

Abstract
Cytosine methylation in the genome of Drosophila melanogaster has been elusive and controversial: Its location and function have not been established. We have used a novel and highly sensitive genomewide cytosine methylation assay to detect and map genome methylation in stage 5 Drosophila embryos. The methylation we observe with this method is highly localized and strand asymmetrical, limited to regions covering ∼1% of the genome, dynamic in early embryogenesis, and concentrated in specific 5-base sequence motifs that are CA- and CT-rich but depleted of guanine. Gene body methylation is associated with lower expression, and many genes containing methylated regions have developmental or transcriptional functions. The only known DNA methyltransferase in Drosophila is the DNMT2 homolog MT2, but lines deficient for MT2 retain genomic methylation, implying the presence of a novel methyltransferase. The association of methylation with a lower expression of specific developmental genes at stage 5 raises the possibility that it participates in controlling gene expression during the maternal-zygotic transition.
Cytosine methylation (5-methylytosine) is an epigenetic modification present on the genomes of many eukaryotes (Suzuki and Bird 2008; Zemach et al. 2010). In vertebrates, methylation is present at CG dinucleotides over most of the genome, excepting CG-rich regions known as CpG islands that are commonly promoters or enhancers; this and other evidence gave rise to the view that methylation functions to suppress transcription initiation (Bird and Wolffe 1999). Genomewide analysis in a variety of unicellular and multicellular eukaryotes has shown that cytosine methylation occurs in multiple sequence contexts (CG, CHG, CHH) and in many species is found in mosaic patterns that mark certain regions of the genome, frequently gene bodies (Suzuki and Bird 2008; Feng et al. 2010; Zemach et al. 2010). This variety of patterns and contexts suggests that methylation may have multiple functions, but the scope of these functions is not clear.
Drosophila was long thought to lack cytosine methylation, but identification of a Drosophila homolog of DNA methyltransferase 2 (MT2, also known as DNMT2) (Hung et al. 1999; Lyko et al. 2000b) prompted studies that established the presence of small amounts of genomic cytosine methylation in early embryogenesis (Gowher et al. 2000; Lyko et al. 2000a; Kunert et al. 2003). Despite this, the pattern and function of cytosine methylation in Drosophila has never been clear, and recently considerable doubt has been expressed as to the presence of any significant quantity of genomic methylation (Goll et al. 2006; Schaefer and Lyko 2010a; Zemach et al. 2010; Raddatz et al. 2013). Inactivation of Mt2 does not visibly alter embryonic development (Kunert et al. 2003), although it can suppress position-effect variegation (Phalke et al. 2009). No clear pattern of genomic cytosine methylation has been established by any study, and genomewide bisulfite sequencing studies of wild type Drosophila embryos at an average depth of 5.6-fold (Zemach et al. 2010) and 32-fold (Raddatz et al. 2013) did not report methylated cytosines. The finding that MT2 can methylate tRNAs has led to speculation that its primary function in Drosophila is RNA methylation (Goll et al. 2006; Schaefer et al. 2010; Durdevic et al. 2013), and that genomic cytosine methylation is random and spurious (Schaefer and Lyko 2010b). The evidence that MT2 methylates RNAs does not demonstrate an absence of genomic cytosine methylation, which could be driven either by MT2 or by an unknown methyltransferase. The true extent and pattern of genomic cytosine methylation has remained obscure because the methods that have been used to detect it either could not localize methylation on the genome or had a sensitivity that was insufficient to detect methylation present on only a small subset of genomic copies.
We developed a method to both detect and localize low-abundance genomic methylation on a genomewide scale by enriching for methylated DNA and bisulfite sequencing the enriched material. Application of this method establishes the presence and location of genomic cytosine methylation in the stage 5 Drosophila embryo, indicating that methylation is dynamic between the unfertilized oocyte and stage 5. Retention of genomic cytosine methylation in Mt2−/− embryos implies that Drosophila must carry another DNA methyltransferase that has hitherto escaped identification.
Results
Mapping of cytosine methylation on the stage 5 Drosophila genome
We designed a strategy, which we call MeDIP-Bseq, to enrich for rare cytosine methylation in Drosophila (Fig. 1). Previous studies have indicated that cytosine methylation is most abundant at embryonic stage 5 (2–3 h post-fertilization) (Lyko et al. 2000a). We enriched sonicated stage 5 genomic DNA for methylcytosine by immunoprecipitation with an antibody to 5-methylcytosine. The immunoprecipitated DNA was then bisulfite converted and Illumina sequenced to obtain direct evidence for the presence of methylation. We obtained 28,607,102 paired reads (Supplemental Table S1) and aligned them to the Drosophila genome using the bisulfite mode of Novoalign. After bisulfite conversion, the two strands of DNA are noncomplementary and can be identified: Pairs of aligned reads were assigned to either the forward (CT) or reverse (GA) strand, and reads that could not be assigned unambiguously to one strand were discarded (Supplemental Table S2). We counted the number of converted and unconverted cytosines in the sequence reads for all cytosine positions on the reference CT and GA strands. Because only the two ends of each fragment were sequenced, it is expected that some methylcytosines present in immunoprecipitated fragments will not be part of the sequenced portion. Nevertheless, 18.1% of the alignments contained at least one unconverted cytosine (Table 1).
Figure 1.

MeDIP-Bseq strategy. MeDIP was performed on sonicated DNA to enrich for fragments containing methylcytosine (closed circles). The enriched immunoprecipitate was subjected to bisulfite conversion, which converts unmethylated cytosine (open circles) to uracil while methylcytosine is spared from conversion. The immunoprecipitated and bisulfite converted DNA was Illumina sequenced.
Table 1.
