

Abstract
Differentially expressed, cell type-specific effector gene sets hold the key to multiple important problems in biology, from theoretical aspects of developmental gene regulatory networks (GRNs) to various practical applications. Although individual cell types of interest have been recovered by various methods and analyzed, systematic recovery of multiple cell type-specific gene sets from whole developing organisms has remained problematic. Here we describe a general methodology using the sea urchin embryo, a material of choice because of the large-scale GRNs already solved for this model system. This method utilizes the regulatory states expressed by given cells of the embryo to define cell type and includes a fluorescence activated cell sorting (FACS) procedure that results in no perturbation of transcript representation. We have extensively validated the method by spatial and qualitative analyses of the transcriptome expressed in isolated embryonic skeletogenic cells and as a consequence, generated a prototypical cell type-specific transcriptome database.
Developmental gene regulatory network (GRN) analysis has focused mainly on the revelation of genomically encoded upstream control circuitry by which embryogenesis and organ formation processes are spatially encoded. Ultimately, in each region of each body part, downstream batteries of morphogenetic and differentiation effector genes are activated. However, in few cases has it thus far been possible, on a global scale, to link experimentally validated upstream developmental GRNs with whole sets of effector genes that are differentially expressed in given cell types. The functional properties of every cell type of course depend directly on their particular effector gene sets. However, with the exception of a small number of well-known differentiation gene batteries, we are in fact mainly ignorant of the genomically encoded circuitry by which cells control expression of the genes that produce their functional characteristics. For both practical and theoretical reasons, this is a project of major interest and importance, but it requires the solution of a general technological problem: how to isolate the multiple cell types from a given organism, required for genome-wide identification of cell type-specific effector gene sets.
The embryo of the sea urchin Strongylocentrotus purpuratus (Sp) provides a special advantage for this problem in that its upstream GRNs are already extensively understood (Peter and Davidson 2009; Peter et al. 2012). Because in its mode of development differentiation occurs precociously (Davidson 2006), the effector gene sets expressed by its various types of cells could in principle be linked directly to the solved upstream GRNs. We have therefore developed a method that enables the physical isolation of virtually any cell type (or embryonic territory), so that the difference in their effector gene expressions can eventually be compared on a global scale with the intent to distinguish specific effector gene sets. Cell identity is defined by regulatory state, i.e., the sum total of transcription factors expressed at effective levels in each nucleus. Thus, we exploit the control apparatus that governs the expression of key regulatory genes unique to the GRNs of the desired cell types in order to physically isolate these cell types for sequencing. The approach combines multiple existing technologies to accomplish this objective. In outline, the method described in this work consists of transgenic embryo generation; disaggregation of the developing embryos to individual cells in such a way that transcript populations are not disturbed; fluorescence activated cell sorting (FACS) to isolate cells of a given regulatory state; RNA-seq to comprehensively profile the transcriptome of the isolated cell type; and computational operations terminating in comparative transcriptome analysis.
Here we present the methodology we have worked out in detail, with the obvious understanding that it will be applicable to other experimental systems as well. We have focused on the experimental validation of every major step of the procedure by applying it to the differentiating skeletogenic cells of the sea urchin embryo, a cell type for which the genomically encoded mechanism of cell fate specification has been resolved in detail (Supplemental Fig. 1; Supplemental Note 1; Oliveri et al. 2008). This cell type has previously been isolated by other means and subjected to some transcript sequence analysis (Zhu et al. 2001; Livingston et al. 2006; Rafiq et al. 2012). These prior studies have enabled us to utilize results reported in the literature for the validation of our current methodology. In addition, by RNA in situ hybridization we corroborated a large number of otherwise unstudied transcripts that according to our analysis should be skeletogenic cell specific, and showed that indeed they are. A comprehensive, ontologically organized online database of skeletogenic cell effector genes is a valuable resource that emerged as a consequence of this study.
