpG sites were methylated in the H1 hESC cell line, and 61/62 loci of the somatic mutations overlapped with these loci, significantly more than expected by chance (P-value = 5.7 × 10−5) (Table 2). From this, we conclude that the somatic mutations are indeed more likely to occur at methylated CpG sites. Since the somatic mutations largely overlap with the methylated CpG sites of H1 hESC stem cells, they likely occurred in a cell type with a methylation signature resembling a stem cell rather than a GM12878 differentiated lymphoblastoid cell.
Distribution of the variant allele frequency suggests that somatic mutations were in two clones
The VAF distribution based on the Ion Torrent PGM reads for the 201 confirmed somatic mutations (Table 1) shows two well-resolved peaks at VAF values of 0.22 and 0.32, which is corroborated by fitting a mixture of Gaussians (Fig. 5; Supplemental Table S10; Methods). After multiplying by 2 to correct for assumed heterozygosity of the somatic mutations, this implies that two clones were present, comprising ∼44% and ∼64% of the peripheral blood cells, respectively. The sum of these percentages is appreciably larger than 100%: 106.2%–112.8%, 95% CI (Supplemental Table S10; Supplemental Material SR9). Thus, the smaller clone at VAF = 0.32 was most likely subsidiary to the larger one at VAF = 0.22, representing the mutations from a more recent subclonal expansion. The remaining ∼36% of the cells were presumably present in much smaller clones that were below our detection limit. The characteristics of variants in the two peaks were similar (Supplemental Material SR10; Supplemental Fig. S7).
Figure 5.

Gaussian fit for VAF values of 201 confirmed somatic mutations. (Gray) Histogram of VAF values of 201 mutations confirmed by Ion Torrent PGM; (red) Gaussian fit of clone A; (green) Gaussian fit of clone B; (blue) Gaussian fit of background mutations; (black) resultant mixture of Gaussians.
No disease in W115 blood
Dominant clones in the blood have been associated with leukemia. However, there were no clinical signs of leukemia at the time of W115’s death. Moreover, no gross chromosomal abnormalities characteristic of leukemia were detected in W115’s WBCs by CGH array analysis or in the sequencing data (Supplemental Fig. S8), thereby excluding the presence of CML, most forms of precursor B-ALL, and AML (Simons et al. 2012).
Unknown germline variants detected in DNA repair genes
W115 may have had germline variants that altered DNA repair mechanisms. (Possibly) damaging effects on protein function, which may also favorably modify protein function, were predicted for variants in the BRCA1, POLL, RAD50, PKD1L1, DCLRE1A, CCNH, EXO1, LIG4, BRCA2, CHAF1A, XRCC1, RNF168, and WRN genes, which all had population allele frequencies >0.1, indicating that these variants commonly occur. Furthermore, W115 had homozygous novel variants in BRCA2, XRCC1, and PKD1L1 that were predicted to modify protein function (Supplemental Table S11). To what extent these variants contribute to DNA repair needs to be further analyzed with functional tests.
Discussion
Here we report, for the first time, a “per clone” somatic mutation burden estimate in truly uncultured blood cells. We estimate that approximately 450 somatic mutations, mostly novel to dbSNP, accumulated in the nonrepetitive genome of a hematopoietic stem cell (HSC) clone of a 115-yr-old woman, indicating that about 600 somatic mutations accumulated in the whole genome of this HSC clone. Since we did not assess the mutation-prone repetitive genome and applied extensive filters to the data, these estimates should be considered lower bounds.
The somatic mutations detected in blood were not tumor-derived, and only a few were detected at minimal frequencies in other tissues, suggesting that they represent the fraction of blood infiltration in these tissues. Together, this indicates that the mutations were confined to the blood compartment.