Methylated regions in the Drosophila genome
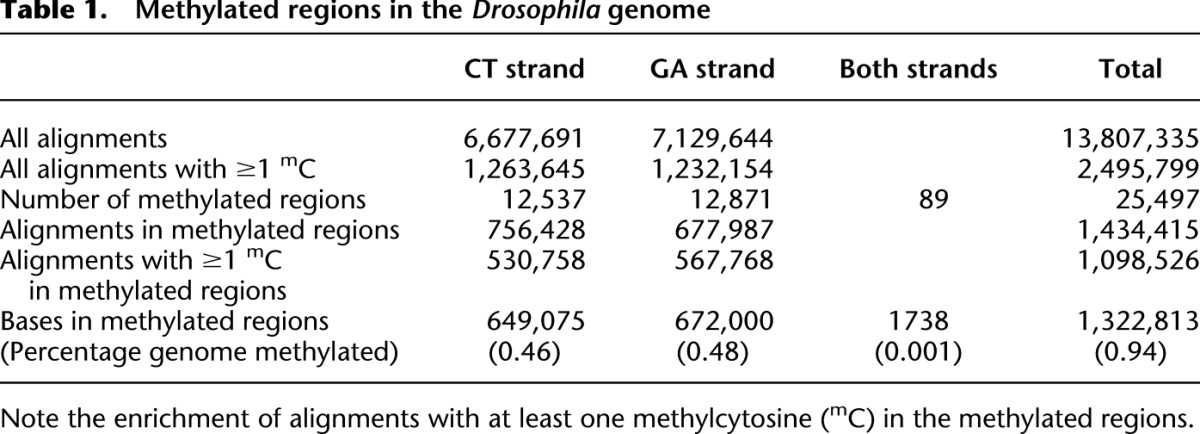
Our goal was to identify regions with a high density of alignments containing unconverted cytosines and to remove the background of unmethylated alignments. To identify the most frequently methylated cytosines, we retained only those positions at which the ratio of methylcytosine over total cytosine was at least 0.1 and at which at least three reads had a methylated cytosine at that position (Supplemental Fig. S1). To identify regions with a relatively high frequency of methylation, we divided the genome into contiguous 25-base segments; in each segment we counted the number of methylated cytosines that passed the steps above. Segments lacking any methylcytosines were discarded, and the remaining segments were merged if they were abutting each other. Finally, we considered as methylated those segments whose aligned reads contained a total of at least 25 methylated cytosines; we will refer to these segments as “methylated regions” or simply “regions.” We tested multiple values for the various parameters used to identify the methylated regions (Supplemental Fig. S1). For each set of parameters, we calculated the correlation coefficient between the number of methylated cytosines and the number of alignments within a region. We chose the set of parameters that optimizes the correlation coefficient, that is, the set of parameters that removes from the analysis the largest number of unmethylated (background) alignments while retaining methylated alignments.
This procedure yielded 25,497 methylated regions that make up 0.94% of the Drosophila genome. Forty-four percent of all alignments containing at least one methylated cytosine are within these regions, and 77% of alignments within these regions contain at least one methylated cytosine (Table 1). Our procedure selected the most highly methylated regions of the Drosophila genome. However it is possible that other regions are also methylated, but at a level too low to be detected by MeDIP-Bseq at the depth of sequencing we used: If a region were methylated in only one or a few cells in the embryo, we might not detect it. Our procedure produced regions in which there is a strong correlation between numbers of reads (enrichment by immunoprecipitation) and the amount of methylation within those reads (revealed by bisulfite sequencing) (Supplemental Fig. S2, bottom right, correlation coefficient = 0.667). This methylation is highly localized: 97% of the regions are 75 bases or less (Supplemental Fig. S3). Methylation is present in similar amounts on the CT and GA strands but is highly asymmetric: It is present on only one strand in >99.6% of the regions (Table 1; Fig. 2B). However, when large genomic intervals of 100 kb are considered, high density of methylation on one strand correlates to high density on the other strand (Fig. 2A). We note a weak anticorrelation of exon density and methylation (Fig. 2C). The heterochromatic chromosomes appear to have low levels of methylation relative to the euchromatic chromosomes, but this could be due to the difficulty of aligning reads to these chromosomes. Most striking is the greater overall density of methylated regions on chromosome X (Fig. 2A). These results indicate that the Drosophila genome is methylated but in a pattern unlike that seen in any other eukaryotic species to date.
Figure 2.

Genomewide distribution of methylation. (A) For both CT (red) and GA (green) strands, the density of methylated regions was calculated with Fseq using a feature length of 100 kb. Density of methylated regions on the CT and GA strands is plotted on the vertical axis with the chromosome displayed horizontally. The blue lines below each chromosome represent exon density calculated in the same way. The scale of the vertical axis is the same on all the plots. This reveals that density of methylation is highest on the X chromosome and lowest on the heterochromatic chromosomes. The density on the CT and GA strands is roughly symmetrical at this large scale. (B) Detailed example of the strand-asymmetric distribution of methylated regions along a 50-kb genomic interval containing two annotated genes. Regions methylated on the CT strand are shown in red, and regions methylated on the GA strand are in green. (C) Anticorrelation between the density of methylated regions and the density of exons over 100-kb genomic intervals. The scatterplot displays the density distribution of methylated regions over 100-kb intervals on the x-axis, and the exon density distribution over the same size intervals on the y-axis . The intensities of the distributions of methylated regions and exons are percentile-normalized. The bottom left corner shows that many regions of low exon density are nearly devoid of methylation. The concentration visible across the upper right half indicates that regions of high exon density tend to have low methylation density, and regions of high methylation density tend to have low exon density.