Results
General approach for the isolation of multiple cell types and regulatory state territories from the developing sea urchin embryo
Approximately 100 recombineered BAC constructs have been created in our laboratory, consisting of differentially expressed regulatory genes tagged with reporter sequences (http://www.spbase.org/SpBase/recomb_bac/recomb_bac_table.php). These encode fluorochromes such as GFP, and this extensive resource underlies our present approach. Thus, available recombineered BACs express accurately in essentially every cell type and developmental territory of the Sp embryo up to gastrulation and in many post-gastrular regulatory state domains as well. In principle, they collectively provide the opportunity to isolate cells of any given regulatory state by FACS. Flow cytometry has been commonly used to isolate particular cell types expressing fluorescent markers from different organisms over the last several decades. However, the thrust of this work has been to find a way to physically obtain not one desired cell type of interest, but rather any or all of the diverse regulatory states represented in the developing embryo at different stages. Given recombineered BACs are introduced into multiple Sp zygotes by microinjection (Fig. 1, Step 1). Transgenic embryos are cultured until they reach the desired developmental stage, at which point they are monitored for accurate reporter expression, then disaggregated en masse to individual cells (Fig. 1, Step 2; Supplemental Fig. 2) and sorted on the expressed BAC fluorescence (Fig. 1, Step 3). The mRNA of fluorescent cells is amplified, sequenced, quantified, and compared to that of nonfluorescent cells (Fig. 1, Step 4), thus providing cell type-specific effector gene sets. Omitting the recovery of nonfluorescent cells will nevertheless yield a territory-specific transcriptome but divest the ability to distinguish effector genes.
Figure 1.

Steps required for attaining cell type-specific transcriptomes. (1) Microinject a group of zygotes with an artificial chromosome genetically engineered to express a fluorescent reporter in the cell type of interest; (2) culture transgenic zygotes until they have reached the desired developmental stage, then disaggregate the embryos to individual cells; (3) run the cells through flow cytometry in order to segregate populations of cells by way of FACS; and (4) isolate, amplify, sequence, and quantify the pool of mRNA from each population in order to calculate relative enrichment for each gene. (AU) Arbitrary units; (BACs) bacterial artificial chromosomes.
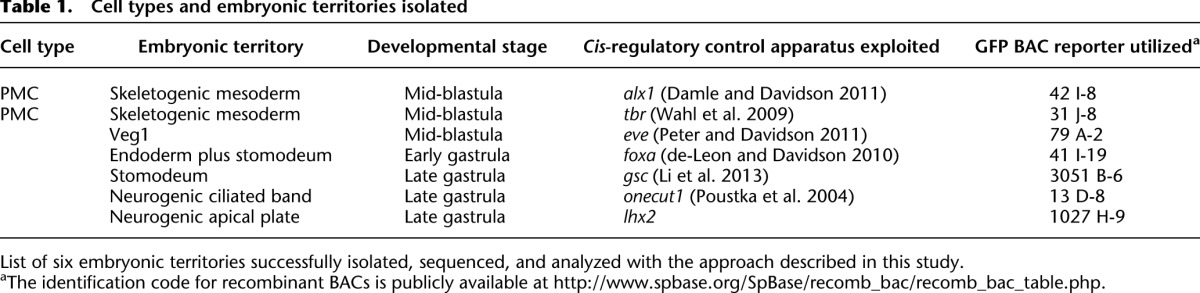
The general applicability of this method is illustrated in Table 1, which shows six different embryonic regulatory state domains that have thus been isolated and characterized from three different embryonic stages, on the basis of BAC expression controlled by a cis-regulatory apparatus specifically active in each. This report is focused on the newly designed methodological aspects that were required. With the exception of the skeletogenic primary mesenchyme cell (PMC) isolates that were here used for detailed validation of the procedures, comparative studies pertaining to effector gene sets from other cell types will be reported elsewhere.
Table 1.
Cell types and embryonic territories isolated
Isolation of cells does not induce change in gene expression
We first set out to determine whether normal gene expression was perturbed due to stress resulting from the disaggregation procedure per se. Using the nCounter Analysis System (Geiss et al. 2008), no perturbation was detectable. This is shown in an experiment in which we measured the expression levels of 181 regulatory genes in cells from disaggregated embryos and compared these to the levels of expression of the same genes in undisturbed whole embryos (Fig. 2A1). For none of the genes assessed was a change in gene expression observed to exceed the error of the measurements; the cells recovered at the end of the procedure do not differ in terms of gene expression profile from intact embryos, as judged by key regulatory genes expressed during this period. In order to evaluate the variance among biological replicates, we compared the expression levels for each of the 181 regulatory genes between different batches of disaggregated embryos spawned and fertilized on separate days (Fig. 2A2). As before, in no instance did the expression level of any gene deviate beyond error. These experimental controls validate the disaggregation and FACS procedures we have developed in order to achieve high-throughput extraction of individual cells from Sp embryos.
