

Abstract
Aberrant DNA hypomethylation may play an important role in the growth rate of glioblastoma (GBM), but the functional impact on transcription remains poorly understood. We assayed the GBM methylome with MeDIP-seq and MRE-seq, adjusting for copy number differences, in a small set of non-glioma CpG island methylator phenotype (non-G-CIMP) primary tumors. Recurrent hypomethylated loci were enriched within a region of chromosome 5p15 that is specified as a cancer amplicon and also encompasses TERT, encoding telomerase reverse transcriptase, which plays a critical role in tumorigenesis. Overall, 76 gene body promoters were recurrently hypomethylated, including TERT and the oncogenes GLI3 and TP73. Recurring hypomethylation also affected previously unannotated alternative promoters, and luciferase reporter assays for three of four of these promoters confirmed strong promoter activity in GBM cells. Histone H3 lysine 4 trimethylation (H3K4me3) ChIP-seq on tissue from the GBMs uncovered peaks that coincide precisely with tumor-specific decrease of DNA methylation at 200 loci, 133 of which are in gene bodies. Detailed investigation of TP73 and TERT gene body hypomethylation demonstrated increased expression of corresponding alternate transcripts, which in TP73 encodes a truncated p73 protein with oncogenic function and in TERT encodes a putative reverse transcriptase-null protein. Our findings suggest that recurring gene body promoter hypomethylation events, along with histone H3K4 trimethylation, alter the transcriptional landscape of GBM through the activation of a limited number of normally silenced promoters within gene bodies, in at least one case leading to expression of an oncogenic protein.
A hallmark of many cancers is the decrease in 5-methylcytosine in genomic DNA relative to non-neoplastic cells or tissue, termed global hypomethylation (Feinberg and Vogelstein 1983; Gama-Sosa et al. 1983; Wild and Flanagan 2010). Although the degree of global hypomethylation can be severe, it is not uniformly distributed across the cancer genome. In colorectal cancer, for example, large hypomethylated domains coincide with late replication, attachment to the nuclear lamina, and partially methylated domains (PMDs) in somatic cells (Berman et al. 2011). In many cancers, demethylation of tandem and interspersed repeats contributes to global hypomethylation, consistent with repeats containing more than half of the 28,217,448 CpGs in the human genome (Rollins et al. 2006). Certain subfamilies of transposons harbor sequences with enhancer activity that is regulated by epigenetic mechanisms in a cell-type specific fashion (Xie et al. 2013). However, normally methylated single copy sequences including those within gene bodies are also hypomethylated in cancer. In glioblastoma (GBM), global hypomethylation is found in ∼80% of primary tumors (Cadieux et al. 2006). The level of hypomethylation varies between individual tumors, ranging from levels seen in normal brain to ∼50% of normal, reflecting demethylation of ∼10 million CpG sites per tumor cell on average. The most severely hypomethylated GBMs with transcriptional activation of the putative oncogene MAGEA1 are also the most proliferative, suggesting increased tumor aggressiveness (Cadieux et al. 2006). Furthermore, LINE-1 hypomethylation, potentially reflecting global hypomethylation, is more pronounced in GBM compared with more indolent lower-grade glioma (Zheng et al. 2011).
DNA demethylation of specific regulatory elements in cancer genomes can contribute to the up-regulated expression of the associated gene. A prototypical example is the 5′ promoters of cancer-germline (CG) antigen genes (also known as cancer-testis antigen genes) that are methylated and repressed in somatic cells but demethylated in the germline and some cancers, thereby allowing CG gene transcription (De Smet et al. 1996). CG promoter hypomethylation and gene activation are accompanied by a gain in H3K4me3 and histone acetylation (James et al. 2006; Rao et al. 2011). In GBM, severely hypomethylated and hyperproliferative tumors are associated with demethylation and transcriptional activation of the CG gene MAGEA1, a putative oncogene (Cadieux et al. 2006; Monte et al. 2006). The CG genes SOHLH2, SSX2, SSX4B, SSX8, SSX9, and PAGE5 are also recurrently hypomethylated in GBM (Wu et al. 2010). Promoter hypomethylation is associated with transcriptional activation at other single-copy genes, for example IGF2/H19 (associated with loss of imprinting) (Cui et al. 2002; Ito et al. 2008), CA9 (Cho et al. 2001), and SRPX2 (Oster et al. 2013). As ∼98% of 5′ promoter CpG islands (CGIs) are unmethylated in brain (Maunakea et al. 2010), they are more frequently targets of aberrant hypermethylation in cancer (Costello et al. 2000; Zardo et al. 2002). Thus, potential targets of promoter hypomethylation in cancer may include CGI promoters in gene bodies and non-CGI promoters that are methylated, or differentially methylated, in somatic cells (Weber et al. 2007; Meissner et al. 2008; Maunakea et al. 2010).
Hypomethylation also has the potential to activate cryptic promoters that are rarely active in normal somatic cells. Interspersed transposable elements and endogenous viruses contain promoters and other functional elements that may have been co-opted into tissue-specific regulatory networks in normal cells (Wang et al. 2007; Lynch et al. 2011; Xie et al. 2013). These sequences are a potentially vast reservoir of normally silenced, mostly unannotated promoters that might only become active in the aberrant epigenetic environment in cancer cells. For example, hypomethylation of a LINE-1 promoter is associated with activation of an alternate transcript of the MET oncogene in bladder tumors and adjacent normal tissue (Wolff et al. 2010). In non-Hodgkins lymphoma, hypomethylation of an endogenous retrovirus (ERV) in the THE1B promoter activates transcription of the adjacent CSF1R proto-oncogene (Lamprecht et al. 2010). The extent of hypomethylation-associated transcription of endogenous viral and transposon sequences in GBM is unknown.
Recent genome-wide studies of cancer methylomes have provided new insights into the distribution and potential molecular consequences of hypomethylation. However, the contribution of hypomethylation to transcriptional up-regulation, and which genes are affected, remains unclear. Hypomethylated domains found using 5× coverage shotgun bisulfite sequencing in colon cancer are associated with extreme expression variability of the genes within (Hansen et al. 2011). In contrast, in a breast cancer cell line, hypomethylated blocks were associated with gene silencing and repressive chromatin (Hon et al. 2012). In another colon cancer study, focal hypomethylation at enhancers located 5′ of TACSTD2 or within B3GNTL1 was associated with increased expression of these genes (Berman et al. 2011). These studies, however, did not distinguish primary transcripts from overlapping alternative transcripts and did not address alternative promoter usage.
Identification of recurrent hypomethylation that overlaps directly with specific regulatory elements, and evaluating changes in the associated transcripts, is one strategy to further distinguish functional hypomethylation events in cancer from the potentially large number of unproductive passenger epimutations. Here we investigated recurrent, focal hypomethylation in GBM, in annotated and previously unannotated promoters. We mapped full DNA methylomes for five GBMs, encompassing the three most common expression subtypes (proneural, classical, and mesenchymal) and including tumors with canonical genetic alterations (e.g., EGFR amplification in GBM 2, CDKN2A/B homozygous deletion in GBMs 2–5, PDGFRA amplification in GBM 5). Our data point to a role for recurrent gene body hypomethylation in activating normally silenced alternate promoters and demonstrate up-regulation of the associated alternative transcripts, and in one case, a protein isoform that is oncogenic.
Results
Genome-wide identification of differentially methylated regions (DMRs) using the M&M algorithm
Methylated DNA immunoprecipitation and sequencing (MeDIP-seq) and methyl-sensitive restriction enzyme sequencing (MRE-seq) (Harris et al. 2010; Maunakea et al. 2010) were used to map DNA methylomes for five newly diagnosed GBMs (Supplemental Table S1). Unlike bisulfite that conflates methylcytosine and hydroxymethylcytosine, MeDIP provides a definitive measurement of 5-methylcytosine only (Jin et al. 2010). Here we identify regions of hyper- and hypomethylation genome-wide with M&M, a method that takes into account local CpG density to normalize, scale, and integrate MeDIP- and MRE-seq data into a single relative measurement that can be compared between samples (Zhang et al. 2013). In contrast to Infinium arrays and reduced representation bisulfite sequencing (RRBS) which assay 2%–10% of the methylome, M&M analysis of MeDIP-seq and MRE-seq calls methylation differences in 500-bp windows across ∼80%–90% of the 28 million CpG sites. Because copy number can confound sequencing-based methylation methods relying on variation in read density (Laird 2010; Robinson et al. 2010), we performed array-CGH on each GBM and normalized MeDIP- and MRE-seq signals across the genome by estimated copy number (Methods; Supplemental Fig. S1A–R). We also analyzed global mRNA expression by microarray to identify potential functional effects of recurrent epigenetic alterations, and used the Illumina Infinium HumanMethylation27 array for large-scale validation of our M&M results. Based on copy number and expression profiles, the three most common GBM expression subtypes (Verhaak et al. 2010) are represented in our small sample set (proneural, classical, and mesenchymal) (Supplemental Table 1). Methylation changes in the rare IDH1 mutant, G-CIMP positive GBM class, which represent 5%–10% of all GBM, have been described elsewhere using Infinium arrays (Noushmehr et al. 2010).