Validation and quantification of cytosine methylation
Our method identifies many small regions of the Drosophila genome that appear to carry cytosine methylation, but the pattern is so unusual as to create doubt as to its validity. Doubts may be heightened by the complexity of the method and the necessity for bioinformatic analysis. Furthermore, the method is not absolutely quantitative: The methylcytosine immunoprecipitation enrichment step prevents us from estimating the level of cytosine methylation in the segments we have identified. If the results of the procedure are valid, they should allow the targeted detection of methylation at specific locations on the Drosophila genome with bisulfite sequencing of PCR amplicons; this would also produce a precise measure of the amount of methylation at a targeted site. To this end, we used extremely deep sequencing of selected regions that were PCR amplified from bisulfite-converted genomic DNA. PCR primers were designed to prime completely converted (i.e., unmethylated) DNA. Although this primer design is biased against methylation in the primer binding sites, the highly localized pattern of methylation suggests that the primer binding sites are unlikely to be methylated, and this design provides a stringent bias against amplification of DNA in which bisulfite conversion has failed. As a control for bisulfite conversion, we spiked unmethylated lambda phage DNA into the Drosophila genomic DNA prior to bisulfite conversion and amplified and sequenced two segments of the phage genome; this indicates that average conversion is >99% (Supplemental Fig. S4). Deep sequencing of 66 bisulfite PCR amplicons to a depth greater than 10,000× confirms cytosine methylation in the identified segments and further demonstrates the highly localized pattern of methylation (Fig. 3A; Supplemental Figs. S5, S6).
Figure 3.

Direct amplification of bisulfite-converted DNA confirms methylation patterns and the presence of methylation in the absence of MT2. Four regions selected from the 66 that were analyzed (the full set is displayed in Supplemental Figs. S5, S8). Methylated regions identified by MeDIP-Bseq were PCR amplified from bisulfite converted DNA and Illumina sequenced to at least 10,000× coverage. Each dot represents one cytosine position. (A) bisulfite PCR (green); MeDIP-Bseq (purple). The y-axis at the left indicates the percentage of methylated cytosines in the bisulfite PCR; the y-axis at the right indicates the number of methylated cytosines detected by MeDIP-Bseq. Although the MeDIP-Bseq analysis is not quantitative, bisulfite PCR demonstrates the proportion of methylated cytosines at a given position, as well as the pattern of methylation of the amplified region. There is good agreement in the pattern of methylation detected by the two methods. (B) bisulfite PCR (green) (same data as A); unfertilized oocyte (brown); Mt2 deficient (Mt299) (red); EP(2)GE15695 (Mt2 wild type; strain provided by F. Lyko) (blue). The y-axis indicates the percenage of methylated cytosines in the bisulfite PCR. Methylation is also present in flies deficient for the DNA methyltransferase MT2 and at some loci in unfertilized oocytes. (C) Wild type EP(2)GE15695 (Mt2 wild type; stage 5 DNA provided by G. Reuter) (light blue); Mt2 deficient (Mt2149) (orange).
In many of the bisulfite resequenced regions, >10% of sequence reads showed evidence of methylation, and a few were much higher. This indicates that such regions could be detected by shotgun bisulfite sequencing if sufficient sequencing depth were achieved. Two previously published studies used whole genome bisulfite sequencing to conclude that methylation is absent from the Drosophila genome (Zemach et al. 2010; Raddatz et al. 2013). Comparison of our data with data from the first study (Zemach et al. 2010) indicates that sequence coverage was generally too low to detect these methylated segments. The second study (Raddatz et al. 2013) has sufficient coverage for most of the resequenced regions (Supplemental Table S3; Supplemental Fig. S7); only three of the regions with sufficient coverage show no evidence for methylation in that data set.
Since methylation is most abundant in the early stages of Drosophila development, but clearly is not present on all alleles, we considered the possibility that methylation is present at these sites in the unfertilized oocyte, and that the proportion of methylated alleles is diluted by genome replication thereafter. We prepared DNA from unfertilized Drosophila oocytes and assayed methylation of the 66 validated regions by deep bisulfite sequencing. This revealed that some of the regions that are methylated at stage 5 are more methylated in the unfertilized oocyte, whereas most are unmethylated (Fig. 3B; Supplemental Fig. S8). This finding indicates that methylation is a dynamic process in Drosophila development, and that a de novo DNA methyltransferase activity must be present post-fertilization.
Thus we were able to use the genomewide methylation map we generated to identify regions that prove to be methylated when subjected to deep bisulfite resequencing. This methylation cannot be an artifact of bisulfite nonconversion caused by sequence characteristics within the methylated regions: Most regions found to be methylated at stage 5 lack methylation in unfertilized oocytes, indicating that DNA in those regions can be efficiently converted by bisulfite treatment.
Genomic cytosine methylation is present in the absence of Mt2
MT2 is the only DNA methyltransferase homolog known in Drosophila. We and others had assumed that it must be responsible for any genomic methylation. To confirm this assumption, we prepared DNA from stage 5 Drosophila embryos of strains lacking a functional Mt2 gene (Jurkowski et al. 2008; Schaefer et al. 2010) and bisulfite resequenced the same loci used to validate methylation. This analysis revealed that cytosine methylation is still present in the MT2-deficient flies, albeit in some cases with an altered pattern (Fig. 3B,C; Supplemental Fig. S8). Although the fine details of the location vary slightly among the several sequenced strains, the presence of methylation at a site in the first sequenced strain (Canton-S) correlates with methylation at or very near that site in the Oregon R strain and the two MT2-deficient lines used in resequencing experiments. We conclude from this evidence that cytosine methylation on the Drosophila genome does not require MT2.
Distribution and sequence context of cytosine methylation
Methylated regions in the Drosophila genome are depleted from unique sequences and transposable elements and enriched in low complexity sequences, particularly simple sequence repeats (Supplemental Fig. S9A). However, methylation in simple sequence repeats is confined to specific classes, particularly those that are depleted of guanine, and is not found at all in most others (Supplemental Fig. S10). Similarly, methylated regions are enriched in some low complexity repeats, particularly C-rich and CT-rich, and nearly absent from others (Supplemental Fig. S9C). Among transposable elements, only the I element is not depleted of methylation (Supplemental Fig. S9B). Although one report has suggested heavy methylation of Invader4, a member of the Gypsy family of LTR retrotransposons (Phalke et al. 2009), bisulfite resequencing of the Invader4 LTR did not detect any methylation above our threshold of 1.2% (not shown).
