

Abstract
It is commonly known that mammalian microRNAs (miRNAs) guide the RNA-induced silencing complex (RISC) to target mRNAs through the seed-pairing rule. However, recent experiments that coimmunoprecipitate the Argonaute proteins (AGOs), the central catalytic component of RISC, have consistently revealed extensive AGO-associated mRNAs that lack seed complementarity with miRNAs. We herein test the hypothesis that AGO has its own binding preference within target mRNAs, independent of guide miRNAs. By systematically analyzing the data from in vivo cross-linking experiments with human AGOs, we have identified a structurally accessible and evolutionarily conserved region (∼10 nucleotides in length) that alone can accurately predict AGO–mRNA associations, independent of the presence of miRNA binding sites. Within this region, we further identified an enriched motif that was replicable on independent AGO-immunoprecipitation data sets. We used RNAcompete to enumerate the RNA-binding preference of human AGO2 to all possible 7-mer RNA sequences and validated the AGO motif in vitro. These findings reveal a novel function of AGOs as sequence-specific RNA-binding proteins, which may aid miRNAs in recognizing their targets with high specificity.
The Argonaute protein family is a class of conserved and essential effectors in constituting the RNA-induced silencing complex (RISC) in animals for post-transcriptional gene silencing (Liu et al. 2004; Hock and Meister 2008; Djuranovic et al. 2011). This protein family is typically subdivided into two groups: The Argonautes (AGOs) interact with small interfering RNAs (siRNAs) and microRNAs (miRNAs), whereas the PIWI proteins interact with the PIWI-interacting RNAs (piRNAs) (Djuranovic et al. 2011). In human, the AGO protein family includes AGO1–4, which incorporates siRNAs or miRNAs as guide strands into the RISC (Liu et al. 2004; Peters and Meister 2007; Hock and Meister 2008). Structurally, the AGO protein consists of four domains, including the N-terminal, PAZ, MID, and PIWI domains (Peters and Meister 2007; Djuranovic et al. 2011). The PAZ and MID domains anchor the 3′ and 5′ ends of the small RNAs, respectively (Lingel et al. 2004; Ma et al. 2004; Parker et al. 2005; Mi et al. 2008; Frank et al. 2010), while the PIWI domain shares substantial similarities with ribonuclease H for the endonucleolytic activity (Song et al. 2004). Specifically for miRNAs, the ribonucleoprotein complex RISC centered on AGO usually binds to the 3′ untranslated regions (3′ UTRs) of the target messengers recognized by the miRNA guide strand, and subsequently triggers destabilization or translational inhibition of the target mRNAs (Bartel 2009). It is known that miRNAs usually recognize their mRNA targets through seed complementarity, where the 3′ UTR target sites (or sometimes coding regions) are base-paired with the miRNA’s seed region (the second through the seventh nucleotides along the mature miRNA’s 5′ ends) (Grimson et al. 2007; Bartel 2009). Structural analysis of Thermus thermophilus AGO revealed that the duplex formed by the target sequence and the miRNA seed was placed near the center of the ternary complex in proximity to AGO’s endonucleolytic PIWI domain (Wang et al. 2008).
Seed complementarity alone has very limited power in identifying real miRNA targets through a genome scan. Furthermore, recent AGO crosslinking and coimmunoprecipitation experiments (AGO-CLIP) have revealed extensive AGO-bound mRNAs devoid of complementarity with miRNA seeds (Chi et al. 2009; Hafner et al. 2010; Leung et al. 2011; Helwak et al. 2013). These observations indicate that (1) other factors beyond the seed complementarity may regulate the specificity of miRNA target recognition, or (2) AGO protein might behave like generic RNA-binding proteins, which directly recognize their own targets independent of miRNA’s guidance. The latter notion is bolstered by recent structural modeling of the AGO–miRNA–mRNA ternary structure, which suggested that it is the arginine residues in AGO’s nucleotide-binding channel, rather than the miRNA–mRNA duplex, that play a more significant role in stabilizing AGO binding to target mRNAs (Wang et al. 2010). It is also consistent with the crystal structure of human AGO2, where residues protruding into the RNA-binding groove control the trajectory of the target RNA along the entire protein (Elkayam et al. 2012). More direct evidence was revealed from an AGO2-CLIP experiment in mouse embryonic stem cells where the miRNA biogenesis pathway has been presumably deactivated by knocking out the mouse Dicer gene (Dicer1) (Leung et al. 2011). This study showed that the coimmunoprecipitated fragments with mouse AGO2 were enriched for a G-rich motif, whose perturbation was associated with downstream gene expression (Leung et al. 2011). However, such an association does not necessarily imply a physical interaction between mouse AGO2 and the G-rich motif. First, recent high-throughput experiments have shown that RNA motifs recognized by RNA-binding proteins are typically AU-rich for structurally accessibility (e.g., UGUAHAUA for the pumilio protein) (Ray et al. 2013); it thus remains undetermined how the G-rich motif would fit with the general principle. Second, and more importantly, an in vitro assay is required to validate the physical binding affinity between AGO2 and its recognition motif, which serves to exclude other confounding cofactors in vivo. To this date, it remains unclear whether AGO proteins are indeed a class of RNA-binding protein with its own specificity for mRNA sequence recognition.
In this work we aim to uncover the principles for AGO mRNA targeting in human cells. Independent of miRNA’s regulation, we identified an ∼10-bp region immediately upstream of the AGO crosslinking site that can accurately distinguish AGO-bound mRNAs from those unbound sequences. This region is structurally accessible, evolutionarily conserved, and significantly enriched for a sequence motif, whose binding to human AGO2 was further validated in vitro using RNAcompete assay (Ray et al. 2009, 2013). Collectively, these findings demonstrate the RNA-binding specificity of human AGO without guidance by small RNAs.