To discover differentially methylated regions (DMRs), we compared each GBM to normal brain samples from two individuals separately and identified 500-bp windows, excluding sex chromosomes, which were significantly different in both GBM-normal comparisons. The two normal brains (frontal cortex from two adult males) were very similar though not identical (Supplemental Fig. S2A; Maunakea et al. 2010). Using M&M at a stringent threshold of Q < 10−13, we identified between 343–2288 hypermethylated and 4–2124 hypomethylated DMRs across the five GBMs (Fig. 1A). We also used a less stringent cutoff of Q < 10−5 and found additional DMRs with similar relative frequencies among tumors (Supplemental Fig. S2B). Proneural GBM 5 presented an unusual profile of frequent hypermethylation, within range of the other GBMs, but sparse hypomethylation; however, none of the GBMs were of the glioma-CpG island methylator phenotype (G-CIMP), based on Infinium array data at diagnostic loci (Noushmehr et al. 2010). Hierarchical clustering of all probes indicated that the pattern of GBM 5 methylation was most closely related to normal brain of the five GBMs (data not shown). Histopathological analysis showed ∼95% tumor cell purity for GBM 5, and the magnitude of hypermethylation was similar compared with the other GBMs, suggesting that the infrequent hypomethylation in this tumor is likely not due to significant admixtures of normal cells.
Figure 1.

Discovery and validation of DMRs in GBM. (A) Number of hypermethylated and hypomethylated DMRs at Q < 10−13 in five primary GBMs. The expression subtype (Verhaak et al. 2010) of each tumor is shown below. (n.d.) Not determined. (CL) Classical. (PN) Proneural. (MES) Mesenchymal. (B) Delta beta values (GBM-normal brain) from Infinium HumanMethylation27 arrays, plotted for individual CpGs within hyper- and hypomethylated DMRs (Q < 10−13) for each of five GBMs. For GBM 5, there were no Infinium CpGs within hypomethylated DMRs. (n.d.) No data. (C,D) Validation of gene body hypomethylation in TRPM5 by bisulfite cloning and sequencing. In C, the location of hypomethylation is shown within the TRPM5 gene body. Normalized MeDIP-seq coverage is graphed in brown for normal brain and blue for GBM. For MRE-seq, normalized read counts at single CpGs are in green. For GBM tracks, MeDIP-seq and MRE-seq values are normalized by copy number and sequencing depth. In D, bisulfite PCR, cloning, and sequencing results are shown for 20 CpGs in the region “bisulfite PCR amplicon” in C. Each row is a clone and each column is a CpG site. Asterisks indicate CpGs with MRE sites.
For large-scale validation of the M&M candidate DMRs, we plotted delta beta values (GBM-normal brain) for each Infinium HumanMethylation27 CpG site within a DMR (Fig. 1B; Supplemental Fig. S3). For each GBM, a very consistent overall methylation change was observed in the Infinium data for both hyper- and hypomethylated DMRs, even though the arrays measure individual CpG sites while M&M estimates methylation in 500-bp windows, and despite the differences in detection of hydroxymethylation by these two methods. We further validated several individual loci by the “gold standard” of bisulfite PCR, cloning and Sanger sequencing. We chose a hypomethylated locus within the TRPM5 gene body and a hypermethylated locus at the BCL2L11 promoter and in both cases confirmed the M&M DMR calls (Fig. 1C,D; Supplemental Fig. S4A,B). We also tested the accuracy of M&M in detecting DMRs at uniquely mapping sequences derived from transposons. At two loci examined (one MER52A ERV and one LTR1B), hypomethylated DMRs were confirmed by bisulfite sequencing (Supplemental Fig. S5A–D).
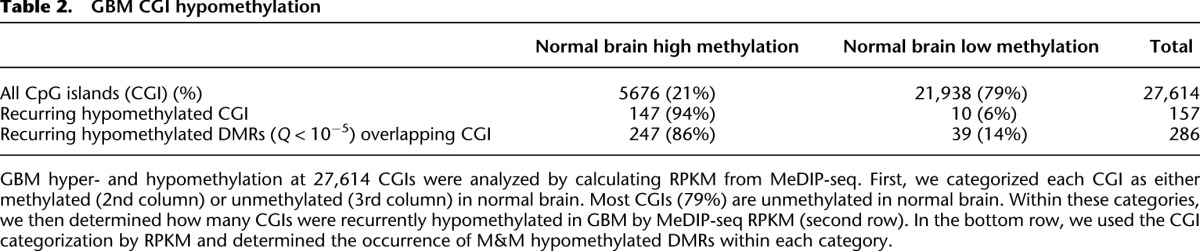
To highlight functionally relevant methylation alterations, we identified DMRs shared between two or more GBMs (Table 1). Recurrent hypermethylated and hypomethylated DMRs showed striking differences in genomic distribution. Recurring hypermethylation showed strong enrichment for CGIs (40–50×) and moderate enrichment for promoters, 5′-untranslated regions (UTRs), and exons (Fig. 2). In contrast, recurring hypomethylated DMRs were relatively infrequent in CGIs and promoters compared with hypermethylation. To some degree, these patterns reflect the genomic distribution of methylation in brain and other normal somatic tissues, i.e., most of the genome is highly methylated except for discrete, almost completely unmethylated regions corresponding to CGIs. In colon cancer, partially hypomethylated domains (PMDs), encompassing up to half the genome, have been reported to coincide with nuclear lamina attachment regions (LADs) (Berman et al. 2011; Hansen et al. 2011). In our GBM data, 29.5% of recurring hypomethylated DMRs localize within LADs, compared with a genomic background of 36.9%. We analyzed MeDIP reads per kilobase per million mapped reads (RPKM) in 20- and 50-kb windows genome-wide but did not detect large-scale differences between GBMs and normal brain (not shown). Although some broad regions of low-magnitude hypomethylation might be below the threshold of detection, the focally hypomethylated DMRs we identified are primarily not associated with LADs. We also examined methylation alterations at CGIs by MeDIP-seq RPKM, independent from the M&M analysis. By this approach, 147/157 (94%) of recurring hypomethylated CGIs in GBM were strongly methylated in normal brain (Table 2). Approximately 68% of these hypomethylated CGIs were located within gene bodies. Together, these data show that normally methylated gene bodies and intergenic regions, which are sites of alternate promoters, enhancers, and other regulatory elements, are commonly affected by hypomethylation in GBM.
Table 1.
Recurring DMRs present in at least two GBMs

Figure 2.

Enrichment for genomic features among recurring hyper- and hypomethylated DMRs at Q < 10−13 and Q < 10−5. Enrichment was calculated using the background distribution of CpG-containing 500-bp windows on autosomes that were also used for M&M analysis. Statistically significant enrichment (or depletion) was calculated using a binomial test, and those significant at P < 0.001 are indicated with asterisks. The definitions of genomic features are the following: intergenic: regions between 3′ transcription end site (TES) of a gene to the 5′-most TSS of next gene; gene body: 5′-most TSS of a gene to 3′-most TES of the same gene; CGIs: from UCSC annotation; promoter: −2.5 to +0.5 kb from the 5′-most TSS. 5′ UTRs, exons, introns, and 3′ UTRs were defined from the RefSeq database. Note that some of these genomic features are not mutually exclusive.
Table 2.
GBM CGI hypomethylation
Functional analysis of recurring hypermethylated DMRs using GREAT (McLean et al. 2010) uncovered highly significant enrichments for genes bound by Polycomb complex proteins and marked by H3K27me3 in embryonic stem cells (Supplemental Table S2; Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007). This corroborates similar results in GBM obtained by other methods (Martinez et al. 2009; Wu et al. 2010). Significant enrichment was also observed for gene sets affected by CGI promoter hypermethylation in other cancer types.