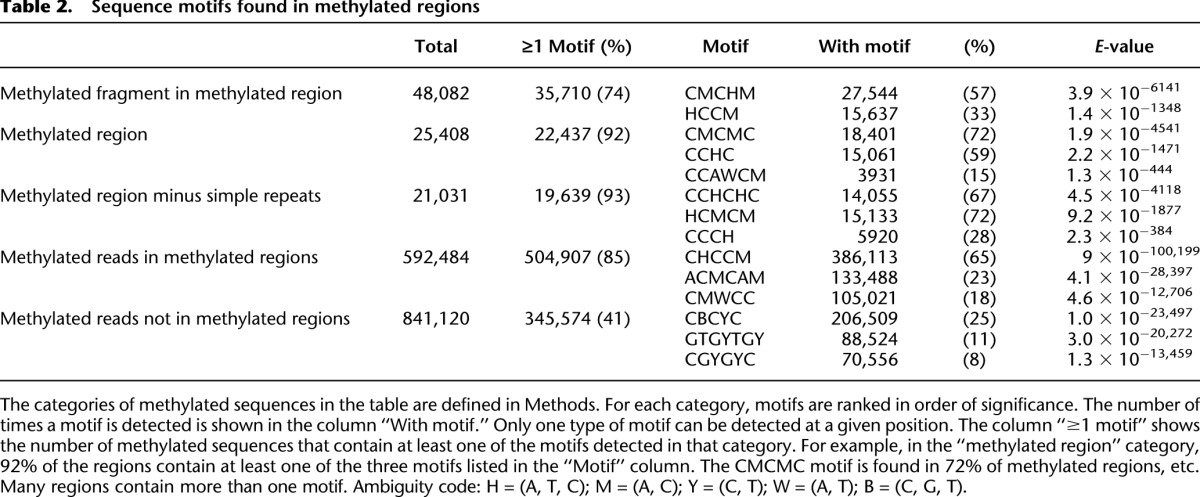
This pattern of methylation suggests that it may be found at specific sequence motifs. We obtained all short sequence fragments with evidence of cytosine methylation and used DREME (Bailey 2011) to identify motifs associated with methylation. This method identifies two enriched motifs (Fig. 4) that together are present in 74% of the short sequences used by DREME (Table 2). These motifs lack Gs and are CA- and CT-rich. DREME analysis also produces motifs enriched for CA and CT when the sequences of entire methylated regions are used (Fig. 4; Table 2). When methylated regions overlapping simple sequence repeats are excluded from the DREME analysis, highly similar motifs are found (Fig. 4; Table 2). DREME analysis of those methylated reads that are outside of methylated regions produces a different set of motifs, some of which are guanine-rich (Fig. 4; Table 2), suggesting the possibility of another, rarer, pattern of methylation.
Figure 4.

Sequence motifs in which methylation occurs. The motifs were identified by DREME analysis of different types of methylated sequences. The row at the top was derived using sequences identified within methylated regions as containing a high density of methylated cytosine; the first motif is found in 57% of these methylated fragments, and the second motif in 33% of fragments (Table 2). The second row was derived using the entire sequence of each of the 25,319 methylated regions. These motifs are highly similar to the motifs shown in the top row, which were derived from fragments (short sequences) within methylated regions. The third row was derived using the same entire methylated regions as in the top row, but excluding all regions overlapping with a simple sequence repeat. The similarity between the top and second rows indicates that the motifs associated with methylation are not confined to simple sequence repeats. The fourth row was derived using only sequence reads that contain at least one methylated cytosine, and align to a methylated region. Again, these motifs are similar to the others above, further corroborating the association of the motif with methylation; all these motifs are devoid of guanine. The enrichment of methylated regions in simple sequence repeats is largely explained by the presence of these motifs in some simple sequence repeats (Supplemental Fig. S10). Finally, the bottom row was derived using sequence reads that do not align to a methylated region but contain at least one methylated cytosine. The contrast between these motifs and those derived from methylated reads within methylated regions (fourth row) supports the specificity of the motif associated with methylated regions. It may also suggest the presence of a much rarer type of methylation associated with a different sequence motif.
Table 2.
Sequence motifs found in methylated regions
Functional correlates of genomic cytosine methylation
To characterize the relationship of methylation with genes and gene expression, we determined the location of methylated regions with respect to annotated features in the Drosophila genome. Methylated regions were significantly enriched in introns and intergenic sequences and depleted from coding regions and promoters (Supplemental Fig. S11A). They are more likely to be found at a greater distance from transcription start sites than expected based on a random distribution (Supplemental Fig. S11B), and are not associated with any chromatin features characterized by modENCODE (Supplemental Fig. S12; The modENCODE Consortium 2010).
To ask if methylation within genes was correlated with their expression, we used Cufflinks (Trapnell et al. 2010; Roberts et al. 2011) to reanalyze RNA-seq data generated by modENCODE (Graveley et al. 2011). We found an average expression of 25.4 fragments per kilobase of exon per million fragments mapped (FPKM) for the 600 genes with a high density of methylated regions, and 32.9 FPKM for the 8490 genes without methylation, corresponding to a 23% lower expression of genes containing methylated regions (P = 0.049, Wilcoxon two-sided test). Gene Ontology analysis of the same sets of methylated and unmethylated genes shows that the methylated genes are highly significantly enriched for terms associated with organ and anatomical structure development and transcription factors (Table 3). This evidence suggests that, as in other eukaryotic systems, cytosine methylation of the Drosophila genome may participate in gene regulation.
Table 3.
GO terms associated with genes having a high density of methylation

Discussion
These results provide the first unequivocal evidence of cytosine methylation at specific sites in the Drosophila genome and raise new questions about the origin and function of genomic methylation in this species. The relative rarity of methylation in Drosophila, even at stage 5 where it has been shown to be most abundant (Lyko et al. 2000a), has made it a subject of controversy. Some have recently suggested that it may be absent, or its presence is spurious and a reflection of nonspecific activity by MT2 (Marhold et al. 2004; Schaefer and Lyko 2010a; Raddatz et al. 2013); the near-normal phenotype of flies lacking functional MT2 has supported this point of view (Kunert et al. 2003; Phalke et al. 2009; Schaefer and Lyko 2010b). Although we find that methylation can be present on a substantial proportion of alleles within methylated regions, methylation is virtually never present on more than half of the alleles. Thus, although methylated regions make up ∼1% of the Drosophila genome, the proportion of methylated cytosines at stage 5 is much less than this, consistent with previous estimates derived from studies of bulk methylation (Gowher et al. 2000; Lyko et al. 2000a; Marhold et al. 2004).