Results
We analyzed the mRNAs crosslinked to AGO proteins from a PAR-CLIP experiment (Hafner et al. 2010) in human HEK293 cells, which significantly improved the conventional crosslinking experiments by allowing identification of the crosslinking sites on the associated RNA fragments. The experiment immunoprecipitated human AGO1–4 and revealed similar sets of mRNAs bound by these homologous proteins. A 41-nucleotide (nt) region of these AGO-associated mRNA fragments was selected for analysis, in which the fragments were all centered at the crosslinking sites at the 21st nucleotide with a characteristic T-to-C transition. These fragments have been subjected to maximal subtraction of background crosslinking (Hafner et al. 2010).
Because miRNAs usually act on the 3′ UTRs of their target mRNAs and sites in coding regions (CDS) only mediate marginal repression (Hafner et al. 2010), here we limited our analysis to the 5495 AGO-crosslinked mRNA fragments derived from 3′ UTRs unless otherwise mentioned. Among these sequences, 2243 were “site-containing” with at least one confident miRNA binding site identified by Hafner et al. (2010), while the remaining 3252 were “non-site-containing,” devoid of confident miRNA binding sites. For the latter, it is possible that there existed unknown noncanonical miRNA binding sites, and thus we also designed separate control experiments and an in vitro assay to ensure that our observation from PAR-CLIP was indeed independent from miRNA-mediated regulation (see below). Notably, all of the identified miRNA binding sites in PAR-CLIP were exclusively localized in the ∼10-bp region immediately downstream from the crosslinking sites.
To unravel the sequence patterns for AGO binding, it is critical to generate a set of negative controls that is comparable with the AGO-bound mRNAs. Due to the disparate expression levels, variable UTR length, or biased molecular functions, random sampling from the genome background or the unbound sequences was inappropriate in this study. Instead, we used a method requiring a one-to-one match (for both site-containing and non-site-containing sequences) between each positive and negative sample to control for those potential confounders. Briefly, for each positive sample, its negative counterpart of the same length was randomly sampled from the same 3′ UTR (guarantees equal expression level and molecular function) that (1) was not associated with AGO in PAR-CLIP; (2) centered on a thymine (T) mimicking the T-to-C transition at the crosslinking site; (3) had similar CG dinucleotide frequency as the matched positive samples; and (4) had similar G content as the matched positive samples (P ≥ 0.05, Wilcoxon rank sum test). The last criterion aimed to overcome the G-depletion bias of PAR-CLIP sequences due to the use of RNase T1 (Kishore et al. 2011).
The predictability of AGO–mRNA interactions
We postulated that if AGO recognizes mRNAs in a sequence-specific manner, identifiable sequence features should distinguish the AGO-bound fragments from those unbound sequences. Thus we devised an approach that first identifies the defining elements for AGO binding, followed by a systematic characterization of this element for AGO binding specificity. To exclude the effects of miRNA-mediated regulation, we first considered 3252 AGO-bound fragments that were devoid of miRNA binding sites (i.e., non-site-containing sequences, defined above). These positive samples were matched with their respective negative counterparts using the protocol described above. Two-thirds of the sequences were randomly chosen to train a support vector machine (SVM), and the remaining one-third of the sequences were then examined as a blind test set to determine the predictability of AGO binding. The prediction accuracy was measured by the area under the receiver operating characteristics (AUC [area under curve]). Higher AUC indicates a greater predictive power of AGO binding, whereas AUC = 0.5 is equivalent to a random guess. Our predictions are solely based on learning the differences in sequence composition between the positive and negative samples, and the learning process was achieved by constructing an SVM classifier, whose input was the mRNA fragment sequences orthogonally encoded into binary strings (for details, see Methods). We found that those AGO-bound sequences (devoid of miRNA binding sites) showed marked distinguishability from the matched non-AGO-bound transcripts, with AUC = 0.75 on the independent test set (Fig. 1A, solid curve). Our analysis further revealed that the observed distinguishability could not be fully explained by the distinct dinucleotide composition profiles (the frequencies for the 16 different dinucleotides) between the positive and the negative samples, whose AUC only reached 0.58 (Fig. 1A, dashed curve). This observation suggests that AGO can specifically recognize a particular set of sequences without the guidance of miRNAs. Interestingly, when the same procedure was performed on the 2243 AGO-bound sequences with at least one confident miRNA binding site (site-containing sequences), the prediction accuracy on the blind test set was further enhanced (AUC = 0.86) (Fig. 1B, solid curve), suggesting that the presence of miRNA binding sites might further enhance the predictability of the target mRNAs. Since the predictive power does not directly result from the dinucleotide composition (AUC = 0.58 and 0.63, respectively, in Fig. 1A,B), there must exist other sequence characteristics beyond dinucleotide composition that have defined the binding specificity of AGO, independent of the presence of miRNA binding sites.
Figure 1.

AUC scores for the blind test of predicting AGO-associated sequences that do not have (A) or do have (B) miRNA binding sites. AUC scores were computed using sequence alone (solid curve) or using the dinucleotide composition within the 41-nt regions (dashed curve). The thin dashed line is a reference for a lack of predictive power.
Dissecting sequence features underlying AGO binding
We reasoned that if there existed any region in a relatively constant position defining AGO-binding specificity, other regions outside this area would give no, or at least substantially reduced, predictive power for AGO binding. We then extended the original 41-nt AGO-crosslinked sequences to a 81-nt window by mapping the mRNA fragments back onto their reference UTR sequences, and the 81-nt sequences were again centered on the observed crosslinking site (at the 41st nucleotide). This was to test the existence of further elements outside the original 41-nt AGO-associated RNA fragments. We used a 15-nt sliding window to scan the 81-nt region to locate signals for AGO binding, and fed these truncated sequences into an SVM classifier constructed using the same procedures described above (also see Methods). The 15-nt window was slid along the 81-nt region, and at each position an AUC was calculated on the blind test set to indicate the distinguishability between the positive and the negative samples. The samples were randomly split into 70% and 30% for training and testing, respectively, and the site-containing and non-site-containing mRNAs were considered separately.