Because of the strong focus on hypermethylation in cancer, less is known about the genes and pathways affected by hypomethylation. GREAT enrichments for hypomethylated DMRs from individual GBMs were diverse, with relatively few commonalities between these tumors with shared GBM histology but differing molecular subtype. A highly significant hypomethylation enrichment, common to four of five individual GBMs and also significant in recurring hypomethylated DMRs, was for an ∼3.4-Mb region of chromosome 5p15 that contains 26 genes and is recurrently amplified in breast cancer (Supplemental Table S3; Nikolsky et al. 2008). This region is not amplified in our five GBMs, but encompasses a large number of hypomethylated DMRs including a recurrent hypomethylated DMR in the gene body of TERT, encoding telomerase reverse transcriptase which plays a critical role in telomere length and tumorigenesis.
Because promoter hypomethylation is hypothesized to be one mechanism leading to oncogene overexpression (Hoffman 1984), we carefully examined the promoter methylation status of GBM oncogenes. We did not detect hypomethylation at the 5′ promoters of 16 prototypical oncogenes that are recurrently activated by genetic mechanisms in GBM (McLendon et al. 2008), though many of these have 5′ CGIs that are unmethylated in normal brain (Supplemental Fig. S6A). These findings are consistent with the genomic distribution of hypomethylated DMRs (Fig. 2), which suggest that CGIs and 5′ promoters are rarely targets of hypomethylation. We confirmed the absence of oncogene promoter hypomethylation in a large GBM data set from The Cancer Genome Atlas (TCGA). Analysis of Infinium HumanMethylation27 methylation data for 292 TCGA GBMs showed that oncogene CGI promoters are almost always fully unmethylated in both normal brain and GBM, but at a few regions are in fact hypermethylated in a subset of GBMs, e.g., at a portion of the CCND2 promoter marked by Polycomb-associated H3K27me3 in embryonic stem cells (Supplemental Fig. S6B,C). It is unlikely that this hypermethylation inhibits transcription, as it is confined to the part of the CGI that is distal from the transcription start site (TSS). For non-CGI promoters, the TCGA data show high or low methylation, depending on the CpG site assayed, but at similar levels in GBM and normal brain.
As aberrant hypomethylated DMRs were rare at 5′ promoters, we hypothesized that gene body and intergenic regulatory elements, including alternate promoters, could be targets of hypomethylation. We plotted average ENCODE DNase I hypersensitivity (DHS) across 10.5-kb windows centered on the complete set of recurring hypomethylated DMRs (Q < 10−13) (Supplemental Fig. S7). Hypomethylated DMRs were enriched for DHS, centered near the middle of the DMRs and rapidly dropping to background in flanking regions. The DHS enrichment is likely driven by colocalization of a subset of hypomethylated DMRs with regulatory elements, as well as the innate regulatory capacity of many CpG-containing sequences.
Tumor-specific hypomethylation of alternative promoters in gene bodies
The genomic distribution of hypomethylation and the enrichment of hypomethylated DMRs for epigenetically defined regulatory regions suggested that activation of alternative promoters in gene bodies could be one consequence of GBM hypomethylation. We utilized annotated gene body promoters from the UCSC Known Genes (Hsu et al. 2006), which encompasses a greater number of transcripts compared with RefSeq. Gene body promoters were defined as −2 to +0.5 kb relative to all UCSC TSS located 3′ of the 5′-most TSS for the same gene. Seventy-six genes were sites of recurring gene body promoter hypomethylation, determined by the presence of at least one hypomethylated DMR (Q < 10−5) in two or more GBMs. Nearly all (71/76; 93%) were not associated with any significant methylation change at the corresponding 5′ promoter of the same gene (neither hypo- nor hypermethylation). Three genes (TERT, TP73, DIO3OS) had concurrent 5′ hypermethylation in at least two GBMs with gene body promoter hypomethylation, though the 5′ hypermethylation was focal and did not encompass the core promoter. The other two genes, LSP1 and DMKN, had hypomethylated promoters in the gene body and 5′ region.
Tumor-specific hypomethylation at candidate alternative promoters in gene bodies
In addition to the above recurrently hypomethylated gene body promoters, cryptic promoters not previously associated with the 5′ ends of RefSeq or UCSC Known Genes transcripts might be novel targets of GBM hypomethylation. We used chromatin states defined by the ChromHMM algorithm, which defines regulatory elements based on combinatorial histone modifications (Ernst et al. 2011), to identify putative promoters. We annotated each 500-bp genomic window by the presence of the following chromatin states in at least one ENCODE cell type: (1) active, weak, or poised promoter (Ernst et al. 2011, states 1–3), (2) active or weak/poised enhancer (states 4–7), or (3) insulator (state 8) (Methods). We found that 364/4498 (8.1%) of recurring hypomethylated DMRs (Q < 10−5) occurred at ChromHMM-defined promoters (Fig. 3A). Many hypomethylated DMRs are potential enhancers or insulators, and some overlap with apparently multifunctional loci, with different regulatory functions depending on cell type.
Figure 3.

Recurrent DNA hypomethylation of previously unannotated promoters in GBM. (A) ChromHMM-defined promoters, enhancers, and insulators from nine ENCODE cell lines overlap with recurrent GBM hypomethylated DMRs (Q < 10−5 in at least two of five GBMs). (B–D) Promoter assays for DNA sequences within hypomethylated DMRs that were not associated with the 5′ ends of RefSeq or UCSC transcripts, but had a promoter state assigned by ChromHMM in at least one ENCODE cell type. Each bar shows fold induction (±SD) relative to pGL3-Basic empty vector control. pGL3-SV40 promoter is used as a positive control, shown immediately to the right of the empty vector. These candidate promoters were tested in HEK293 cells and two GBM cell lines, U87 and LN229. Asterisks indicate P < 0.05, one-tailed t-test. (E) Previously unannotated alternative promoter in the body of SIGLEC11 showing GBM hypomethylation and overlapping promoter state by ChromHMM. The region cloned for luciferase assay is shown in blue, and human ESTs suggesting transcription initiation are shown below (black). LTR repeats from UCSC RepeatMasker track, including the HERV3 element in which the promoter is embedded, are indicated with gray. The location of a transposon-associated CAGE tag cluster (Faulkner et al. 2009) is shown by the purple hatch mark. H3K4me3 ChIP-seq signal in GBMs 1, 2, and 5 is shown at the bottom. (F) qRT-PCR with primers located within human EST BM977347 (see E). Fold expression ± SD relative to normal brain is graphed for a representative experiment. (NB) Normal brain.
We used the ChromHMM annotations to detect hypomethylated novel promoters identified solely by promoter chromatin state that were >2 kb intragenic or intergenic from the 5′ or 3′ boundaries of RefSeq and UCSC Genes transcripts. The presence of ENCODE transcription factor binding sites, DNase I hypersensitivity, and 5′ ends of GenBank mRNAs and ESTs at four such loci strongly suggested promoter function, which we tested experimentally. Three of these loci were located within gene bodies and a fourth was intergenic (Supplemental Fig. S8A,B).
We cloned each of the four candidates into the pGL3-basic reporter vector and quantified promoter activity by luciferase assay in HEK293 cells and two GBM cell lines, LN229 and U87. The three putative gene body promoters exhibited significant promoter activity in all cell lines (Fig. 3B–D). The TNXB gene body promoter showed particularly strong activity in U87 and LN229 cells, with the smaller of two cloned fragments harboring activity even higher than the SV40 positive control in U87 GBM cells. TNXB encodes an extracellular matrix glycoprotein of the tenascin family that is expressed in GBM, possibly with a role in promoting neovascularization (Hasegawa et al. 1997).
The SIGLEC11 gene body promoter is located within a HERV3 ERV repeat (Fig. 3E). Human TSS profiling using cap analysis of gene expression (CAGE) tag sequencing had previously identified this region as a site of transposon-associated transcriptional initiation (Faulkner et al. 2009). The CAGE reads supporting a TSS came from the MCF7 breast cancer cell line (not shown). ChIP-seq for H3K4me3 identified a peak in GBM 5 and slight enrichment in GBMs 1 and 2 at the region overlapping the confirmed promoter. Three human ESTs, including one from an ovarian tumor, suggested transcription initiation within SIGLEC11 intron 7 (Fig. 3E). Because these do not overlap with known SIGLEC11 exons, the EST sequences allowed us to design RT-PCR primers specific for the putative gene body transcript. We first confirmed with conventional endpoint PCR complete lack of amplification from reverse transcriptase-negative controls for each of our GBMs and normal brains, using two independent primer pairs (data not shown). We then used qRT-PCR to quantify relative expression of the alternate transcript, again including RT-negative controls for all samples. We found that GBM 1, which was partially hypomethylated at the gene body promoter, expressed the alternate transcript almost 16-fold higher than normal brain (Fig. 3F). Interestingly, the other hypomethylated GBMs did not show up-regulated expression, suggesting that additional factors are required for efficient transcription initiation. These data show that novel or cryptic promoters are targets of GBM hypomethylation and can drive expression of normally silenced gene body transcripts from transposons.