The presence of cytosine methylation on the Drosophila genome has been controversial. Although traces of methylcytosine were found in previous studies (Gowher et al. 2000; Lyko et al. 2000a; Marhold et al. 2004), no pattern has emerged from the data and many have concluded that any methylation is spurious (Schaefer and Lyko 2010b) or artifactual (Raddatz et al. 2013). Recently, two groups have used whole genome bisulfite sequencing to conclude that Drosophila lacks genome methylation (Zemach et al. 2010; Raddatz et al. 2013). The key difference between these two studies and ours is the enrichment of methylcytosines by immunoprecipitation prior to bisulfite conversion and sequencing. This allowed us to identify methylated regions and confirm them with deep bisulfite resequencing; the sensitivity of this combination of methods is superior to whole genome bisulfite sequencing at the depths used in the two previous studies. Our data is nevertheless fully compatible with these prior reports. Our reanalysis shows that the Zemach et al. (2010) data have insufficient depth to detect the methylation, whereas the Raddatz et al. (2013) data, when their depth of coverage is sufficient, support the presence of cytosine methylation at the same locations identified by our study (Supplemental Table S3). Furthermore, Lyko and colleagues observed the same enrichment of methylation at CA and CT dinucleotides that we do (cf. motifs in Fig. 4; Lyko et al. 2000a) and the same lack of dependence of cytosine methylation on MT2 activity (Raddatz et al. 2013).
Our findings refute conclusions that “Drosophila melanogaster lack detectable DNA methylation patterns” and that “residual unconverted cytosine residues shared many attributes with bisulfite deamination artifacts” (Raddatz et al. 2013). Analysis of the MeDIP-Bseq data identifies a defined DNA methylation pattern, and methylated regions are confirmed by targeted bisulfite sequencing at great depth in multiple strains. Artifactual nonconversion in the bisulfite reaction is ruled out by (1) the extremely low nonconversion rate of control DNA (Supplemental Fig. S4); and (2) the absence of the methylated cytosines at many sites in unfertilized oocytes, so that nonconversion cannot be related to an inability of the bisulfite reagent to convert cytosines in some specific sequence contexts. As noted above and in Supplemental Table S3, the Raddatz data set contains evidence for methylation at the regions we validated with bisulfite resequencing. Although the depth of sequencing in that whole genome bisulfite sequencing data set was insufficient to unambiguously identify methylation present on a small proportion of DNA strands in a stage 5 embryo with ∼6000 nuclei, one can find evidence of methylation when one knows where to look for it. Our resort to enrichment by immunoprecipitation explains why we were able to identify a pattern, and bisulfite resequencing at great depth confirms the identification of methylated regions.
Our results make necessary a reevaluation of the role of cytosine methylation in Drosophila. It is present at stage 5 and in the unfertilized oocyte in specific locations in association with specific sequence motifs. It is dynamic: Methylation is removed from some sites and added to others between the oocyte and stage 5. The nondependence of this methylation on MT2 means that the phenotype of Mt2 mutants cannot be used to draw conclusions about the role of genomic methylation in Drosophila. A more definite judgment of the importance of genomic cytosine methylation will require flies lacking DNA methylation, but this must await identification of the novel methyltransferase activity, which either has no discernible sequence relationship to the known DNA methyltransferases (Goll and Bestor 2005) or is harbored in one of the unsequenced portions of the Drosophila genome.
Consistent with previous reports of cytosine methylation in Drosophila (Lyko et al. 2000a; Kunert et al. 2003), we find that methylation occurs in CA- and CT-rich contexts. However our results show that, in the relatively highly methylated regions we have identified, methylation is found at specific 4- to 5-base sequence motifs that lack guanine (Fig. 4); these motifs are found in ∼75% of methylated regions (Table 2). When highly methylated regions are excluded from the analysis, different, guanine-rich motifs are associated with the 56% of methylated reads that do not align within methylated regions (Fig. 4). This reconciles our findings with bulk studies indicating some methylation in a CG context (Lyko et al. 2000a), but such methylation does not occur in high local concentrations that allow reliable detection of patterns related to annotated features.
Although the data presented here establish the presence of genomic methylation in Drosophila, they are by no means comprehensive, and it is quite possible that more regions are methylated at other stages in development and/or in small subsets of nuclei at stage 5. The regions we have identified are those with the highest density of methylation and in which methylation is most common in the embryo as a whole. We speculate that methylation may affect many more sites in the Drosophila genome but in only a very small proportion of nuclei in the embryo; this methylation might occur in sequence contexts very different from the ones we have found. It is also possible that methylation is present at other stages of development, even in the adult, but again in cell types and tissues that make up only a very small proportion of the organism. Detection of this methylation is challenging and might require sequencing at extreme depths.
The pattern of methylation revealed by our analysis is unlike that seen to date in other species (Suzuki and Bird 2008; Feng et al. 2010; Zemach et al. 2010), further complicating its interpretation. The recent comparative survey of eukaryotic methylation revealed a general conservation of DNA methylation in gene bodies (Zemach et al. 2010). Our method precludes the quantitative evaluation of methylation used in the comparative survey, but analysis of the genomic distribution of methylated regions shows a modest enrichment of methylation in intronic and intergenic sequences, and a depletion at coding and promoter sequences (Supplemental Fig. S11). Neither the patterns described in the comparative studies, nor the one we detect in Drosophila, suggest an obvious function for genomic methylation. Another unusual aspect of the Drosophila methylation is that when methylated regions are compared in different strains, the precise location of methylation varies by several nucleotides within a region, suggesting that it is not tightly controlled.