As shown in Figure 2, A and C, for the 81-nt sequences without confident miRNA binding sites (i.e., non-site-containing sequences), the AUC score peaked at the 31st position (covering the region between 31 and 45), which was similar to the highest AUC score (0.75) when using the full-length 41-nt sequences (shown in Fig. 1A). We also scanned the site-containing transcripts with confident miRNA binding sites; in this case, the region from the 35th nucleotide to the 50th nucleotide exhibited the highest AUC (Fig. 2B,D), similar to AUC = 0.86 when using the full-length 41-nt sequences (shown in Fig. 1B). Therefore, these two sequence windows (in the extended 81-nt sequences), which both encompassed the crosslinking sites at the 41st position, have contained the most defining information for AGO target recognition. It is clear from Figure 2, A and B, that the peripheral sequences at position 1–20 and 50–81 (where AUC ≃ 0.5) generally did not provide information for AGO binding.
Figure 2.

Dissecting sequence features underlying AGO target recognition for RNA fragments that have or do not have miRNA binding sites. (A,B) (Blue curve) AUC for predicting AGO binding using a 15-nt window on the independent test set sliding over the 81-nt sequences. Structural accessibility of each nucleotide is shown at the bottom (above the x-axis), where green, red, and blue represent unchanged, elevated, or reduced accessibility in comparison with the negative samples (false discovery rate <0.01). Statistical significance was determined by the paired Wilcoxon rank sum test between the positive and negative samples. (C,D) The discriminative power for the most defining regions (left column, corresponding to the peaks on the blue curves), the most accessible regions (middle column), and the subregions of the most defining regions after the crosslinking sites (right column). (E,F) Seventeen-way phastCons scores (y-axis indicates cross-species evolutionary conservation) of each nucleotide for an RNA fragment without (E) and with (F) miRNA binding sites. It is clear that AGO-associated sequences (the positive samples in red) tend to be more conserved than the negative samples (in blue). The highly accessible regions immediately flank the peaks, indicating elevated conservation for these regions of high accessibility. Error bars, 1 SEM.
Since these two regions were the most informative in predicting AGO binding for site-containing and non-site-containing transcripts, we hypothesized that AGO (or with its cofactors) might physically interact with these regions. Such a direct interaction between a protein and an mRNA requires that the region on the mRNA should have high structural accessibility. In the literature, the accessibility of mRNA molecules to proteins is defined as the probability of the RNA nucleotide being single-stranded in the RNA secondary structure, i.e., being unpaired with other nucleotides of the same RNA, which alone is highly predictive of the binding between RNAs and RNA-binding proteins in vivo (Li et al. 2010). We used the same method and computed the accessibilities for each nucleotide along the 81-nt mRNA sequences (see Methods) for both AGO-bound sequences and their matched negative controls. As shown in Figure 2, A and B (bottom green bar), for both the site-containing and non-site-containing sequences, we observed that an ∼10-nt region immediately upstream of the crosslinking site (at the position 41) showed substantially elevated accessibility compared to its corresponding regions on the non-AGO-bound sequences (false discovery rate [FDR] < 0.01). To exclude any boundary effects when comparing site accessibility between positive and negative samples, we also further extended the 81-nt sequences to 201 nt (centering on the crosslinking sites) and again found only the same regions (for both non-site-containing and site-containing sequences, respectively) showing the elevated structural accessibility.
Interestingly, for both site-containing and non-site-containing sequences, the accessible region largely overlapped with the most defining 15-nt window for AGO binding (the AUC peaks in Fig. 2A,B). Specifically, the region encompassing the 31st to 45th nucleotides was the most defining for AGO binding for the non-site-containing sequences, which fully has covered the accessible region from 32–40 (Fig. 2A,C), and the inclusion of the accessible nucleotides in this region was not expected by chance (P = 1.12 × 10−10, hypergeometric test). Similarly, for the site-containing sequences, six accessible nucleotides were included in the 15-nt window most defining for AGO binding, a stronger enrichment than random expectation (P = 0.007, hypergeometric test). Taken together, the openness of the ∼10-nt window immediately upstream of the crosslinking site is likely to mediate the AGO–mRNA interaction, independent of miRNA-mediated regulation.
Our analysis further revealed that these ∼10-nt regions of higher accessibility can effectively distinguish the AGO-bound mRNAs from those not bound by AGO. The average AUC was 0.64 for non-site-containing sequences (Fig. 2C) from a threefold cross-validation, and 0.68 for site-containing sequences (Fig. 2D). These two AUC scores were qualitatively comparable, suggesting that the defining feature for AGO binding in the 10-nt accessible region is likely shared between the site-containing and non-site-containing sequences. However, the prediction power of using this 10-nt accessible region was lower than that of using the most defining 15-nt sequences (represented by the peaks in Fig. 2A,B, corresponding to the positions of 31–45 for the non-site-containing sequences and 35–50 for the site-containing sequences). This observation suggested that the regions within the 15-nt window at the position of 42–45 for the non-site-containing sequences (Fig. 2A, the 41st nucleotide is the crosslinking site) or 42–50 for the site-containing sequences (Fig. 2B) might contain independent information for predicting AGO binding. This was clearly shown in Figure 2C, where, for non-site-containing sequences, the region from 42–45 reached AUC scores similar to the upstream region (32nd to 40th) (P ≥ 0.05), but was still lower than the 15-nt sequence (31st to 45th). For the site-containing sequences, however, the downstream region from 42 to 50 alone could achieve AUC scores similar to using the full-length 15-nt sequence (35th to 50th) (Fig. 2D), indicating that the region 42–50 might play a predominant role in defining AGO binding. This observation was anticipated simply because all the miRNA binding sites were located within the region of 41–50, and the observed predominance should be attributed to the enriched sequence complementarity in this region with miRNA seeds that were most abundantly expressed in this cell line.