Acquisition of H3K4me3 in primary GBM at a subset of recurrently hypomethylated DMRs
In addition to examining the relationship between histone modifications from ENCODE cell lines and hypomethylated DMRs in GBM, we sought to directly link DNA hypomethylation with promoter-associated histone modification profiled in the same primary GBM tissues. We performed tissue-ChIP-seq for H3K4me3 on GBMs 1, 2, and 5. We used MACS (Zhang et al. 2008) with a 1% false discovery rate and P-value cutoff of 1 × 10−10 to identify regions with localized H3K4me3 enrichment in GBM 1 (68,675 peaks), GBM 2 (57,748 peaks), and GBM 5 (47,719 peaks). We confirmed the quality of the ChIP-seq data with CHANCE (Supplemental Fig. S9; Diaz et al. 2012). In addition, we found that 51%–55% of GBM H3K4me3 peaks overlap with the ChromHMM promoter state in at least one of the nine ENCODE cell lines analyzed in Ernst et al. (2011) (Supplemental Fig. S10).
We identified 200 loci with both H3K4me3 peaks and DNA hypomethylation (Q < 10−5) in total over three GBMs (DNA hypo/K4me3 loci). Of these, 133 were within gene bodies. Most were specific to a particular tumor, although nine were shared between two GBMs (Supplemental Fig. S11). GREAT functional analysis identified significant enrichment for multiple cancer-related gene sets (Supplemental Table S4). We examined mRNA expression levels of genes with gene body DNA hypo/K4me3 loci by Affymetrix GeneChip Human Gene 1.0 ST arrays. In each GBM, we observed both increased and decreased gene expression relative to normal brain, but overall these genes showed increased expression (Fig. 4A). In GBMs 1 and 5 the increased expression was statistically significant (P = 2.9 × 10−6 and P = 0.02, respectively). The partial association between DNA hypo/K4me3 loci and expression is likely because not all DNA hypo/K4me3 loci are promoters driving expression of alternate transcripts of their “host” genes. Some might drive antisense expression or splice to distal downstream exons. In addition, measurements of gene expression by array might not capture the most relevant exons for measuring expression of specific alternate transcripts, which are more precisely distinguished by manual, transcript-specific qRT-PCR or RNA-seq.
Figure 4.

A subset of recurrent DNA hypomethylation coincides with H3K4me3 in the same primary tumor. (A) Gene expression change for genes nearest each DNA hypo/K4me3 locus, which, taking into account the direction of transcription, can be located upstream (5′), within the gene body, or downstream (3′). Hypomethylated DMRs at Q < 10−5 and H3K4me3 peaks (P < 1 × 10−10) were used to define DNA hypo/K4me3 loci. The Affymetrix GeneChip Human Gene 1.0 ST expression changes (GBM-NB) for the nearest RefSeq gene are shown as box plots on a log2 scale. The number of genes analyzed in each box plot is given along with the labels at the bottom. Statistically significant gene expression changes were determined using a t-test at P < 0.05 (asterisks above box plots). A total of 19,628 Entrez genes were analyzed on the array. (B) Recurring colocalization of DNA hypomethylation and H3K4me3 at the Delta GLI3 transcript promoter. Individual hypomethylated DMRs are shown under the MRE. (C) Quantification of Delta GLI3 transcript abundance by isoform-specific qRT-PCR. Fold expression ± SD relative to normal brain is graphed for a representative experiment. (NB) Normal brain.
Twenty-two of the 76 recurrently hypomethylated gene body promoters we identified above also had H3K4me3 peaks in at least one GBM (Supplemental Fig. S12). The promoter of Delta GLI3, a transcript variant of GLI3 (glioma-associated oncogene family zinc finger 3), was one example and showed recurrent colocalization of DNA hypomethylation and H3K4me3 (Fig. 4B). Full-length GLI3 mRNA expression was up-regulated by expression microarray (data not shown). All five GBMs had increased copy number for the entire chromosome 7, which could partially but not fully account for the increased expression. For example, GBM 5 had a 2.7-fold increase in GLI3 expression and only a 1.2-fold copy number increase relative to diploid. The gene body promoter had H3K4me3 peaks in GBMs 1, 2, and 5, and was partially hypomethylated in all five GBMs, with hypomethylated DMRs found in GBMs 2 through 5 (Fig. 4B). We confirmed that the gene body promoter hypomethylation was recurrent by analysis of TCGA HumanMethylation450 methylation array data from 126 GBMs (Supplemental Fig. S13A,B). Since our microarray data were not specific for the Delta GLI3 transcript, we performed isoform-specific qRT-PCR to quantify Delta GLI3 in the GBMs with both hypomethylation and H3K4me3 at the Delta GLI3 promoter. We found increased mRNA expression in GBMs 1 and 5 relative to normal brain (Fig. 4C). GBM 5 showed the highest expression and we observed H3K27ac enrichment at the Delta GLI3 promoter in GBM 5, consistent with epigenetic activation (RJA Bell, J Song, JF Costello, unpubl.). This DNA hypo/K4me3 locus is evolutionarily conserved (a phastCons conserved element in primates, mammals, and vertebrates) and is a site of DHS, transcription factor binding, and ChromHMM promoter state in some ENCODE cell lines (not shown). GLI3 encodes one of a family of three zinc finger domain transcription factors that signal in the Sonic Hedgehog pathway. The Delta GLI3 mRNA contains a 1521 a.a. open reading frame for an in-frame GLI3 alternate protein, lacking the N-terminal region compared with the 1580 a.a. full-length GLI3, but otherwise identical to the full-length protein. The function of the delta GLI3 isoform, and whether it differs from full-length GLI3, is not known.
Tumor-specific gene body hypomethylation is associated with increased expression of an oncogenic isoform of TP73
We investigated in depth a hypomethylated gene body CGI promoter in the TP53-related gene TP73. This alternative promoter was recurrently hypomethylated, with the strongest hypomethylation overlapping the alternate TSS in GBM 3 (Fig. 5A). TP73 encodes multiple protein isoforms with opposing tumor suppressor and oncogenic functions, depending on alternate promoter usage and splicing (Rufini et al. 2011). The transcript produced from the gene body promoter, DeltaN TP73, encodes the oncogenic deltaNp73 protein, a dominant negative inhibitor of p73 and p53 signaling (Ishimoto et al. 2002). We validated gene body promoter hypomethylation by bisulfite PCR, cloning, and Sanger sequencing (Fig. 5B). In two normal brains and GBM 1, the gene body promoter was highly methylated (>90% average methylation across 12 CpGs). GBM 3 was severely hypomethylated (34% average methylation) and nine of the 15 sequenced clones were completely or nearly completely unmethylated, consistent with a transcriptionally active, open chromatin state at these alleles in a significant fraction of the tumor. Infinium methylation array data from the same GBMs were concordant with MeDIP/MRE data and bisulfite sequencing (Fig. 5C; data not shown).
Figure 5.

TP73 gene body CGI hypomethylation associated with activation of the oncogenic DeltaN TP73 isoform. (A) Strong focal hypomethylation in GBM 3 detected by MeDIP-seq and MRE-seq. (B) Bisulfite sequencing for two normal brains (top) and GBMs 1 and 3 (bottom). Asterisks indicate CpGs with MRE sites. (C) Infinium HumanMethylation27 array data at the DeltaN TP73 promoter. Percent methylation for four CpGs, shown schematically at the top, is graphed below. (Gray points) 292 TCGA-assayed GBMs. (Black squares) GBM 3. (Black triangles) Normal brains. (D) Quantitative RT-PCR for the oncogenic DeltaN TP73 mRNA isoform. Fold expression ± SD relative to the average of two normal brains is graphed for a representative experiment. (NB) Normal brain. (FNSC) Cultured fetal neural stem cells. (U87) GBM cell line. (GBM5 cult.) Short-term serum-grown culture of GBM 5. (E) Western blotting with anti-deltaNp73 (top) and anti-GAPDH control (bottom).