We asked if methylation participates in the regulation of gene expression in Drosophila, as it does in vertebrates (Lienert et al. 2011; Stadler et al. 2011), and observed modestly but significantly lower levels of expression in genes with relatively high density of methylation. However the association between methylation and gene expression could be stronger than indicated by this analysis. The allelic heterogeneity we observe raises the possibility that specific patterns of methylation could characterize distinct subsets of cells in the embryo. If this were the case, methylation of a gene in a subset of cells might affect its expression only in those cells, but the apparent impact of methylation on expression would appear to be small because expression is determined using the entire embryo. The association of methylation with gene expression is also suggested by the observation that the X chromosome is more heavily methylated than all other chromosomes. In Drosophila, the X chromosome is subject to gene dosage compensation through the up-regulation of X-linked genes in males (Baker et al. 1994; Deng et al. 2011). Although it is tempting to speculate that the higher density of methylation on the X chromosome is involved in dosage compensation, it is not clear how methylation would contribute to the process.
We find that genes with relatively high methylation density are significantly enriched for functions associated with organ and anatomical structure development (Table 3), which taken together with the findings discussed in the preceding paragraph suggests that genes with a role in later stages of development may be suppressed by methylation at stage 5. Embryonic stage 5 in Drosophila coincides with the beginning of zygotic gene expression (Foe and Alberts 1983; Tadros and Lipshitz 2009). The relative abundance of cytosine methylation at this stage, its apparent effect on gene expression, and its association with genes that play a role in development, prompt us to speculate that methylation participates in gene regulation at the maternal-zygotic transition.
This study provides a clear answer to the question of whether genomic methylation is present in Drosophila, but it raises several new questions. Although cytosine methylation is present, the pattern is unique among species studied to date. We find a correlation between methylation and gene expression levels, but a large fraction of methylated regions are far from genes. Furthermore the mechanism by which methylation might regulate gene expression is obscure, and methylation is not clearly associated with epigenetic modifications mapped by modENCODE (Supplemental Fig. S12; The modENCODE Consortium 2010). Genomic methylation occurs in the absence of MT2, implying that Drosophila has a cryptic DNA methyltransferase, and that no conclusions regarding the role of genomic methylation can be drawn from Mt2 mutant flies. Because no mutant lacking the novel methyltransferase has been characterized, the role of genomic methylation in Drosophila is once again an open question.
Methods
Immunoprecipitation and bisulfite conversion of methylated DNA
Genomic DNA from synchronized stage 5 (2–3 h) embryos of Canton-S Drosophila melanogaster was a kind gift from Mark Biggin and Xiaoyong Li. Methylated DNA fragments, obtained by sonication of 2 μg of stage 5 DNA (fragment size range: 100–700 bp), were immunoprecipitated by overnight incubation at 4°C with 5 μg of monoclonal antibody against 5-methylcytidine (Eurogentec) following the manufacturer’s instructions. Antibody-DNA complexes were recovered with Dynabeads M-280 Sheep anti-Mouse IgG (Dynal Biotech), washed twice in IP buffer, once in high salt buffer, and twice in TE buffer. Washed complexes were incubated overnight with Proteinase K. Immunoprecipitated DNA was collected from the supernatant with the QIAquick PCR purification kit (Qiagen) and converted with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen) following the manufacturer’s instructions.
PCR amplification and sequencing library construction
DNA is single-stranded following bisulfite conversion. To make DNA double-stranded and generate enough material for constructing a sequencing library, we performed a random-primed PCR amplification. Bisulfite converted DNA was subjected to a round of linear amplification using an adapter combined with a random 9-nucleotide (nt) primer [GTTTCCCAGTCACGGTC(N)9], purified, and PCR-amplified using primers against the adapter sequences. The PCR reaction was purified with MinElute PCR Purification Kit (Qiagen) and digested with HpyCH4III (NEB) to remove the adapter sequences. The digest was resolved on a 2% agarose gel; the smear in the range of 100–500 bp was excised, purified with the Gel Extraction Kit (Qiagen), and used for library construction with the ChIP-Seq Sample Prep Kit (Illumina) according to the manufacturer’s instructions. The library was sequenced on an Illumina GAII to collect paired-end reads of 36 nt each.
Read calling and alignment
Illumina raw data were processed with Illumina’s Pipeline 1.3.2, skipping the first two cycles (corresponding to the portion of the HpyCH4III recognition site that was left after restriction digestion) to facilitate cluster identification. The first 9 nt of each read, corresponding to the C in the adapter following the HpyCH4III site and the first eight bases of the random primer, showed higher error rates than the remaining 25 nt and were trimmed prior to sequence alignment. Paired reads were aligned to the Drosophila reference genome assembly (version dm3, downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/dm3/bigZips/), using all the chromosomes except Uextra, with Novoalign v2.07.11 (http://Novocraft.com) run in bisulfite mode. Novoalign was run with the following options: -t 90 -h 120 -r A 3 -k. These options allow a set of paired reads to align with up to (approximately) three mismatches over the combined length of the two reads and report up to three alignments for reads generating more than one optimal alignment. The forward (CT) and reverse (GA) strands of DNA become noncomplementary following bisulfite conversion. Novoalign in bisulfite mode can align bisulfite converted sequences against a reference genome in a single pass that aligns against both CT and GA in silico indexed reference sequences.
Identification of methylated cytosines
Unconverted (methylated) cytosines will be read as “C” if the sequence is derived from the CT strand and as “G” if the sequence is derived from the GA strand. Conversely, a “C” from the GA strand and a “G” from the CT strand correspond to a guanine in the reference sequence and must be ignored in the analysis. The loss of complementarity following bisulfite conversion makes it possible to determine if a methylated cytosine is on the CT or the GA strand. This is important if methylated sequences are asymmetric (i.e., non-CG methylation). To determine if a sequence alignment is derived from the CT or GA strand, we implemented the following decision tree:
If an alignment has only CT or GA mismatches, it is assigned to the respective strand if the corresponding number of CT or GA mismatches is at least 2; otherwise, the read is considered ambiguous and further processed in Step 3.