Evolutionary conservation of the highly accessible regions
We next examined the level of evolutionary conservation for each nucleotide along the 81-nt sequence for these AGO-bound sequences (Fig. 2A,B), followed by a comparison with the matched negative samples not bound by AGO. We quantified evolutionary conservation based on the UCSC phastCons score (Siepel et al. 2005) derived from multiple genome comparisons (phastCons 17way), and assigned each nucleotide a score between zero and one, where a higher score indicates elevated evolutionary conservation. We compared the average phastCons scores of the positive samples against those of the negative samples across each nucleotide; statistical significance was determined by the paired Wilcoxon rank sum test. As seen in Figure 2, E and F, regardless of the presence of miRNA binding sites, the positive samples showed marked elevation in evolutionary conservation compared to the negative samples. This trend, particularly pronounced for those AGO-bound fragments depleted of miRNA binding sites (Fig. 2E), strongly argued that AGO binding is not promiscuous and is of substantial biological significance. More interestingly, also for this group of positive samples, a significant elevation in phastCons scores was clearly seen, peaking at the position of 30 (Fig. 2E, red curve). This trend was absent in the corresponding negative samples (Fig. 2E, blue curve). We emphasize that these positive samples were devoid of miRNA binding sites, and this region encompassed the highly accessible region from positions of 32–40 (Fig. 2A), suggesting strong selective constraint on these regions of high accessibility for AGO binding independent of miRNA’s involvement. We also confirmed this trend on the mRNAs that have miRNA binding sites (shown in Fig. 2F), where there was a clear increase in phastCons scores between the accessible positions from 31 to 40. However, the peak was shifted to the crosslinking site at the position 41, which should be explained by the strong evolutionary conservation on miRNA binding sites exclusively located between position 41 and the 50 (downstream from the crosslinking site at 41). Taken together, this observed strong purifying selection on the highly accessible region suggested that AGO might indeed physically interact with this region to recognize its targets.
Uncovering a sequence motif in the region of high accessibility
Since the accessible region alone can predict AGO binding (Fig. 2C,D), we next explored the enriched motif in this region followed by experimental validation to confirm its binding affinity with AGO in vitro. We first considered the non-site-containing sequences to exclude any potential effects from miRNA binding sites, and performed a discriminative motif discovery by comparing motif occurrence between the positive and the matched negative samples within the accessible region between the 32nd and 40th nucleotide positions (Fig. 2A). We implemented the software DEME for this purpose due to its improved performance and specific design for discriminative motif discovery (Redhead and Bailey 2007). Motifs and their corresponding position-specific scoring matrices (PSSMs) were estimated from 70% of the samples based on a probabilistic model and were subsequently tested for their discriminative power on the remaining 30% of the samples in a blind test. With this we identified an overrepresented 5-mer motif in this accessible region (shown in Fig. 3A).
Figure 3.
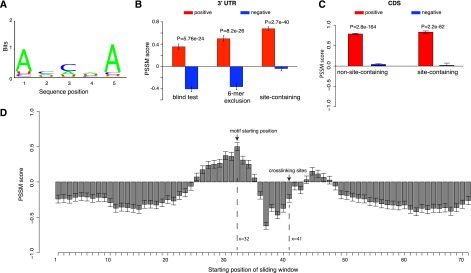
AGO recognizes its targets in a sequence-specific manner within the highly accessible region upstream of the crosslinking site. (A) The mRNA fragments that are depleted of miRNA binding sites have an overrepresented sequence motif within the ∼10-nt accessible regions. (B) Discriminative power of the motif among three independent test sets. (Left) A subset of the AGO-bound mRNAs that was not used in deriving the motif; note these mRNAs were depleted of miRNA binding sites. (Middle) The same data set after removing sequences with at least one 6-mer match to any miRNA seed region. (Right) AGO-bound mRNA sequences that are highly enriched for miRNA binding sites. Overrepresentation of the motif in each data set was assessed by the sum of the position-specific scoring matrix (PSSM) scores for its letters. The more positive scores indicate a greater enrichment for the sequences resembling the motif, while the negative scores indicate the depletion of this motif. (C) Discriminative power of the motif among the AGO-bound sequences in coding regions with (site-containing) and without (non-site-containing) downstream miRNA binding sites. (D) Nine-nucleotide sliding window scan for the AGO-bound sequences that were not used to derive the AGO motif. These sequences were centered at the crosslinking site at position 41. The x-axis is the starting position of the sliding window, and the y-axis is the PSSM score for the AGO motif inside each sliding window. The motif is strongly favored in the ∼10 nt immediate upstream (x = 32) of the crosslinking sites (x = 41) and is disfavored in most other regions (receiving negative PSSM scores). Error bars, 1 SEM.