TP73 gene body promoter hypomethylation was associated with up-regulation of DeltaN TP73 mRNA. DeltaN TP73 was ∼13-fold overexpressed in GBM 3 compared with normal brain by isoform-specific qRT-PCR, and was higher than GBMs without hypomethylation (Fig. 5D). DeltaN TP73 expression was low, similar to normal brain, in fetal neural stem cells (FNSC) as well as established GBM cell lines and in a short-term culture of GBM 5. DeltaN TP73 mRNA was also dramatically increased during tumor progression in two of two patients with secondary GBM relative to their patient-matched, indolent low-grade gliomas resected years earlier (Q = 4.9 × 10−6 and Q = 1.3 × 10−6, RNA-seq data [Johnson et al. 2013]).
We next determined whether hypomethylation-associated mRNA up-regulation resulted in expression of the deltaNp73 protein. DeltaNp73 has a unique N terminus and lacks the transactivation domain found in full-length p73, but shares a C-terminal localization domain with full-length p73, accounting for its dominant negative function. Western blotting with a deltaNp73-specific antibody revealed expression in multiple GBMs including GBM 3, but undetectable or very low deltaNp73 in two normal brains, GBM cell lines, and primary GBM cultures (Fig. 5E). As deltaNp73 was expressed in GBMs without hypomethylation-associated DeltaN TP73 expression, gene body hypomethylation may be one of multiple mechanisms influencing the deltaNp73 protein level in GBM. Another mechanism involves alternate splicing of transcripts initiating from the 5′ promoter, which can also produce a transcript encoding the deltaNp73 protein (Ishimoto et al. 2002). Our qRT-PCR primers are specific for gene body promoter transcript, and do not amplify the alternately spliced 5′ transcripts.
Hypermethylation of the 5′ promoter and hypomethylation of the gene body promoter of TP73 are recurrent but not correlated
Although TP73 gene body hypomethylation was pronounced in one of our five GBMs, we hypothesized that it might be a recurrent epimutation detectable in a larger tumor cohort. To address this question, we analyzed TCGA HumanMethylation27 array data from 292 GBMs. The TP73 gene body promoter is assayed by the array at four CpG sites including three in the hypomethylated region that we previously validated with bisulfite sequencing (Fig. 5B,C). In addition, four of nine 5′ promoter probes interrogate a region where we observed recurring hypermethylation of ∼1 kb of the 3191-bp 5′ CGI (Supplemental Fig. S14A). We plotted methylation levels for these 13 TP73 probes for the entire cohort of 292 TCGA primary GBMs (Fig. 5C; Supplemental Fig. 14B). Both gene body hypomethylation and 5′ hypermethylation were highly recurrent events with varying magnitude. However, hypomethylation and hypermethylation were not correlated (r = 0.12, Pearson correlation), consistent with the nearly complete lack of methylation change at the 5′ end of 71 out of 76 genes with recurring gene body promoter hypomethylation that were discovered by M&M analysis. These data strongly suggest that different mechanisms underlie hypomethylation of alternative promoters in gene bodies compared with DNA methylation changes in 5′ promoters.
GBM-specific expression of a TERT alternate transcript from a hypomethylated gene body promoter
TERT encodes telomerase reverse transcriptase and is located within a broad region of recurring GBM hypomethylation on chromosome 5p15. Functional enrichment analysis by GREAT found that individual and recurring hypomethylated DMRs significantly enriched for a gene set corresponding to recurrent 5p15 genomic amplification in breast cancer (Nikolsky et al. 2008), and numerous hypomethylated DMRs are found in this region (Fig. 6A). Up-regulation of TERT mRNA leads to increased telomerase activity in most cancers, facilitating telomere maintenance, unlimited proliferation capacity, and immortalization (Meyerson et al. 1997; Shay and Bacchetti 1997). TERT mRNA is expressed, and telomerase activity is detected in the majority of primary GBM (Le et al. 1998; Lotsch et al. 2013). A SNP (rs2736100) in the TERT gene body is associated with susceptibility to glioma in several genome-wide association studies (GWAS) (Shete et al. 2009; Wrensch et al. 2009; Rajaraman et al. 2012). In addition, highly recurrent TERT promoter mutations, potentially generating binding sites for ETS/TCF transcription factors and presumably mediating increased TERT transcription, have recently been reported in GBM, low grade glioma, hereditary and sporadic melanoma, and other cancers (Horn et al. 2013; Huang et al. 2013; Killela et al. 2013).
Figure 6.

GBM-specific expression of an alternate transcript from a hypomethylated promoter in the TERT gene body. (A) GBM individual hypomethylated DMRs (Q < 10−5) within a ∼3.4-Mb region that is recurrently amplified in breast cancer and is enriched by GREAT analysis of recurring and individual hypomethylated DMRs. The location of the breast cancer copy number amplicon is shown in gray at the bottom. (B) GBM hypomethylation at a TERT gene body promoter, which is located near the glioma risk SNP rs2736100. GBMs 1, 2, and 4 had hypomethylated DMRs (Q-values are 10−14, 10−14, 10−21). (C) Infinium HumanMethylation450 array data for two CpGs in the recurrent hypomethylated DMR at the Delta TERT promoter. (Gray points) 126 TCGA-assayed GBMs. (Black triangles) TCGA normal brain. The percentage of GBMs at each CpG with methylation <80% (red line) is shown in red text. (D) Delta TERT mRNA isoform abundance quantified by qRT-PCR. Fold expression ± SD relative to the average of two normal brains is graphed. (NB) Normal brain. (FNSC) Cultured fetal neural stem cells. (U87) GBM cell line. (GBM5 cult.) Short-term serum-grown culture of GBM 5. (E) Detection of the transcription initiation site within the TERT hypomethylated gene body by 5′-RACE.
Three GBMs had a hypomethylated DMR (Q < 10−5) at a TERT gene body promoter, and increased MRE-seq was observed at this region in all five GBMs (Fig. 6B). Two CpGs are present on the Infinium HumanMethylation450 array within this recurring hypomethylated DMR, and we plotted methylation levels at these CpGs for 126 GBMs assayed by the TCGA (Fig. 6C). Forty-two percent (CpG #1) and 56% (CpG #2) of GBMs had <80% methylation, whereas normal brain was >90% methylated at both sites. The UCSC Known Genes annotates an alternate TERT mRNA, which we term Delta TERT, initiating within the hypomethylated gene body CGI, supported by a full-length cDNA from Burkitt’s lymphoma (Fig. 6B). To determine if a spliced mRNA is transcribed from this putative gene body promoter, we performed exon-joining RT-PCR with primers in the 3′ UTR and putative 5′ UTR to amplify the entire coding portion of Delta TERT from GBM cDNA (Supplemental Fig. S15A,B). The exon splicing structure matched the UCSC Genes alternate transcript, with exons 7 and 8 (numbering based on the full-length transcript) spliced out. We next designed isoform-specific qRT-PCR primers to quantify expression of Delta TERT and observed elevated expression in GBMs 1, 3, and 5 (Fig. 6D). Its expression was low, similar to normal brain, in cultured FNSCs and in the GBM cell line U87. We also performed 5′-rapid amplification of cDNA ends (5′-RACE) on GBM 3, which showed elevated expression by qRT-PCR and for which we had abundant high-quality RNA. We detected gene body transcription initiating from a single TSS within the CGI (Fig. 6E). No RACE product was observed in a negative control reaction performed on FNSC RNA. Together, the 5′-RACE, exon-joining RT-PCR, and qRT-PCR data indicate recurrent expression of Delta TERT from a hypomethylated gene body promoter in GBM. The alternate transcript includes an open reading frame encoding a putative short TERT protein of 329 amino acids, which shares the C terminus with full-length TERT but lacks part of the N-terminal reverse transcriptase domain (Supplemental Fig. S15C). One of the three catalytic aspartic acid residues critical for reverse transcriptase activity, D712, is absent from the putative short protein, likely compromising the ability to maintain telomere ends. Alternatively, as recently described for full-length TERT, it may have a function unrelated to telomere maintenance such as transcriptional regulation (Park et al. 2009).