Somatic mutations routinely occur in the blood genome
These somatic mutations accumulated in a cell type with a methylation signature resembling an embryonic stem cell—possibly an HSC (Broske et al. 2009). If we assume that it took 115 years for all of the roughly 450 somatic mutations to accumulate in the nonrepetitive genome of one HSC, then with a constant mutation rate, this amounts to about four mutations per year or about three mutations per division, given that HSCs self-renew once every 25–50 wk (Catlin et al. 2011). This is in line with the finding that exomes from three clones from seven healthy individuals acquired 0.13 mutations per year (Welch et al. 2012), which extrapolates to about five mutations per year in the nonrepetitive genome. Likewise, the vast majority of the somatic mutations detected in HSC clones from patients with myelodysplastic syndrome were harmless events randomly distributed in the genome (Walter et al. 2012). Therefore, it is likely that somatic mutations are routinely acquired in HSCs during normal aging. Note, however, that comparison of mutation rates for any clone/genome is subject to uncertainty because one cannot determine when each mutation was acquired and how the analyzed clones expanded during a lifetime. Also, inconsistencies between sequencing techniques and downstream analyses complicate an accurate comparison of mutation numbers.
Chromatin organization influences the genomic susceptibility to acquire somatic mutations
Somatic mutations detected in the healthy blood compartment did not map to coding regions and are not predicted to confer selective advantage on the growth pattern of their host cell. The mutations were enriched in mutation-prone sites, such as methylated cytosines and sites accessible by surrounding solvents. They occurred in regions that were not evolutionarily conserved and in AT-rich heterochromatin and gene-poor sequences such as LADs (genomic regions that attach to the nuclear lamina to secure the spatial orientation of the chromosome in the nucleus). In these regions, mutations are unlikely to have deleterious effects on cellular fitness. In contrast, they were depleted in regions where mutations may lead to favorable or adverse effects on cellular fitness, such as in actively transcribed gene-rich regions. The mutation spectrum thus resembles that of mutations that occurred in germ cells and persisted in the offspring population, often with no pathogenic effects (dbSNP) but distinctly different from that of disease-associated mutations (ClinVar). These results are in agreement with recent findings that chromatin organization influences the genomic susceptibility to acquire somatic mutations (Michaelson et al. 2012; Schuster-Bockler and Lehner 2012).
Thus, it appears that the many somatic mutations accumulated in the healthy HSC compartment were harmless passenger mutations that occurred at nondeleterious genomic regions.
W115 blood compartment was oligoclonal
The majority (∼65%) of the healthy blood compartment of W115 was populated by the offspring of two HSC clones, one of which was likely derived from the other. A possible explanation for this oligoclonality may be found in the extremely short telomere lengths (TLs) of W115’s peripheral blood cells. Telomere attrition to critical lengths has been associated with the replicative senescence of somatic cells (Frenck et al. 1998). Although the TLs of W115’s blood cells were in line with normal telomere shortening in blood as a function of age, a 17-fold reduction in TL relative to birth (proxied by brain tissue) is extreme (Frenck et al. 1998). The very long lifetime of W115 may have allowed many HSCs to reach critically short TLs, leading to their disappearance from the HSC pool (Orford and Scadden 2008).
Possible implications of oligoclonality for the immune system
According to a recent model (Catlin et al. 2011), roughly 11,000 HSCs reside in the marrow, of which only 1300 are actively generating WBCs, implying that most of the HSCs are quiescent. The composition of the HSC compartment changes significantly during a lifetime and consists of several types of HSCs. Each has its own differentiation requirements and self-renewal programs and is subject to stem cell exhaustion (Roeder et al. 2008; Forsberg et al. 2012). Although percentages differ widely between individuals and with age, ∼35% of the peripheral WBCs are of lymphoid origin (T and B lymphocytes and NK cells), and ∼65% are of myeloid origin (Stulnig et al. 1995). The most common myeloid cells, granulocytes, are the immediate progeny of actively contributing HSC clones since they have a half-life of only 6–8 d. As a consequence of the finite lifespan of HSCs, the short-lived myeloid and lymphoid WBCs may have been continuously generated by the offspring of only a few HSC clones that were still active at the time of W115’s death. In contrast, T lymphocytes, which make up ∼25% of the WBCs, are generated in the thymus, where they are seeded by a limited number of HSC clones (Weerkamp et al. 2006) and can expand by homeostatic peripheral proliferation. Because thymus function and output rapidly decrease with age (Montecino-Rodriguez et al. 2013), most of the T cells may have originated decades ago from HSC clones that were active then. Although data from more subjects is needed to provide further support for these hypotheses, it would not be surprising if in very old individuals only a few active HSCs clones were left to contribute to the T cell pool, and that T cell-mediated immunity is upheld by peripheral T cells that are offspring from older HSCs.