We tested this motif within the region (32nd and 40th nucleotide positions) on three independent test sets for different purposes. Following conventional procedures, the sum of the motif PSSM scores across its positions was used to quantify the similarity between this motif and any given 5-mer sequence, and higher positive scores indicate more resemblance of this motif. The test sets were designed to perform the following comparisons: (1) the blind test set not used for training (Fig. 3B, left column); (2) the blind test set after subtracting AGO-bound sequences with any 6-mer match to any miRNA seeds (Fig. 3B, middle column); and (3) the AGO-bound sequences containing at least one confident miRNA binding site (site-containing sequences) (Fig. 3B, right column). The second step was to maximally eliminate any unknown sites potentially interacting with miRNAs, which served to ensure that the uncovered motif was indeed not affected by miRNA binding sites. In all the comparisons, we consistently observed that the AGO bound sequences were always more highly scored than these matched unbound samples. Interestingly, in the third comparison, this motif performed very well when applied to the highly accessible region on the site-containing AGO-bound mRNAs: positions 31–40, upstream of the crosslinking site (Fig. 2B). Note that these miRNA binding sites were localized downstream from the crosslinking site at the 41st position. Therefore this observation suggests a possible synergistic interaction between AGO and miRNA binding for target recognition (described below). Taken together the motif in this accessible region represents an RNA recognition motif by AGO, which is independent of miRNA-mediated regulation. Although the motif was derived from 3′ UTR sequences, we further asked whether the same motif could be extrapolated to coding sequences (CDSs) that were bound by AGO in the PAR-CLIP data set. Following the procedures described above, the CDSs coimmunoprecipitated with AGO were divided into site-containing and non-site-containing, and each was then matched by their negative CDS counterparts (used the criteria above). We then scanned the region upstream of the crosslinking sites in the CDS fragments and observed that the AGO-bound sequences (both site-containing and non-site-containing) were consistently highly scored relative to the sequences not bound by AGO (P < 1 × 10−50, Wilcoxon rank sum test) (Fig. 3C). This observation suggests that this AGO motif was not only manifested in 3′ UTRs but also in CDS.
This motif was derived from the sequences only within the accessible region at 32–40 (Fig. 2A). We reasoned that if this motif is indeed specifically recognized by AGO, such motif enrichment should be absent from sequences outside of this motif region (Fig. 2A). We then used a 9-nt sliding window (matching the length of positions 32–40) (Fig. 2A) to scan the AGO coimmunoprecipitated fragments from the blind test set (non-site-containing sequences, not used to derive the AGO motif) (Fig. 3B). Sequences within the sliding window were then scored by the PSSM for the AGO motif. As shown in Figure 3D, the PSSM score profiles peaked exactly at the expected motif position, starting at the position of 32 (the starting position of the structurally open region) (Fig. 2A), and this motif is strongly disfavored by many other regions receiving negative PSSM scores, particularly regions encompassing the crosslinking site (Fig. 3D). Therefore, these patterns, inside and outside the motif region, collectively define AGO–mRNA interaction, where the motif only lies in the structurally open region (∼10 nt upstream of the crosslinking site) for AGO recognition.
Since AGO2 is a highly conserved protein with amino acid identity ∼99.1% between human and mouse (Supplemental Fig. S1), we next tested whether our observations from human also hold in mouse. To this date, there is no available AGO2 PAR-CLIP experiment in mouse; we thus validated our observations on the conventional mouse HITS-CLIP data (Chi et al. 2009), in which 294 AGO2 HITS-CLIP crosslinking sites in 3′ UTRs have been previously predicted (Zhang and Darnell 2011). The procedure was then repeated on these HITS-CLIP fragments by matching each positive HITS-CLIP fragment with its respective negative sequences by randomly sampling a nonoverlapping fragment from the same 3′ UTRs with almost identical nucleotide composition. Again we observed that the expected motif region (∼10 bp immediately upstream of crosslinking sites) showed a significant increase in the PSSM of the AGO motif (derived from human PAR-CLIP, P < 2.4 × 10−2, Wilcoxon rank sum test) (Supplemental Fig. S2). We also shuffled the nucleotides within the motif region to counteract against any potential biased nucleotide composition, and again we observed that the real mouse sequences were highly scored by the AGO PSSM (P = 8.9 × 10−3, Wilcoxon rank sum test). Taken together, this comparison demonstrated that the observed AGO motif is likely a conserved regulatory mechanism shared between human and mouse. More importantly, the consistency between PAR-CLIP and HITS-CLIP further indicated that our observation was unlikely biased by a particular platform.
We also noticed recent literature suggesting a potential bias in base composition for the PAR-CLIP system (Kishore et al. 2011). To ensure that the discovered motif was not resultant from any systematic bias with PAR-CLIP protocol, we performed a set of additional controls. To account for any potential bias in base composition for the PAR-CLIP data, we generated a set of negative samples with the same base composition with the positive samples by randomly shuffling the nucleotide positions for positive sequences. Again we found this motif significantly distinguished the positive from the negative sequences (Supplemental Fig. S3A). We next compared our AGO motif with other RNA-binding proteins studied on the same PAR-CLIP platform, including PUM2, QKI, and IGF2BP1-3. PUM2’s signature motif is highly distinct from the AGO motif, thus expectedly receiving negative scores (P < 5.7 × 10−7, Wilcoxon rank sum test) (Supplemental Fig. S3B). This suggests our motif was not promiscuous on the PAR-CLIP platform. To some extent, resemblance was observed among the motifs for the AGO, QKI, and IGF2BP proteins. For example, IGF2BP fragments received positive scores when scanned with QKI’s signature motif and also with our AGO motif. However, this observation does not suggest that these proteins recognize a similar set of sequences. In fact, in our in vitro assay (data shown below), AGO binding affinity was only significantly correlated with its own motif rather than the signature motifs of the QKI and IGF2BP proteins (Supplemental Fig. S4). Moreover, the in vivo PAR-CLIP coimmunoprecipitated sequences for these individual proteins were also nonoverlapping, confirming that the binding specificities of each motif for individual proteins were highly specific.