Discussion
Recent studies have described the patterns of DNA hyper- and hypomethylation in cancer, but in most cases it is not yet clear which, if any, events are drivers of tumor phenotypes versus which ones have little or no effect on transcription, and may be passenger events. Here we sought to more fully understand hypomethylation in GBM, especially how it relates to regulation of alternate promoters. We have recently highlighted the importance of tissue and cell-type specific DNA methylation in gene bodies in the regulation of alternative transcripts (Maunakea et al. 2010). We identified focal hyper- and hypomethylation with M&M, normalizing for copy number, and validated methylation changes with both Infinium array and bisulfite sequencing. Although promoter hypomethylation did not occur at oncogenes that are commonly genetically altered in GBM, we found that alternate promoters in bodies of a small number of genes with potential oncogenic function are targets of recurrent GBM hypomethylation, and in some cases show concurrent gain of H3K4me3.
Many genome-wide methylation studies examine promoters defined by the 5′ ends of transcripts annotated in databases such as RefSeq (Pruitt et al. 2007). However, there is accumulating evidence for widespread transcription and regulatory elements throughout a majority of the genome that are not yet well annotated and which in some cases might be driven by novel promoters (Faulkner et al. 2009; Djebali et al. 2012; The ENCODE Project Consortium 2012). We used chromatin states defined by combinations of histone modifications to identify previously unannotated promoters that are hypomethylated in GBM. We confirmed promoter activity in GBM cell lines for gene body promoters in TNXB, PAX7, and SIGLEC11. The SIGLEC11 gene body promoter is located within an ERV-derived repeat, and we found that a gene body transcript can be up-regulated in a subset of GBMs with hypomethylation at this locus. This highlights the potential functional importance of epigenetic alterations at transposons in GBM. Our approach used chromatin state information from ENCODE cell lines and likely includes only a subset of all possible promoter regions, but suggests that using complementary epigenomic data in addition to conventional annotations will provide a more complete understanding of epigenetic (and genetic) alterations.
We noted many loci where GBM hypomethylation colocalizes with ENCODE ChromHMM enhancers. In chronic lymphocytic leukemia (CLL), a subset of gene body hypomethylation colocalized with enhancers, correlating in some cases with gene expression changes (Kulis et al. 2012). In colon cancer, 662 focally hypomethylated elements were enriched for TAF1 and other transcription factor binding in ENCODE cell lines, suggesting that these hypomethylated loci function as promoters or enhancers (Berman et al. 2011). In future studies, comparing the recurrent DNA hypomethylated loci to GBM enhancer profiles assayed by ChIP-seq with H3K27ac and H3K4me1 would be of great interest, especially since enhancers show strong cell type specificity.
We generated sample-matched H3K4me3 ChIP-seq profiles to explore the relationship between promoter-associated histone modifications and DNA hypomethylation. GBM promoters were determined by H3K4me3 peak calling, although some peaks might be associated with low transcriptional activity. We hypothesized that the dual presence of DNA hypomethylation and H3K4me3 would more strongly enrich for functional hypomethylation events. Most hypomethylated DMRs did not colocalize with H3K4me3 assayed in the same tumor. However, 200 hypomethylated DMRs did colocalize with H3K4me3 in the same tumor, and we found increased expression of genes containing dual DNA hypo/K4me3 loci in their gene bodies. The GLI3 oncogene contained a gene body promoter with recurrent DNA hypomethylation and H3K4me3, and we found that the Delta GLI3 transcript expressed from this promoter was up-regulated in two GBMs with these dual epigenetic marks.
In contrast to our findings, a breast cancer methylome study found that some hypomethylation was allelic with the non-hypomethylated allele occupied by repressive chromatin marks H3K9me3 or H3K27me3, associated with gene silencing (Hon et al. 2012). We did not examine H3K9me3 or H3K27me3, but our data show that a limited number of hypomethylated loci are associated with gene activation rather than repression. The genetic and epigenetic contexts are likely important for determining the effect of individual hypomethylation events, which might have diverse consequences from one locus to another, or from one tumor to another.
Finally, we examined in detail the molecular consequences of hypomethylation at gene body promoters within TP73 and TERT, two genes with functions important to cancer. Our study is the first to detect deltaNp73 oncoprotein expression in GBM. DeltaNp73 competes for p53 and full-length p73 binding sites and inhibits activation of apoptosis in cancer cells (Ishimoto et al. 2002). DeltaNp73 retains DNA binding but not transactivation functions, and thus acts as a dominant-negative inhibitor of both p53 and p73 by direct competition for binding to genomic targets. We also uncovered GBM-specific expression of a novel TERT alternate transcript from a hypomethylated gene body promoter. The Delta TERT transcript initiates within intron 2 and encodes a putative delta TERT protein lacking most of the reverse transcriptase domain but gaining a unique N terminus, similar to the general pattern in deltaNp73. We were unable to determine if the delta TERT protein is expressed in GBM due to the lack of a specific anti-C-terminal antibody. Alternatively spliced TERT variants lacking reverse transcriptase activity enhanced cell proliferation and stimulated WNT signaling (Hrdlickova et al. 2012). Full-length TERT physically interacts with the SWI/SNF-related chromatin remodeling protein SMARCA4, and occupies and activates WNT-dependent genes, indicating a role in transcriptional activation (Park et al. 2009). It remains to be determined whether delta TERT is similarly multifunctional and has telomere-independent oncogenic properties.
Our global and gene-specific analyses implicate a subset of gene body promoter hypomethylation in up-regulating alternate transcripts with potential oncogenic consequences in GBM. Some of the expressed RNAs might be transcriptional noise or noncoding RNAs. However, the gene body transcripts initiating within TP73, TERT, and GLI3 all encode known or putative alternate proteins. Methylation-regulated gene body promoters confer protein diversity among normal tissues, and our data suggest that gene body hypomethylation might contribute to the up-regulation of oncogenic protein isoforms in GBM as well.
Methods
MeDIP-seq and MRE-seq
DNA was isolated from frozen tissue and tumor specimens by 55°C overnight digestion in SDS/Tris/EDTA/Proteinase K lysis buffer followed by two phenol/chloroform/isoamyl alcohol extractions then two chloroform extractions, precipitation with sodium acetate and ethanol, one 70% ethanol wash, and resuspension in TE buffer. MeDIP- and MRE-seq were performed as previously described (Maunakea et al. 2010) with the following modifications. For GBM 5, the MRE-seq library was constructed using five restriction enzyme digests (HpaII, AciI, Hin6I, Bsh1236I, HpyCH4IV) instead of the three (HpaII, AciI, Hin6I) used for all other samples. GBM 5 MRE-seq reads were informatically filtered to include only those from the three-enzyme protocol to make the GBM 5 data comparable to other samples.
Processing of MeDIP-seq and MRE-seq data
MeDIP-seq and MRE-seq reads from GBM (this study) and two normal brains (Maunakea et al. 2010) (NCBI Sequence Read Archive accession number SRP002318) were aligned with Bowtie (Langmead 2010) to hg19. Only uniquely mapping reads were used for downstream analyses. For MRE-seq, an additional constraint is that the 5′ end of a read must map to the CpG site within a methyl-sensitive restriction enzyme site. For MRE-seq, duplicate reads with identical start positions and sequences are retained, but duplicates were filtered from MeDIP-seq data to retain only one read. MRE reads were normalized to account for differences in enzyme efficiency and scoring consisted of tabulating reads with CpGs at each fragment end (Maunakea et al. 2010). For display of GBM MeDIP-seq and MRE-seq on the browser, uniquely mapping reads were normalized by aCGH copy number. To normalize for differences in sequencing depth, MeDIP-seq was normalized to 50 M reads and MRE-seq to 20 M reads. The MeDIP-seq tracks show the density of coverage, based on extension of each read to the average fragment size. For MRE-seq, an MRE score was defined for each MRE CpG site as the normalized number of MRE reads that map to the site, regardless of the orientation of the read.