We conclude that there is a vast somatic mutation background, even in a healthy blood compartment, with a spectrum similar to that between generations (dbSNP) and distinctly different from disease-associated mutations. The detected somatic mutations occurred in an undifferentiated cell type but had no favorable or adverse effects on genomic fitness. Moreover, our telomere length measurements suggest that the oligoclonality in the HSC pool of W115 may be a consequence of the finite lifespan of HSCs.
Methods
DNA isolation and telomere length analysis
Details about DNA isolation procedures are described in Supplemental Material SM1. The telomere-to-single copy gene (T/S) ratio was determined as described in Lin et al. (2010) using a real-time PCR assay and using DNA isolated from the Hela cancer cell line as reference DNA. For each tissue, the T/S ratio was measured twice for both the Promega and the Qiagen DNA isolations.
Identification of somatic SNVs and indels
We used SOLiD paired-end sequencing to obtain whole-genome sequences of W115 blood and brain tissues, each with approximately 60× read depth (see Supplemental Material SM2 for further details). Variants were called using both the GATK Unified Genotyper and SAMtools (v0.1.18) (Fig. 2; Supplemental Material SM3). The SNV calls that overlapped between GATK and SAMtools genotyping algorithms were considered most trustworthy. SNVs were passed through high stringency (HS-SNV) and low stringency (LS-SNV) filters. Indels in blood and brain were detected with GATK and BFAST (Homer et al. 2009); the two sets of read counts were filtered to eliminate spurious indel calls. For further descriptions of SNV and indel stringency filters see Supplemental Material SM4, SM5 and Supplemental Figure S9.
Mutation validation experiments
A subset of somatic mutation candidates was validated in all available tissues by targeted sequencing using the Ion Torrent PGM with an average mapped read depth >2000×. Indels that mapped to repeat regions or homopolymer sequences could not be validated with Ion Torrent PGM sequencing and were validated with Sanger sequencing. Details of experimental procedures are described in the Supplemental Material SM6.
Comparison of characteristics of mutations with dbSNP and ClinVar variants
Somatic mutation characteristics were compared in a random set of 10,000 mostly nonpathogenic polymorphisms (dbSNP; http://www.ncbi.nlm.nih.gov/SNP) and with 12,979 single nucleotide mutations implicated in disease (ClinVar; http://www.ncbi.nlm.nih.gov/clinvar). For the comparison of variant characteristics, we applied a consistency filter by including variants that mapped in unique sequences (50-mer mapability track; UCSC) and regions with high read depth (≥20× read coverage in the blood and the brain sequence) (for details, see Supplemental Material SM7). This left 376/382 of the “highly likely” somatic mutations, 7242/10,000 dbSNP variants, and 8189/12,979 ClinVar variants for analysis (Table 2). For each set of variants, we determined the percentage of variants with a characteristic listed in Table 2. Distances between these percentages were compared, taking their associated uncertainty into account. Probability values indicate the probability that the set of somatic mutations is more similar to dbSNP than ClinVar (for further details, see Supplemental Material SM8).