If an alignment has both CT and GA mismatches, it is assigned to the strand that has at least a twofold excess of one type of mismatch; otherwise, the read is considered ambiguous and further processed in Step 3.
If an ambiguous alignment is primary and in a proper pair, the strand assignment of the mate pair is transferred to the ambiguous read; otherwise the ambiguous alignment is discarded.
Alignments that are primary, in a proper pair, and not containing an ambiguous strand assignment, are checked for consistent strand assignment (i.e., both mate pairs are assigned to the same strand). If the assignment is inconsistent, the pair is discarded.
We identified methylated cytosines on the CT and GA strands separately by counting how many alignments contained either a “C” or a “T” at C positions on the CT strand and how many alignments contained either a “G” or an “A” at G positions on the GA strand. From here onward, we refer to C positions on the CT strand and G positions on the GA strand as cytosines. We first selected cytosines with strong evidence of methylation, by retaining only those cytosines at which at least three alignments had an unconverted C and the ratio of C-containing alignments over the sum of C- and T-containing alignments was greater than 0.1. These criteria remove cytosines whose methylation is supported by only one or two reads or where the vast majority of the reads do not support the presence of methylation. These filtering steps remove cytosines with low levels of methylation, which are difficult to distinguish from noise created by conversion or sequencing errors.
Preparation of genomic DNA
Oregon-R wild type were a kind gift from Sarah Siegrist, and EP(2)GE 15695 and Mt299 were a kind gift from Frank Lyko. All stocks are propagated in Bloomington standard food and maintained at room temperature. DNA from stage 5 (2–3 h) embryos was purified using the Gentra Puregene Kit (Qiagen) following the manufacturer’s instructions. We also received stage 5 DNA from EP(2)GE 15695 and Mt2149 strains from Gunther Reuter. Unfertilized oocytes were prepared by crossing Oregon-R wild type females and tud1 bw sp/CyO l(2)DTS5131 males (Bloomington Drosophila Stock Center stock number 1786). tud1 bw sp/CyO l(2)DTS5131 stock was maintained as described above. Oocytes were collected and frozen prior to DNA extraction. About 200–300 μL frozen oocytes were homogenized with a plastic pestle and DNA purified using the Gentra Puregene Kit (Qiagen) following the manufacturer’s protocol with the addition of an extra centrifugation step to remove cell debris.
Bisulfite PCR sequencing
Genomic DNA from synchronized stage 5 (2–3 h) embryos of Oregon R D. melanogaster and equimolar of unmethylated lambda DNA (Promega) were bisulfite treated with the MethylEasy Xceed kit (Human Genetic Signatures). Five to 20 ng of treated DNA was used to amplify specific target regions in the Drosophila and lambda genomes with PCR primers designed to prime completely converted (i.e., unmethylated) DNA. The full list of primers is available in the Supplemental Material. The amplified products were gel purified, pooled in equimolar quantities, and fragmented with Fragmentase (NEB). An Illumina sequencing library was constructed with paired-end adapters following the paired-end protocol. Sequence reads were aligned to a reference sequence consisting of the regions that were targeted for amplification using Novoalign v. 2.07.11 run in bisulfite mode with the -g200 option. After bisulfite conversion, PCR amplifies one of the two DNA strands (labeled “CT” and “GA”), the one against which the PCR primers are designed. Only reads aligning against the correct DNA strand were further processed. The mean median coverage was 99,372 reads (range 13,766–242,551). We used a modified pile-up program, which retains reads with multiple alignments, to determine the degree of methylation at all cytosines. Cytosine positions at which the total count of converted and unconverted cytosines was <90% where discarded as potentially polymorphic. We used the sequences from the lambda phage to assess the efficiency of bisulfite conversion.
Comparison of methylation density with exon density
The density distribution of methylated regions over 100-kb intervals was determined using fseq with the -l100000 -s100 options. The gene annotation for the D. melanogaster dm3 assembly was downloaded in BED format from the UCSC Table Browser table:flyBaseGene. A nonredundant exon file was obtained by collapsing all exons with overlapping coordinates; the file was converted to a density distribution using fseq with the -l100000 -s100 options. The intensities of the distributions of methylated regions and exons were percentile-normalized and compared by scatterplot using the “smoothscatter” package in R.
Identification of sequence motifs in methylated sequences
We used the DREME program (Bailey 2011) to identify sequence motifs enriched in methylated sequences. DREME was modified to search for motifs only on the input strand and not on its reverse complement. The background data set was generated by random permutation of the input data set with shuffleBED from the BEDTools (Quinlan and Hall 2010) suite, run with the -chrom option. Motifs were visualized using the seqLogo package in R. For each data set, only the strand on which methylation is found was used for the DREME analysis; we analyzed the following data sets:
Methylated fragments in methylated regions: the longest nonoverlapping sequences, derived from all methylated regions, which contain one or more methylated cytosines that are at a relative distance from each other not greater than 4 nt, the sequences between these cytosines, and the three bases 3′ and 5′ of the leftmost and rightmost cytosine. If a given cytosine did not have any other methylated cytosine within 4 nucleotides, the fragment contained only the methylated cytosine and the 3 nt to its left and right. Methylated cytosines were identified as described above. These fragments identify the parts of a methylated region with high density of methylation.
Methylated regions: the sequence of the methylated strand of an entire methylated region.
Methylated regions minus simple repeats: same as above but excluding those regions whose sequence overlaps with a simple repeat sequence by >50%.
Methylated reads in methylated regions: all the reads with at least one methylated (unconverted) cytosine and overlapping a methylated region by at least 51%.
Methylated reads not in methylated regions: all the reads with at least one methylated (unconverted) cytosine and not included in the group above.