M&M analysis of MeDIP- and MRE-seq to identify DMRs
The coverage of sequencing data (MeDIP-seq and MRE-seq) and genomic CpG information were calculated in a 500-bp window across the human genome (hg19 assembly), excluding autosomes. Agilent array CGH copy number estimates by segment (see below) were used to scale the MeDIP-seq and MRE-seq counts. We used the seqCBS algorithm (Shen and Zhang 2012) on the MeDIP-seq sequencing data to further refine the segments identified by CBS on the Agilent data, since there is ambiguity in the genomic regions between Agilent markers. SeqCBS segments sequencing data in a manner analogous to CBS based on changes in the ratio of test counts to test plus reference counts. We used MeDIP-seq reads, and not MRE-seq, to refine the genomic positions of copy number segments because its sequencing reads are more evenly distributed across the genome. MRE-seq reads are sparser because they derive from unmethylated CpGs located within MRE recognition sites that generate fragments of a specific size range when digested. To refine the segments for each GBM, we compiled the genomic regions that were between the CGH array probes at the ends of adjacent copy number segments (“gap regions”) and ran seqCBS, using MeDIP-seq reads from GBM and normal brain 1 as test and reference, respectively. SeqCBS analyzes the ratio of test/test + reference to find changepoints. For each gap region, if seqCBS could call a single changepoint, we used this genomic position as the new copy number breakpoint for adjacent segments. If seqCBS could not identify a single changepoint, we used the midpoint between the CGH probes as the new changepoint. In this way, we closed all of the gap regions. Sequencing depth normalization on the MeDIP-seq and MRE-seq data was performed after CNV normalization. The R package methylMnM (http://epigenome.wustl.edu/MnM/; Zhang et al. 2013) was used to identify DMRs by paired comparison between each normal brain sample and each GBM. Only windows on autosomes were considered. Windows with significant Q-values compared with both normal brains were identified as DMRs, at either Q < 10−5 or Q < 10−13. These thresholds were determined by comparing M&M analysis of MeDIP- and MRE-seq on H1 embryonic stem cells to shotgun bisulfite sequencing data from the same cells.
Array-CGH and copy number segmentation analysis
Array CGH was performed on the Agilent 244K platform for each GBM using 500 ng of DNA and following manufacturer’s protocol. Pooled normal DNA, different from the normal brain DNA used for methylation analysis, was used for the reference. All samples passed Agilent’s quality control metrics. We utilized the circular binary segmentation (CBS) method (Venkatraman and Olshen 2007) to estimate copy number. This method splits the genome, one chromosome at a time, into regions of equal number based on changes in the distribution of log tumor to reference values. The estimated copy number in each segment is based on the average of the log ratios in the segment. Before segmentation, the copy number data were GC-normalized. To confirm the accuracy of our copy number segmentation, we downloaded TCGA copy number array data (Agilent and Affymetrix) for GBMs 2 and 3, and compared the segmentation values with our own. Across all chromosomes, the segmentations were highly similar (data not shown).
Illumina Infinium HumanMethylation27 array
A total of 500 ng of genomic DNA per sample was used for Infinium methylation array analysis. Bisulfite conversion was performed with the EZ DNA methylation kit (Zymo Research) and each sample was eluted in 12 μL water. Amplification and hybridization to the HumanMethylation27 BeadChip were carried out according to manufacturer’s instructions at the UCSF Genomics Core Facility. Beta values, representing quantitative measurements of DNA methylation at individual CpGs, were generated with Illumina GenomeStudio software. Beta values were normalized to background and filtered to remove those with low signal intensity. The filtered data were used for all subsequent analysis.
Expression microarrays
High-quality total RNA was prepared by TRIzol (Invitrogen) purification and confirmed by RIN value determined from Agilent Bioanalyzer analysis. Expression analysis was performed on the Affymetrix GeneChip Human Gene 1.0 ST platform at the UCSF Gladstone Genomics Core. Data normalization was performed by the Robust Multi-array Average (RMA) method.
Bisulfite sequencing
Total genomic DNA was bisulfite converted as per the protocol of Grunau et al. (2001), with modified conversion conditions: 95°C for 1 min, 50°C for 59 min for a total of 16 cycles. Locus-specific PCR primers were designed with MethPrimer (Supplemental Table S5; Li and Dahiya 2002). PCR products were TOPO TA cloned into pCR2.1/TOPO (Invitrogen). At least 10 individual colonies were sequenced per sample. DNA methylation patterns and levels were analyzed suing BISMA (Rohde et al. 2010).
Processing of Roadmap Epigenomics histone modification data
H3K4me3 ChIP-seq data of relevant cell types were produced as part of the Roadmap Epigenomics project (Bernstein et al. 2010) and deposited to GEO (GSE16368). Mapped read density was generated from aligned sequencing reads using customized Perl scripts. Read density overlapping DMRs and their 5-kb upstream/downstream regions were extracted at 50-bp resolution as RPKM values, with histone input data subtracted.
ENCODE HMM chromatin state annotation
ChromHMM annotation of nine ENCODE cell lines (Ernst et al. 2011) was obtained from the UCSC Genome Browser (Rosenbloom et al. 2012). The nine cell lines are the following: H1 ESC, GM12878, K562, HepG2, HUVEC, HMEC, HSMM, NHEK, and NHLF. For each DMR, we examined overlapping annotation of “promoter,” “enhancer,” and “insulator” states in these ChromHMM maps. We determined that 3.3% of background genomic windows were potential promoters, 17.3% were potential enhancers, and 1.4% potential insulators.
Genomic features
RepeatMasker annotations, CGIs, UCSC Gene, and refGene coding loci features were all downloaded from the UCSC Genome Browser (Kent et al. 2002; Meyer et al. 2013).
qRT-PCR
Total RNA was isolated by TRIzol. cDNA synthesis was performed with a mix of random hexamer and oligo dT using Moloney Murine Leukemia Virus Reverse Transcriptase (Invitrogen). Real-time RT-PCR was performed with the Opticon2 Continuous Fluorescence Detector (MJ Research) and relative expression levels calculated using the deltaCt method with GAPDH as a housekeeping control. Melting curves were inspected to confirm PCR specificity. Primers are listed in Supplemental Table S5.
Western blotting
GBM primary tumor, GBM cell line, and normal brain lysates were electrophoresed on 4%–20% polyacrylamide with SDS, transferred to PVDF by wet transfer, blocked, and then probed with mouse 1:100 anti-DeltaNp73 (Calbiochem #38C674) and rabbit 1:1000 anti-GAPDH (Cell Signaling # 14C10) antibodies. After incubation with labeled secondary antibodies, a signal was detected by chemiluminescence.
Luciferase promoter assay
Target loci were PCR-amplified from normal human brain DNA with primers containing added restriction sites (Supplemental Table 5). PCR products were purified, restriction digested, and cloned into the pGL3-Basic vector (Promega). One microgram of each vector was cotransfected with 10 ng (1:100) of pRL-TK (Promega) expressing Renilla luciferase by using FuGENE 6 (Roche). After 48 h, firefly luciferase activity was measured by using the Dual-Luciferase reporter assay system (Promega) and normalized against Renilla luciferase activity. The pGL3 basic vector was used as a basal level of luciferase activity, and the pGL3 “promoter” vector containing the SV40 promoter was used as a positive control.
Chromatin immunoprecipitation-sequencing (ChIP-seq)
Chromatin isolated from GBMs 1, 2, and 5 frozen tissue was digested to mononucleosomes with micrococcal nuclease. Histones marked with H3K4me3 were immunoprecipitated with a monoclonal antibody (Cell Signaling, Catalog # 9751) using Sepharose beads coated in Protein A/G, and the DNA purified. An IgG negative control was performed in each experiment and quantitative PCR verified enrichment. Illumina library construction was performed as per manufacturer’s instructions. A total of 75-bp single-end or paired-end sequencing was performed on the Illumina HiSeq. As a control, input DNA from each chromatin preparation was also sequenced. The resulting sequences were quality filtered and mapped back to the human genome using the Burrows-Wheeler Aligner (Li and Durbin 2010). The sequencing libraries were aligned as single-end samples to ensure equal mapping bias across the samples. ChIP enrichment was further verified using CHANCE (Diaz et al. 2012). Peak calling was performed using MACS at a 1% false discovery rate and a P-value < 1 × 10−10 (Zhang et al. 2008).
5′-RACE
Total RNA was isolated by TRIzol (Invitrogen) and used to amplify the 5′ end of the putative Delta TERT mRNA using the Gene Racer kit (Invitrogen) following the manufacturer’s protocol. The TERT gene-specific primers are listed in Supplemental Table 5. The amplification products were gel-purified, cloned into pCR4-TOPO (Invitrogen), and inserts were sequenced.
Data access
MeDIP-seq, MRE-seq, and ChIP-seq data have been submitted to the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/) under accession number EGAS00001000685. Affymetrix GeneChip Human Gene 1.0 ST and Agilent 244K array-CGH data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE49412 and GSE49808.