Enrichment/depletion of variants in functional genomic regions (ENCODE)
Somatic mutations, dbSNP variants, and ClinVar variants were intersected with functional genomic regions tracked by ENCODE. In addition to seven cell-line-independent tracks, we chose to use the 140 GM12878 B-lymphocyte and 103 H1 hESC or H7 human embryonic stem-cell lines because we speculated that somatic mutations in an HSC had a differentiation status between a human stem cell and a fully differentiated lymphocyte (tracks are listed in Supplemental Table S9). To detect enrichment or depletion of a variant set in genomic functional elements, we calculated an “ENCODE score” by summing the track-values at the variant loci. Significance was determined by comparing this value with the ENCODE scores of 1,000,000 equally sized sets of random loci as further described in Supplemental Material SM9. Next, we compared levels of enrichment/depletion between the different variant sets by comparing the ENCODE score between variant collections (for the exact test, see Supplemental Material SM9).
Methylation signature of somatic mutations
Methylation signatures were taken from two whole-genome bisulfite sequencing (WGBS) data sets for H1 hESC and GM12878 cell lines: HAIB Methyl RRBS tracks from ENCODE (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40832). For each cell line, these tracks indicate methylation percentages for each cytosine, in (A) CpG sites and (B) nonCpG sites. A methylation percentage of ≥ 50% was regarded as “methylated,” and only sites that were assessed for both H1 hESC and GM12878 cell lines were included in the analysis (98% overlap).
Mixture of Gaussians fit to the VAF distribution of confirmed somatic mutations
For the 201 confirmed mutations, a mixture of three Gaussians was fit to the VAF distribution using the Matlab gmdistribution.fit function with regularization of 0.0001. The fit was repeated 50 times from different initial guesses using a maximum of 500 iterations (Fig. 5). Two Gaussians fit the mutations within the large clones, whereas the third Gaussian component fit the background mutations.
Detection of germline variants in genes associated with DNA repair
A list of 177 genes associated with DNA repair was taken from http://sciencepark.mdanderson.org/labs/wood/dna_repair_genes.html#Human DNA Repair Genes. All 4880 germline variants in the W115 genome that mapped in these genes and passed the consistency filter were analyzed with SIFT and PolyPhen.
Data access
All sequence data from this study have been submitted to the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega/) under accession number EGAS00001000660.
Competing interest statement
Life Technologies provided financial support for whole-genome sequencing using SOLiD technology and for validation experiments using the Ion Torrent PGM.
Acknowledgments
The authors would like to thank Mrs.Hendrikje van Andel-Schipper: without her contribution this project would not have been possible. We would also like to thank Dr. Q. Waisfisz, Y. Waterham, P.P. Eijk, D. Israeli, F. Rustenburg, Dr. G.T.N. Burger (Bethesda Hospital, Hoogeveen), Dr. J. Hoozemans, P. Poddighe, Dr. A.W. Langerak, M. Rijnen, and Q. Doan. Support for T.J.N., M.A.M., and S.L., as well as for computer time at SDSC was provided by the National Institutes of Health via Grant UL1 TR000109. T.J.N. was also funded by the Scripps Health Dickinson Fellowship.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162131.113.
Freely available online through the Genome Research Open Access option.