Figure 2.

Assemblage of control experiments. (A) nCounter analysis system measurements quantifying the expression level of 181 regulatory mRNAs used to assess the gene expression profile of disaggregated embryos. Red line represents a twofold change in gene expression from moderate abundance onward. (A1) Cells from disaggregated embryos collected by flow cytometry show no change in gene expression when compared to undisturbed whole embryos. (A2) Cells from disaggregated embryos collected by flow cytometry show no change in gene expression among biological replicates. (B) Fixed Sp blastula with PMCs labeled in purple. Labeling was achieved by whole mount RNA in situ hybridization of cah10l mRNA. (C) Live transgenic Sp blastula carrying an artificial chromosome that contains GFP under control of the cis-regulatory apparatus governing tbr expression. (C1) DIC image of transgenic blastula. (C2) Epifluorescent image of the same specimen shown in C1 reveals that GFP is exclusively expressed in PMCs. (C3) Composite image generated by merging C1 with C2. (D,E) Flow cytometry data in the form of 2% probability contour plots. Events falling outside of the lowest contour have been depicted as dots within the graph. A vertical red line demarcates the value beneath which an event likely reflects a technical artifact. (D) The exclusion of nonviable cells required that threshold parameters be calibrated for 7-AAD fluorescence. (D1) Data reflects the variation in size among the cells utilized for 7-AAD calibration. (D2) Untreated cells were used in determining baseline fluorescence, depicted as a horizontal red line. (D3) Following 7-AAD treatment, cells observed to fluoresce above baseline were excluded from further analysis. (E) Segregation of cell populations by FACS required that threshold parameters be calibrated for GFP fluorescence. (E1) Data reflects the variation in size among the cells utilized for GFP calibration. (E2) Cells from uninjected embryos were used in determining baseline fluorescence, depicted as a horizontal red line. (E3) Cells from transgenic embryos were then segregated by FACS, according to their fluorescence relative to baseline. (AU) Arbitrary units.
Cell type-specific skeletogenic transcriptome
Attaining skeletogenic cells from sea urchin blastula
At the blastula stage, cells of skeletogenic lineage undergo an epithelium to mesenchyme transition and ingress into the blastocoel, marking the first morphogenetic event of embryonic development (Wu et al. 2007). Following ingression, the PMCs divide for the last time as they position themselves within the embryo in response to signals from the ectoderm (Duloquin et al. 2007; Knapp et al. 2012). They then form syncytial cables within which the skeletal rods are secreted. Here we focus on PMCs from the Sp blastula stage (Fig. 2B), harvested immediately following ingression into the blastocoel. We have exploited the exclusively PMC-specific cis-regulatory control apparatus of two different regulatory genes, which encode the transcription factors Alx1 and Tbrain (Tbr). The recombineered BACs containing these regulatory systems precisely drive reporter expression in ingressed PMCs, as illustrated here for tbr (Fig. 2C1–3) and for alx1 (Supplemental Fig. 2A). Expression of these same BACs has been extensively characterized in prior cis-regulatory analyses (Wahl et al. 2009; Damle and Davidson 2011). The use of two different reporters to label the same cell type in independent analyses (Table 1) further validates concurrence across replicates, as we see below. We utilized a FACS gating strategy that ensured the exclusive recovery of individual viable cells. Particle-size exclusion removed any cells that had failed to adequately separate from one another as well as any cell fragments. The forward and side scatter parameters (Fig. 2D1) were empirically determined by visually monitoring the recovery of individual cells under the microscope (e.g., Supplemental Fig. 2B). Additionally, cells that incorporate 7-Aminoactinomycin D (7-AAD) were excluded on the basis that this fluorescent compound binds DNA but does not penetrate intact external cell membranes. The threshold for 7-AAD fluorescence was calibrated with reference to control cells never exposed to 7-AAD (Fig. 2D2). The 7-AAD treated cells that fluoresce above the established threshold represent apoptotic or otherwise nonviable cells and reflect the small percentage of cells adversely affected by embryo disaggregation (Fig. 2D3, 4% quadrant). Ultimately, the selected population was subjected to reporter-based cell sorting (Fig. 2D3, 96% quadrant). The threshold for GFP fluorescence was calibrated using cells that do not encode the GFP transgene (Fig. 2E1,2). Individual PMCs that fluoresce above this threshold were thus isolated for downstream analysis (Fig. 2E3, 3% quadrant), whereas those that do not represent all other embryonic cell types as well as PMCs that did not contain the exogenous construct. These unlabeled but undamaged cells served as an internal control (Fig. 2E3, 97% quadrant).