Acknowledgments
We thank collaborators in the NIH Reference Epigenome Mapping Centers. We thank Jonathan Woo, Joanna Philips, Keith Ligon, Michael Barnes, Vy Ngo, and Tali Mazor for assistance, and Melinda Baerwald for manuscript editing. This work was supported in part by NIH NCI NRSA-T32 and NCI NRSA-F32 fellowships to R.P.N. and grants from the National Brain Tumor Society, Accelerate Brain Cancer Cure, the Goldhirsh Foundation, and US NIH grant 5U01ES017154 to J.F.C. B.Z. is supported by NIDA’s R25 program DA027995. J.S. is supported by a Distinguished Scientist Award from the Sontag Foundation. T.W. is supported in part by the March of Dimes Foundation, the Edward Jr. Mallinckrodt Foundation, P50CA134254, and a grant from the Foundation for Barnes-Jewish Hospital. J.F.C. and T.W. are supported by NIH grant 5U01ES017154. J.F.C. and R.P.N. are supported by US NIH grant 1P01CA168540.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.164707.113.
References
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CPE, van Dijk CM, Tollenaar RAEM, et al. 2011. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44: 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieux B, Ching TT, VandenBerg SR, Costello JF 2006. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res 66: 8469–8476 [DOI] [PubMed] [Google Scholar]
- Cho M, Uemura H, Kim SC, Kawada Y, Yoshida K, Hirao Y, Konishi N, Saga S, Yoshikawa K 2001. Hypomethylation of the MN/CA9 promoter and upregulated MN/CA9 expression in human renal cell carcinoma. Br J Cancer 85: 563–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, et al. 2000. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 24: 132–138 [DOI] [PubMed] [Google Scholar]
- Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh C-L, Feinberg AP 2002. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res 62: 6442–6446 [PubMed] [Google Scholar]
- De Smet C, De Backer O, Faraoni I, Lurquin C, Brasseur F, Boon T 1996. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci 93: 7149–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A, Nellore A, Song JS 2012. CHANCE: comprehensive software for quality control and validation of ChIP-seq data. Genome Biol 13: R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, et al. 2009. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–571 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B 1983. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301: 89–92 [DOI] [PubMed] [Google Scholar]
- Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M 1983. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11: 6883–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A 2001. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res 29: E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. 2011. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Yoshida T, Matsumoto K, Katsuta K, Waga S, Sakakura T 1997. Differential expression of tenascin-C and tenascin-X in human astrocytomas. Acta Neuropathol 93: 431–437 [DOI] [PubMed] [Google Scholar]
- Hoffman RM 1984. Altered methionine metabolism, DNA methylation and oncogene expression in carcinogenesis. A review and synthesis. Biochim Biophys Acta 738: 49–87 [DOI] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, et al. 2012. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. 2013. TERT promoter mutations in familial and sporadic melanoma. Science 339: 959–961 [DOI] [PubMed] [Google Scholar]
- Hrdlickova R, Nehyba J, Bose HR Jr 2012. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol Cell Biol 32: 4283–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC Known Genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA 2013. Highly recurrent TERT promoter mutations in human melanoma. Science 339: 957–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S 2002. Possible oncogenic potential of ΔNp73: a newly identified isoform of human p73. Cancer Res 62: 636–641 [PubMed] [Google Scholar]
- Ito Y, Koessler T, Ibrahim AE, Rai S, Vowler SL, Abu-Amero S, Silva AL, Maia AT, Huddleston JE, Uribe-Lewis S, et al. 2008. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum Mol Genet 17: 2633–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SR, Link PA, Karpf AR 2006. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 25: 6975–6985 [DOI] [PubMed] [Google Scholar]
- Jin S-G, Kadam S, Pfeifer GP 2010. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res 38: e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. 2013. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343: 189–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The Human Genome Browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, et al. 2013. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci 110: 6021–6026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. 2012. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 44: 1236–1242 [DOI] [PubMed] [Google Scholar]
- Laird PW 2010. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11: 191–203 [DOI] [PubMed] [Google Scholar]
- Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, Kochert K, Bouhlel MA, Richter J, Soler E et al. 2010. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med 16: 571–579 [DOI] [PubMed] [Google Scholar]
- Langmead B 2010. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics 32: 11.7.1–11.7.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Zhu JJ, Anthony DC, Greider CW, Black PM 1998. Telomerase activity in human gliomas. Neurosurgery 42: 1120–1124; discussion 1124–1125 [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotsch D, Ghanim B, Laaber M, Wurm G, Weis S, Lenz S, Webersinke G, Pichler J, Berger W, Spiegl-Kreinecker S 2013. Prognostic significance of telomerase-associated parameters in glioblastoma: effect of patient age. Neuro Oncol 15: 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Leclerc RD, May G, Wagner GP 2011. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet 43: 1154–1159 [DOI] [PubMed] [Google Scholar]
- Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, Ropero S, Schackert G, Esteller M 2009. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 4: 255–264 [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N, Aldape K, et al. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. 2013. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41: D64–D69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, et al. 1997. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90: 785–795 [DOI] [PubMed] [Google Scholar]
- Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, Rodolfo M, Schneider C 2006. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci 103: 11160–11165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolsky Y, Sviridov E, Yao J, Dosymbekov D, Ustyansky V, Kaznacheev V, Dezso Z, Mulvey L, Macconaill LE, Winckler W, et al. 2008. Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res 68: 9532–9540 [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. 2007. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster B, Linnet L, Christensen LL, Thorsen K, Ongen H, Dermitzakis ET, Sandoval J, Moran S, Esteller M, Hansen TF, et al. 2013. Non-CpG island promoter hypomethylation and miR-149 regulate the expression of SRPX2 in colorectal cancer. Int J Cancer 132: 2303–2315 [DOI] [PubMed] [Google Scholar]
- Park J-I, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, et al. 2009. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460: 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman P, Melin BS, Wang Z, McKean-Cowdin R, Michaud DS, Wang SS, Bondy M, Houlston R, Jenkins RB, Wrensch M, et al. 2012. Genome-wide association study of glioma and meta-analysis. Hum Genet 131: 1877–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, Liu F, Marquez VE, Morgan RA, Schrump DS 2011. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res 71: 4192–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Statham AL, Speed TP, Clark SJ 2010. Protocol matters: which methylome are you actually studying? Epigenomics 2: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, Jeltsch A 2010. BISMA—fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 11: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH 2006. Large-scale structure of genomic methylation patterns. Genome Res 16: 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, Cline MS, Karolchik D, Barber GP, Clawson H, et al. 2012. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res 40: D912–D917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufini A, Agostini M, Grespi F, Tomasini R, Sayan BS, Niklison-Chirou MV, Conforti F, Velletri T, Mastino A, Mak TW, et al. 2011. p73 in cancer. Genes Cancer 2: 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236 [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S 1997. A survey of telomerase activity in human cancer. Eur J Cancer 33: 787–791 [DOI] [PubMed] [Google Scholar]
- Shen JJ, Zhang NR 2012. Change-point model on nonhomogeneous Poisson processes with application in copy number profiling by next-generation DNA sequencing. Ann Appl Stat 6: 476–496 [Google Scholar]
- Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre J-Y, et al. 2009. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41: 899–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB 2007. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23: 657–663 [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D 2007. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci 104: 18613–18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466 [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. 2007. Epigenetic stem cell signature in cancer. Nat Genet 39: 157–158 [DOI] [PubMed] [Google Scholar]
- Wild L, Flanagan JM 2010Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta 1806: 50–57 [DOI] [PubMed] [Google Scholar]
- Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G 2010. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 6: e1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrensch M, Jenkins RB, Chang JS, Yeh R-F, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S, et al. 2009. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 41: 905–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Rauch TA, Zhong X, Bennett WP, Latif F, Krex D, Pfeifer GP 2010. CpG island hypermethylation in human astrocytomas. Cancer Res 70: 2718–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. 2013. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet 45: 836–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zardo G, Tiirikainen MI, Hong C, Misra A, Feuerstein BG, Volik S, Collins CC, Lamborn KR, Bollen A, Pinkel D, et al. 2002. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet 32: 453–458 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, Cheng JB, Li D, Stevens M, Lee HJ, et al. 2013. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res 23: 1522–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Houseman EA, Morrison Z, Wrensch MR, Patoka JS, Ramos C, Haas-Kogan DA, McBride S, Marsit CJ, Christensen BC, et al. 2011. DNA hypermethylation profiles associated with glioma subtypes and EZH2 and IGFBP2 mRNA expression. Neuro Oncol 13: 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]