Experimental validation using the RNAcompete assay
To validate the AGO binding affinity to our motif, we utilized RNAcompete (Ray et al. 2009, 2013) to test in vitro whether the interaction with our motif is biochemically intrinsic to AGO without involving miRNAs or other cofactors in the RISC complex. RNAcompete is a recently developed technique that allows systematic identification of RNA-binding specificities for RNA-binding proteins through its binding reaction with a complete range of k-mers in diverse RNA contexts (structured and unstructured) (Ray et al. 2009, 2013), and each k-mer score can be calculated and normalized (see Methods), with higher scores representing greater recognition specificity by the tested protein. This assay is ideal for our purpose simply because of two facts. First, it enumerates all possible k-mers, which allows us to explore AGO–RNA preferences in an unbiased manner. Second, it is an in vitro system, which will confirm that the observed binding preferences are a natural property of AGO and are not modulated by other cofactors or small RNAs. To this end, we followed previous RNAcompete procedure (Ray et al. 2009, 2013) to determine the binding affinities of the full-length human AGO2 protein across all possible 7-mer sequences (i.e., 16,384 possible 7-mers), which, as demonstrated previously, gives the best performance when assessing protein–RNA binding specificities (Ray et al. 2009).
We chose human AGO2 as a representative AGO protein for RNAcompete validation given its prime importance in the RNAi pathway. AGO2 shares amino acid identity >75% with all other human Argonaute proteins (80% with AGO1, 77% with AGO3, and 76% with AGO4). A previous RNAcompete study has shown that RNA-binding proteins sharing >70% amino acid identity at the RNA-binding domain tend to bind similar RNA sequences, in vitro (Ray et al. 2013), and the AGOs recognize similar targets in vivo (Landthaler et al. 2008; Hafner et al. 2010). Therefore our observation on AGO2 should be extrapolated to other AGO proteins.
All 7-mer RNA oligonucleotides on the RNAcompete platform were scored and normalized for binding to human AGO2, and were subsequently scanned with the PSSM scores for our motif (Fig. 3A). If our motif can be recapitulated by RNAcompete assay, 7-mers with higher affinities with human AGO2 are then anticipated to have greater similarity with the motif, thus receiving higher PSSM scores in our model.
By analyzing 16,384 possible 7-mers from the RNAcompete platform, we observed a significant correlation between RNAcompete scores and PSSM scores from our model, where sequences receiving higher PSSM scores tended to show increased preference by human AGO2, and vice versa (Spearman’s correlation 0.5, P < 1 × 10−5) (Fig. 4A,B for two technical replicates). However, this trend was completely absent when we used a partial AGO2 only involving the PIWI domain, which collectively confirmed that (1) the observed trend was not a systematic bias of the RNAcompete platform, and (2) the observed binding affinity is innate for human AGO2, which does not require the engagement of small RNAs and other cofactors.
Figure 4.
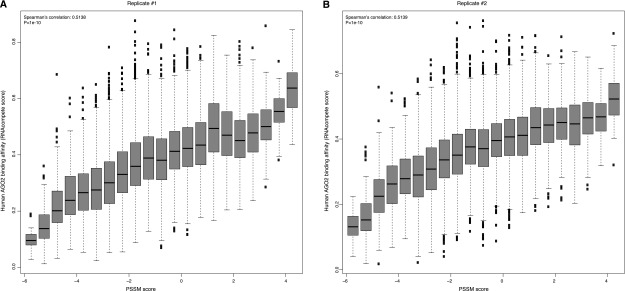
Significant correlation between RNAcompete scores and PSSM scores for the identified motif in replicate 1 (A) and replicate 2 (B). In both panels, the higher RNAcompete scores indicate greater binding specificities of AGO2 measured from the assay. Similarly, higher PSSM scores represent a greater resemblance of the identified motif. It is clear that sequences with high similarity with the identified motif tend to have a higher binding specificity of human AGO2.
The synergistic effect between the AGO motif and miRNA binding sites
Having validated the AGO motif, we further explored its potential function for post-transcriptional gene regulation. Since the AGO motif and miRNA binding sites are localized within the ∼10-bp region upstream of and downstream from the crosslinking site, respectively, we hypothesized that the AGO motif is likely to strengthen miRNA-mediated regulation. We thus only considered the site-containing sequences bound by AGO in PAR-CLIP, and retrieved the miRNA expression data assayed in the same cells (HEK293) where PAR-CLIP was performed (Hafner et al. 2010). We divided the miRNAs according to their expression levels into bins from high (the 10 most highly expressed miRNAs), to moderate (expression ranks from 11 to 20), to low (all the other miRNAs) (Fig. 5). This division was based on the observation that the top 25 most abundant miRNAs account for >75% of the small RNA reads in this particular cell line (Hafner et al. 2010). With this, the AGO-bound sequences were grouped based on the presence of the binding sites (within 10 nt immediately downstream from the crosslinking site, see PAR-CLIP) of miRNAs with maximal expression falling into the three groups from high to low. Interestingly, as shown in Figure 5, the lowly expressed miRNAs indeed required a stronger AGO motif (greater PSSM scores) for their target recognition (P = 5.7 × 10−3, Wilcoxon rank sum test). Thus these strong AGO motifs likely complement weak regulatory interactions for lowly expressed miRNAs.
Figure 5.

Weakly expressed miRNAs require a strong AGO motif (P = 5.7 × 10−3, Wilcoxon rank sum test). Motif strength was quantified by their PSSM scores in the motif region.
Discussion
In this study, we analyzed PAR-CLIP data and uncovered a novel function of the human AGO protein as a class of RNA-binding proteins with its own sequence specificity. We identified the RNA motif for AGO recognition in a structurally accessible region. The substantially elevated evolutionary conservation in this open region containing the motif highlights its physiological importance in mediating AGO binding. The motif uncovered from in vivo data was also subjected to in vitro validation, establishing the AGO’s role in recognizing its own targets without miRNA’s guidance. Particularly, for the AGO coimmunoprecipitated fragments without known miRNA sites, they were enriched for functions including transcription, RNA splicing, and protein localization (FDR ≤ 0.05, hypergeometric test), suggesting a potential role of AGO in these biological processes. More importantly, our analyses also suggested that a true miRNA target might require both AGO binding sites and miRNA binding sites, and the former is likely to complement the binding efficiency from the latter. Overall, our discovery is consistent with a recent structural analysis of AGO binding, in which molecular dynamics and thermodynamic modeling of crystal structures of AGO–miRNA–mRNA ternary complex showed that a set of arginine residues concentrated in AGO’s nucleotide-binding channel contributes significantly more to stabilizing the binding of AGO to mRNAs than the RNA–RNA duplex formed by seed-pairing between mRNA and miRNAs (Wang et al. 2010). Moreover the recently solved crystal structure of human AGO2 lends additional support for this argument, where miRNA’s seed is placed in a narrow portion of AGO2’s RNA binding groove, with AGO2’s residues protruding into the groove to control the target RNA’s relative position along the protein (Elkayam et al. 2012).