Comparison with gene expression data
Mapped RNA-seq reads from 2–4 h embryos (Graveley et al. 2011) were downloaded from modMine (DCCid: modENCODE_2885). Cufflinks was used with bias correction enabled (Trapnell et al. 2010; Roberts et al. 2011) to calculate the abundances of all transcripts in the FlyBase r5.22 annotation; we measured transcript abundance as fragments per kilobase of exons per million fragments mapped (FPKM). We defined a methylated gene set to include all genes with methylation density equal to or greater than 0.5 methylated regions per kb of gene body, and an unmethylated gene set to include all genes that had no methylated regions in their gene body; gene body is defined as the gene length plus 1 kb at the 3′ and 5′ ends. We compared the FPKMs of the methylated and unmethylated gene sets using a two-sided Wilcoxon rank sum test with continuity correction.
GO term analysis
We compared genes with a high density of methylation (methylated region/kb of gene ≥0.5, including 1 kb 5′ and 3′ of the annotated gene) against genes with no methylation using GOrilla (Eden et al. 2009) (http://cbl-gorilla.cs.technion.ac.il/). GOrilla does not automatically correct for multiple testing; the significance threshold of P < 0.01 was adjusted to P < 2.6 × 10−6 with a Bonferroni correction for testing 3841 null hypotheses.
Data access
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE34425. Bisulfite sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession nos. SRR1191286, SRR1191445, SRR1191486, SRR1191487, SRR1191684, and SRR1191728.
Acknowledgments
We thank Mark Biggin and Xiaoyong Li for the Canton-S Drosophila melanogaster DNA; Gunther Reuter and Frank Lyko for the Mt2 mutant strains and DNA; Sarah Siegrist for fly stocks, husbandry and other resources, and technical help and discussions. This work was supported by the NIH grants HL084474 (D.B.), ES016581 (D.I.K.M.), CA115768 (D.I.K.M.). J.D. was a Scholar of the California Institute of Regenerative Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: D.I.K.M. conceived the study; J.D. designed and performed the MeDIP-bisulfite deep sequencing experiments; S.T. performed the fly husbandry, obtained unfertilized oocytes, and carried out bisulfite PCR experiments; S.J.H. constructed the bisulfite-PCR sequencing libraries; S.T., G.M., and D.B. designed and performed bioinformatic analyses; A.R. and L.P. carried out the association between methylation and gene expression; D.I.K.M. and D.B. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162412.113.
References
- Bailey TL 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BS, Gorman M, Marin I 1994. Dosage compensation in Drosophila. Annu Rev Genet 28: 491–521 [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP 1999. Methylation-induced repression–belts, braces, and chromatin. Cell 99: 451–454 [DOI] [PubMed] [Google Scholar]
- Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW, Schlesinger F, Davis CA, Reinke VJ, Gingeras TR, et al. 2011. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet 43: 1179–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durdevic Z, Hanna K, Gold B, Pollex T, Cherry S, Lyko F, Schaefer M 2013. Efficient RNA virus control in Drosophila requires the RNA methyltransferase Dnmt2. EMBO Rep 14: 269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. 2010. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci 107: 8689–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe VE, Alberts BM 1983. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci 61: 31–70 [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH 2005. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481–514 [DOI] [PubMed] [Google Scholar]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH 2006. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–398 [DOI] [PubMed] [Google Scholar]
- Gowher H, Leismann O, Jeltsch A 2000. DNA of Drosophila melanogaster contains 5-methylcytosine. EMBO J 19: 6918–6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung M-S, Karthikeyan N, Huang B, Koo H-C, Kiger J, Shen C-KJ 1999. Drosophila proteins related to vertebrate DNA (5-cytosine) methyltransferases. Proc Natl Acad Sci 96: 11940–11945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowski TP, Meusburger M, Phalke S, Helm M, Nellen W, Reuter G, Jeltsch A 2008. Human DNMT2 methylates tRNAAsp molecules using a DNA methyltransferase-like catalytic mechanism. RNA 14: 1663–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert N, Marhold J, Stanke J, Stach D, Lyko F 2003. A Dnmt2-like protein mediates DNA methylation in Drosophila. Development 130: 5083–5090 [DOI] [PubMed] [Google Scholar]
- Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 43: 1091–1097 [DOI] [PubMed] [Google Scholar]
- Lyko F, Ramsahoye BH, Jaenisch R 2000a. DNA methylation in Drosophila melanogaster. Nature 408: 538–540 [DOI] [PubMed] [Google Scholar]
- Lyko F, Whittaker AJ, Orr-Weaver TL, Jaenisch R 2000b. The putative Drosophila methyltransferase gene dDnmt2 is contained in a transposon-like element and is expressed specifically in ovaries. Mech Dev 95: 215–217 [DOI] [PubMed] [Google Scholar]
- Marhold J, Rothe N, Pauli A, Mund C, Kuehle K, Brueckner B, Lyko F 2004. Conservation of DNA methylation in dipteran insects. Insect Mol Biol 13: 117–123 [DOI] [PubMed] [Google Scholar]
- The modENCODE Consortium. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalke S, Nickel O, Walluscheck D, Hortig F, Onorati MC, Reuter G 2009. Retrotransposon silencing and telomere integrity in somatic cells of Drosophila depends on the cytosine-5 methyltransferase DNMT2. Nat Genet 41: 696–702 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz G, Guzzardo PM, Olova N, Fantappie MR, Rampp M, Schaefer M, Reik W, Hannon GJ, Lyko F 2013. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc Natl Acad Sci 110: 8627–8631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn J, Pachter L 2011. Improving RNA-seq expression estimates by correcting for fragment bias. Genome Biol 12: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Lyko F 2010a. Lack of evidence for DNA methylation of Invader4 retroelements in Drosophila and implications for Dnmt2-mediated epigenetic regulation. Nat Genet 42: 920–921 [DOI] [PubMed] [Google Scholar]
- Schaefer M, Lyko F 2010b. Solving the Dnmt2 enigma. Chromosoma 119: 35–40 [DOI] [PubMed] [Google Scholar]
- Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F 2010. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev 24: 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495 [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476 [DOI] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD 2009. The maternal-to-zygotic transition: a play in two acts. Development 136: 3033–3042 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919 [DOI] [PubMed] [Google Scholar]