Recovery of labeled cells
Three replicate experiments are illustrated—the first using an alx1:GFP BAC (Fig. 3A) and the other two a tbr:GFP BAC (Fig. 3B,C). The fraction of individual cells acquired after embryo disaggregation varied, but not significantly (Fig. 3A1,B1,C1). Of the cells analyzed, viability never fell below 92% for any given replicate (Fig. 3A2,B2,C2). FACS was then used to recover a subset of PMCs on account of their GFP expression (Fig. 3A3,B3,C3, uppermost right quadrant); GFP negative cells were also collected separately (Fig. 3A3,B3,C3, lowermost right quadrant). In transgenic Sp embryos, the incorporation of exogenous DNA delivered to the zygote is mosaic. By the end of the blastula stage, there are 32 PMCs in the Sp embryo, of which typically two express GFP when very low concentrations of BAC DNA are introduced per zygote, as was done here to preclude imperfect development. This suggests that exogenous DNA was incorporated into a cell of skeletogenic lineage at seventh cleavage, when there are 16 such cells. On average, we recovered 5838 PMCs per experiment, which would imply a 25% recovery of labeled cells (for details, see Supplemental Note 2).
Figure 3.

Flow cytometry gating strategy across replicates. (A1–C3) Flow cytometry data in the form of 2% probability contour plots. In all instances, events falling outside the lowest contour have been depicted as dots within the graph. (A,B,C) Each series reflects data from one of the three replicates. (A) Replicate #1 obtained by using an alx1:GFP BAC. Replicate #2 (B) and Replicate #3 (C) obtained by using a tbr:GFP BAC. (A1,B1,C1) Each graph depicts all the events detected by flow cytometry. Events enclosed within the polygonal red line were visually corroborated to constitute individual cells, hence promoted to a second round of analysis. (A2,B2,C2) Each graph reflects the fraction of cells that have incorporated 7-AAD. Consequently, these were excluded from further study. A polygonal red line encloses the cell population promoted to a third and final round of analysis. (A3,B3,C3) Each graph reflects the fraction of viable cells segregated by GFP FACS. Data points shown above the horizontal red line represent PMCs, whereas those below represent a heterogeneous population containing all cell types. Percentage displayed in red at the corner of each quadrant. A vertical red line demarcates the value beneath which an event likely reflects a technical artifact. (AU) Arbitrary units.
RNA-seq analysis and validation of the procedure
mRNA was extracted from isolated PMCs and from the sorted control containing all embryonic cell types, then amplified and sequenced. The sequencing reads were mapped to the Sp genome (Tu et al. 2012). Levels of gene expression within PMC and control populations were estimated by quantifying the number of sequencing reads mapped per gene relative to the total number of sequencing reads from each population. Direct comparison between the resulting gene expression profiles identifies mRNA species that are differentially expressed in PMCs. For example, alx1 and tbr transcripts are clearly distinguished within the PMC population in each of the three replicate experiments (Supplemental Fig. 3A–C). For every gene, a quotient was computed between the levels of expression in PMCs relative to that observed for all cell types. Gene expression levels were averaged across the three replicates to generate a composite gene expression profile for isolated PMC and control cell populations (Supplemental Fig. 3D). Genes were statistically determined to be differentially expressed in PMCs by evaluating the quotient calculated for each gene across all three replicate experiments (significantly enriched transcripts are shown as colored data points that lie above the diagonal in Supplemental Figure 3D) (Anders and Huber 2010). We then used this analysis to test whether known PMC specific genes had indeed been recovered in the statistically identified gene sets in order to obtain a measure of validation of the whole procedure. The identity of all regulatory genes responsible for PMC specification is established; and indeed, all of these were recovered in the statistically enriched fraction (Fig. 4A). Similarly, the identity of many terminal effector genes expressed during PMC differentiation is known from earlier studies (Rafiq et al. 2012), including genes that encode for particular cell-surface proteins as well as others directly involved in skeletogenic biomineralization. These known effector genes were also differentially expressed in a statistically significant manner (Fig. 4B). These results encouraged the following more exacting challenges.