We performed in vitro validation for our AGO motif using our recently developed RNAcompete system, which demonstrated that the AGO–motif interaction should be physical and direct, not confounded by any other factors. This validation fits with our observation made from the in vivo data, where the regions mediating the physical interactions are structurally open. Our additional validation involved using mouse HITS-CLIP data to test the motif seen from human PAR-CLIP. Consistencies between species and platforms further suggested that sequence specificity by AGO is a conserved regulatory mechanism. Importantly, given the extreme conservation between the mouse and human AGO2 (protein sequence identity 99.1%; the minimal substitutions are clustered on the N terminus, not significantly affecting major functional domains) (Supplemental Fig. S1), our in vitro system thus allowed us to test the G-rich motif associated with AGO2 from mouse embryonic stem cells in a previous study (mESC with Dicer−/−) (Leung et al. 2011). We were able to regenerate the significantly enriched 5-mer G-rich motifs (the same length for a comparison with our AGO motif) (Fig. 6A) from the two biological replicates from the original data, but our RNAcompete assay revealed that these G-rich motifs (from both replicates) were strongly disfavored by human AGO2 in vitro (using all probes on the RNAcompete array as background, P < 8.65 × 10−165 for all the comparisons with background, Wilcoxon rank sum test) (Fig. 6B), in sharp contrast with the elevated specificity between AGO2 and its motif discovered in this study. This observation was consistent with structural requirement of sequence-specific RNA-binding proteins, whose recognition motifs are usually AU-rich (Ray et al. 2013). Taken together, the AGO2 motif in this study (Fig. 3A), rather than the G-rich motif from previous work (Fig. 6A), directly and physically interacts with AGO2.
Figure 6.
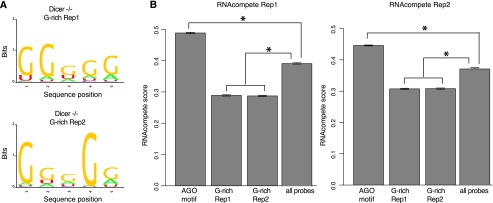
In vitro validation for a previously reported AGO-associated motif. (A) The previously reported G-rich motif was able to be replicated using MEME from the original data (Dicer−/− mutant from mouse embryonic cells, two biological replicates, Dicer−/− replication 1 and 2). (B) The G-rich motifs received the lowest RNAcompete scores for human AGO2 (sharing ∼99.1% protein sequence similarity with mouse) in comparison with all probes on the RNAcompete array and also with the AGO motif in this study. (*) P < 1 × 10−50, Wilcoxon rank sum test.
The observed target recognition specificity by AGO also raises another interesting question that requires further investigation. To this date, AGO is best known as an effector in the RNAi pathway; however, with its own binding affinity independent from small RNA’s guidance, it is also likely that AGO may act as a generic RNA-binding protein to regulate its own cognate targets. Thus it would be important to further study the multifaceted roles of AGO. Particularly considering the fact that the miRNA-independent tethering of AGO proteins to reporter mRNAs could mimic the miRNA-mediated regulation (Pillai et al. 2004), future work is warranted to investigate whether the observed AGO binding preference would contribute to modulating cellular mRNA levels, especially for sequences devoid of miRNA binding sites.
Methods
The PAR-CLIP data sets were retrieved from Hafner et al. (2010). In this study we only considered mRNA fragments that were crosslinked with AGO on their 3′ UTRs and excluded those fragments that were mapped to the protein-coding region. We considered the sequences extended to 81 nt after mapping them back onto the human reference genome (UCSC hg18). The extended sequences beyond the 3′ UTR boundary were discarded from further analysis. For each sequence, we randomly sampled its negative counterpart as a control from the same 3′ UTR and required the negative samples to have comparable dinucleotide frequency with the positive samples. We also required the negative samples be centered on a thymine (T), mimicking the T-to-C transition at the crosslinking site for the positive samples. These procedures served to control for the potential confounding factors for our comparison, including disparate expression levels, unequal 3′ UTR length, or functional bias of the sampled genes. A few positive samples having no appropriate negative correspondence (not satisfying the above criteria, or their 3′ UTRs were too short) were not considered in this study. In the end we retained 3252 AGO-associated mRNA fragments depleted of miRNA binding sites and 2243 mRNA fragments highly enriched for miRNA binding sites downstream from the crosslinking site. When making prediction, we encoded each nucleotide into a binary string, i.e., A→0001, C→0010, G→0100, and T→1000. This procedure thus converted each sequence into a binary string of equal length for both the positive and negative samples. We constructed a support vector machine (SVM) using LIBSVM (http://www.csie.ntu.edu.tw/∼cjlin/libsvm/) with an RBF (radial basis function) kernel. We used the default parameters except setting the parameter C for soft margin to be 1e5. However, varying parameters did not qualitatively affect our results. Nucleotide accessibility in the RNA secondary structure was calculated using RNAplfold (Bernhart et al. 2006), which averages over all the sliding windows of size W to compute the probability of a nucleotide being unpaired with a maximal span of L nucleotides. In these experiments, we used W = 80 and L = 40, which were previously adopted for predicting siRNA and RBP binding (Tafer et al. 2008; Li et al. 2010). We used RNAplfold over other RNA folding programs because of its capability of robustly computing the local base-pairing probabilities without predefining the exact position of sequence window.