References
- Abkowitz JL, Catlin SN, McCallie MT, Guttorp P 2002. Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood 100: 2665–2667 [DOI] [PubMed] [Google Scholar]
- Balasubramanian B, Pogozelski WK, Tullius TD 1998. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc Natl Acad Sci 95: 9738–9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, et al. 2009. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet 41: 1207–1215 [DOI] [PubMed] [Google Scholar]
- Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL 2011. The replication rate of human hematopoietic stem cells in vivo. Blood 117: 4460–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen WF, Brouwer WH, Bijlard E, Kamphuis J, van Linschoten K, Eggens-Meijer E, Holstege G 2008. No disease in the brain of a 115-year-old woman. Neurobiol Aging 29: 1127–1132 [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle ME, Vijg J 2002. Genome dynamics in aging mice. Genome Res 12: 1732–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP 2010. Somatic gene mutation and human disease other than cancer: an update. Mutat Res 705: 96–106 [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Rasi C, Razzaghian HR, Pakalapati G, Waite L, Thilbeault KS, Ronowicz A, Wineinger NE, Tiwari HK, Boomsma D, et al. 2012. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet 90: 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SA 2010. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci (Suppl 1) 107: 1725–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenck RW Jr, Blackburn EH, Shannon KM 1998. The rate of telomere sequence loss in human leukocytes varies with age. Proc Natl Acad Sci 95: 5607–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, Kondeatis E, Nilsson BO, Wikby A, Kipling D, Dunn-Walters DK 2009. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 8: 18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum JA, Pang B, Tullius TD 2007. Construction of a genome-scale structural map at single-nucleotide resolution. Genome Res 17: 947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC 1990. Telomere reduction in human colorectal carcinoma and with ageing. Nature 346: 866–868 [DOI] [PubMed] [Google Scholar]
- Hewakapuge S, van Oorschot RA, Lewandowski P, Baindur-Hudson S 2008. Investigation of telomere lengths measurement by quantitative real-time PCR to predict age. Leg Med (Tokyo) 10: 236–242 [DOI] [PubMed] [Google Scholar]
- Holden C , ed. 2005. Oldest body to science. Science 309: 1670 [Google Scholar]
- Homer N, Merriman B, Nelson SF 2009. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE 4: e7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, et al. 2012. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 44: 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081 [DOI] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. 2013. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, et al. 2012. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 44: 642–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. 2008. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456: 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Epel E, Cheon J, Kroenke C, Sinclair E, Bigos M, Wolkowitz O, Mellon S, Blackburn E 2010. Analyses and comparisons of telomerase activity and telomere length in human T and B cells: insights for epidemiology of telomere maintenance. J Immunol Methods 352: 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. 2009. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K 2013. Causes, consequences, and reversal of immune system aging. J Clin Invest 123: 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ 2005. The influence of age on T cell generation and TCR diversity. J Immunol 174: 7446–7452 [DOI] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al. 2009. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461: 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, Bardeguez A, Gardner JP, Rodriguez P, Ganesh V, Kimura M, Skurnick J, Awad G, Aviv A 2002. Telomere length in the newborn. Pediatr Res 52: 377–381 [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT 2008. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 9: 115–128 [DOI] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. 2011. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147: 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder I, Horn K, Sieburg HB, Cho R, Muller-Sieburg C, Loeffler M 2008. Characterization and quantification of clonal heterogeneity among hematopoietic stem cells: a model-based approach. Blood 112: 4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B 2012. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507 [DOI] [PubMed] [Google Scholar]
- Simons A, Sikkema-Raddatz B, de Leeuw N, Konrad NC, Hastings RJ, Schoumans J 2012. Genome-wide arrays in routine diagnostics of hematological malignancies. Hum Mutat 33: 941–948 [DOI] [PubMed] [Google Scholar]
- Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisen J 2005. Retrospective birth dating of cells in humans. Cell 122: 133–143 [DOI] [PubMed] [Google Scholar]
- Stulnig T, Maczek C, Böck G, Majdic O, Wick G 1995. Reference intervals for human peripheral blood lymphocyte subpopulations from ‘healthy’ young and aged subjects. Int Arch Allergy Immunol 108: 205–210 [DOI] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. 2012. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerkamp F, Pike-Overzet K, Staal FJ 2006. T-sing progenitors to commit. Trends Immunol 27: 125–131 [DOI] [PubMed] [Google Scholar]
- Wei P, Liu X, Fu YX 2011. Incorporating predicted functions of nonsynonymous variants into gene-based analysis of exome sequencing data: a comparative study. BMC Proc 5: S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. 2012. The origin and evolution of mutations in acute myeloid leukemia. Cell 150: 264–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssoufian H, Pyeritz RE 2002. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet 3: 748–758 [DOI] [PubMed] [Google Scholar]