Figure 4.

Cell type-specific transcriptome. (A–D) Scatterplots compare transcriptome data unique to PMCs (GFP+) with that of all cell types (GFP−). Each data point reflects the relative mRNA abundance of a gene as estimated by the number of sequencing reads that map to its locus. The values in the graph reflect measurements taken across three independent replicates. Data points that fall above the diagonal indicate augmented levels of expression among PMCs, relative to other cell types. Genes statistically determined to be differentially expressed are shown as data points colored either red or blue to indicate an associated P-value of less than 0.01 or 0.05, respectively. Data points corresponding to genes of interest have been encircled in order to distinguish them from the rest of the data set. In select cases, a label revealing gene identity accompanies encircled data points. (A) The cohort of transcription factors enriched among PMCs is in accordance with, and expands upon, the known PMC regulatory state. (B) Genes directly involved in biomineralization are overrepresented in the catalog of PMC enriched transcripts, illuminating the skeletogenic fate these cells acquire. (C) Marked data points reflect the subset of PMC enriched transcripts for which spatial expression has been corroborated by RNA in situ hybridization. (D) Marked data points reflect genes reported in the literature to be enriched within PMCs.
Corroboration of highly enriched genes
We next asked whether significant transcript enrichment, according to these procedures, in fact constitutes a reliable guide to PMC-specific expression. Thus, 52 sequences were chosen at random from among the most highly enriched transcript subset, reflecting expression levels that range across three orders of magnitude, and RNA in situ hybridization was used to determine the spatial domain of expression of these genes within the embryo (Fig. 4C; Supplemental Fig. 4A1,2; Supplemental Table 1). For each embryo, microphotographs were taken at different focal planes in order to resolve individually labeled cells, and multiple embryos were observed for each transcript to assess possible variations in gene expression. Every one of the 52 genes was found to be expressed in PMCs. This result provides strong validation for the usefulness of these procedures. Figure 5A1–4 and Supplemental Figures 5–12 illustrate PMC-specific expression pertaining to the data points marked in Figure 5B and Supplemental Figure 4A2, respectively. Several genes, in addition to being expressed in PMCs, were also expressed from an alternate cell type (e.g., Supplemental Fig. 12J1,2; shown here at a later stage of development, where distinct domains of expression become more apparent). Finally, we mapped onto our data set genes reported to be PMC-specific according to earlier large-scale studies (Fig. 4D; Supplemental Table 2). As before, all were enriched, although a minority failed to cross the threshold of statistical significance, which is intrinsically conservative (cf. data points encircled in black to those encircled in red or blue within Fig. 4D). As a case in point, the collagen encoding transcript fcolf (SPU_013557) showed sixfold enrichment in our data set, but the calculated P-value of 0.07 fell just shy of statistical significance despite having been previously identified in PMCs during a genome-wide analysis of biomineralization related proteins (Livingston et al. 2006).
Figure 5.

Corroboration of cell type specificity. (A1–A4) Four of the genes tested for cell type-specific expression. Fixed Sp blastula with PMCs labeled in purple. Labeling was achieved by whole mount RNA in situ hybridization of cah10l (A1), msp130r1 (A2), p16 (A3), and 3apcol (A4). (B) Scatterplot identical to those described in Figure 4. Data points corresponding to the genes shown in A1–A4 have been encircled in order to distinguish them from other differentially expressed genes and labeled accordingly. Supplemental Table 1 lists all genes corroborated in a similar manner and the supporting data are shown in Supplemental Figures 4–12.
Skeletogenic cell-specific effector gene transcriptome
Since enrichment by these criteria constitutes a convincing indication of PMC-specific developmental gene expression, this analysis suffices for construction of a complete PMC effector gene transcriptome. Based on this, further analyses have been carried out as summarized graphically in Supplemental Figures 13–20. In each of these figures, panels (1) reveal the subset of PMC-specific transcripts that encode for proteins of a particular function and ontological category. Panels (2) reveal the fraction of all genes of that category in the Sp genome that are expressed differentially in PMCs. The category of uncharacterized genes is similarly treated in Supplemental Figure 21. The ontological PMC-specific effector gene database constitutes an online resource made available in its entirety at http://www.spbase.org:3838/cellspecific/; for specifics see Supplemental Note 3.