We retrieved a 17-way phastCons score from the USCS Table Browser (Karolchik et al. 2004), and the statistical comparison was performed using the paired Wilcoxon rank sum test for the paired positive and negative samples. Sequence motif was identified using DEME, which was specifically designed for discriminative motif discovery (Redhead and Bailey 2007).
The mRNA fragments crosslinked to PUM2, QKI, IGF2BP1, IGF2BP2, and IGF2BP3 were also generated by PAR-CLIP (Hafner et al. 2010). We also tested the motif on HITS-CLIP data (Zhang and Darnell 2011) on mouse brain tissues. The corresponding negative sequences were generated with the same protocol as performed on PAR-CLIP data, where each positive sequence was paired with a negative sequence from the same 3′ UTR with almost the same base composition. The crosslinking sites were predicted by CIMS (Zhang and Darnell 2011), and we considered the expected motif region, ∼9-bp immediate upstream of the CIMS crosslinking sites.
The RNA pool generation, RNAcompete pull-down assays, and microarray hybridizations were performed as previously described (Ray et al. 2009) with the following exceptions. The GST-tagged AGO2 (20 pmol) and RNA pool (1.5 nmol) were incubated in 1 mL of binding buffer (20 mM Hepes at pH 7.8, 80 mM KCl, 20 mM NaCl, 10% glycerol, 2 mM DTT, 0.1 μg/μL BSA) containing 20-μL glutathione sepharose 4B (GE Healthcare) beads (prewashed three times in binding buffer) for 30 min at 4°C, and subsequently washed four times for 2 min with binding buffer at 4°C. The microarrays were subsequently imaged and analyzed. Each batch of experiments was represented with a matrix where rows correspond to probes and columns are the pull-down intensities of the RBPs in that batch. First, low-quality spots due to spatial trends or other problems related to image analysis are discarded. Next, quantile normalization was applied to calibrate multiple arrays. Background signal and other nonspecific binding effects are removed by subtracting from each spot the row median value across all the experiments and dividing by the robust estimate of standard deviation calculated across all the experiments. Finally, the same robust Z-score transformation is applied to each column (i.e., experiment). The 7-mer scores for each column (experiment) are calculated by assigning zero to all the probes with scores below the median (column median). The score for a 7-mer is then equal to the trimmed mean (5%) of the scores of the probes that contain that 7-mer.
Data access
The RNAcompete data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE55122.
Acknowledgments
We thank Dr. Xiao Li for advice in computing the structural accessibility of RNA sequences. J.L. thanks Dr. Zhihai Ma at Stanford Genetics for helpful comments on this work. We thank the anonymous reviewers for their insightful suggestions. We acknowledge funding support from an NSERC Discovery Grant (grant number 327612) and an Ontario Research Fund (Global Leadership Round in Genomics and Life Sciences). This work is dedicated to the memory of S.Z.
Author contributions: J.L. conceived the project. J.L., T.K., and Z.Z. designed the experiments. R.N. cloned and purified AGO2 full-length and PIWI proteins. D.R. performed the RNAcompete experiment. J.L. and T.K. performed the analysis. Z.Z. and T.R.H. supervised this project. J.L. and Z.Z. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162230.113.
References
- Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart SH, Hofacker IL, Stadler PF 2006. Local RNA base pairing probabilities in large sequences. Bioinformatics 22: 614–615 [DOI] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460: 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R 2011. A parsimonious model for gene regulation by miRNAs. Science 331: 550–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L 2012. The structure of human argonaute-2 in complex with miR-20a. Cell 150: 100–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F, Sonenberg N, Nagar B 2010. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 465: 818–822 [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, Tollervey D 2013. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock J, Meister G 2008. The Argonaute protein family. Genome Biol 9: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M 2011. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 8: 559–564 [DOI] [PubMed] [Google Scholar]
- Landthaler M, Gaidatzis D, Rothballer A, Chen PY, Soll SJ, Dinic L, Ojo T, Hafner M, Zavolan M, Tuschl T 2008. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA 14: 2580–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA 2011. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol 18: 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Quon G, Lipshitz HD, Morris Q 2010. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA 16: 1096–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingel A, Simon B, Izaurralde E, Sattler M 2004. Nucleic acid 3′-end recognition by the Argonaute2 PAZ domain. Nat Struct Mol Biol 11: 576–577 [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441 [DOI] [PubMed] [Google Scholar]
- Ma JB, Ye K, Patel DJ 2004. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429: 318–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, et al. 2008. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133: 116–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Roe SM, Barford D 2005. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature 434: 663–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G 2007. Argonaute proteins: mediators of RNA silencing. Mol Cell 26: 611–623 [DOI] [PubMed] [Google Scholar]
- Pillai RS, Artus CG, Filipowicz W 2004. Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA 10: 1518–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR 2009. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol 27: 667–670 [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead E, Bailey TL 2007. Discriminative motif discovery in DNA and protein sequences using the DEME algorithm. BMC Bioinformatics 8: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Smith SK, Hannon GJ, Joshua-Tor L 2004. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305: 1434–1437 [DOI] [PubMed] [Google Scholar]
- Tafer H, Ameres SL, Obernosterer G, Gebeshuber CA, Schroeder R, Martinez J, Hofacker IL 2008. The impact of target site accessibility on the design of effective siRNAs. Nat Biotechnol 26: 578–583 [DOI] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ 2008. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456: 921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Ma Z, Yang W, Ai C 2010. Mechanism of microRNA-target interaction: molecular dynamics simulations and thermodynamics analysis. PLoS Comput Biol 6: e1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB 2011. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29: 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]