Discussion
The method we present here has been thoroughly validated by the data included in this report. Relying on a large body of previous experience that demonstrates the fidelity of recombineered BAC expression in sea urchin embryos, we were able to utilize the expression of BAC reporters to accurately identify and then isolate various cell types of the sea urchin embryo (Table 1). Figure 2A1 shows that the disaggregation procedure that we have arrived at does not perturb the population of transcripts present since the expression level of nearly 200 regulatory genes in disaggregated embryos cannot be distinguished from that of whole embryos in any statistically detectable manner. We then showed in four different ways, using isolated skeletogenic cells as a test case for validation, that the genes our analysis indicated to be expressed cell-specifically, in fact are. This gene catalog was obtained from cell populations sorted by virtue of expression from two different BAC knock-ins—one derived from the alx1 regulatory gene and one derived from the tbr regulatory gene (colored blue and red, respectively, in Supplemental Fig. 1), both of which are controlled by the double-negative gate activation circuit of the skeletogenic lineage (Oliveri et al. 2008). Strikingly, these two samples produced qualitatively identical enriched gene repertoires (Supplemental Fig. 3A–C). First, we showed that previously identified regulatory genes of the skeletogenic GRN are all found within the enriched gene set (Fig. 4A); note that most of these transcripts are in the rare sequence class, and yet they were satisfactorily recovered. Second, we observed in the statistically enriched category a complete set of biomineralization and cell-biology genes of known function that had been shown in earlier studies to be expressed exclusively in skeletogenic cells (Fig. 4B; Supplemental Figs. 14, 22, 23; Supplemental Note 4). Third, we showed that a large group of other skeletogenic effector genes that had been identified by sequence in isolated skeletogenic cells from another study are almost all recovered in our enriched category (Fig. 4D; Supplemental Table 2; Zhu et al. 2001; Rafiq et al. 2012). Fourth, we tested by RNA in situ hybridization a large set of otherwise uncharacterized genes randomly selected from our enriched gene list, since by this method expression in the skeletogenic cells is easily visualized due to the discrete and unique disposition of these cells within the embryo (Fig. 5; Supplemental Figs. 4–12). All the genes tested were demonstrated to be expressed in skeletogenic cells.
Table 1 presages the general power and usefulness of this methodological approach. We will now be able to define the specifically expressed effector genes of any desired cell type and of every desired ontological category in N-dimensional comparisons. Although comparison between the transcriptome of a given isolate and the whole embryo transcriptome, as in this paper, suffice to identify genes specifically expressed in the isolated cell type, they cannot exclude genes that are also expressed in another cell type so long as it is a minor component of the embryo overall. The ultimate definition of effector gene sets expressed exclusively in each cell type requires comparisons among many cell type isolates. As Table 1 demonstrates, such is now easily in our sights.
On the genome-wide scale that transcriptomes afford, most large data sets available of this sort derive from tissue culture cell lines, which are, to say the least, imperfect representatives of normal developmental states. Certain aspects of our approach have been exploited in C. elegans to isolate a large number of cell types, many neuronal, from the whole animal; and their expressed gene sets have been compared on tiling arrays (Spencer et al. 2011). A generally similar approach to that of the above has also been taken to study the regulatory control of the C. elegans intestine (Haenni et al. 2012). More often, comparable strategies have been used to isolate and study individual differentiating cell types of particular interest, e.g., myoblast subtypes in Drosophila (Estrada et al. 2006) and hematopoietic cells in zebrafish (Cannon et al. 2013). The new methodology described here is of particular significance in the current context of sea urchin embryo regulatory molecular biology. Thus, (1) regulatory states of almost all the territories of the whole embryo are characterized; (2) almost all regulatory state domains are represented in extant recombineered BACs; and (3) more than half the embryo up until gastrulation is already encompassed in a large-scale developmental GRN (Li et al. 2012, 2013, 2014; Ben-Tabou de-Leon et al. 2013; Materna et al. 2013). Thus, application of the method described in this paper promises to provide a first global index of differential use of effector genes throughout a developing organism. Furthermore, as specific effector gene sets are identified, it will lead directly to vertical network architecture that causally joins effector gene expression to the upstream GRNs.
In conclusion, the procedures described in this paper open the way to advances that will be very important for understanding the control of specialized cells and downstream developmental regulatory circuitry in general. Even in this first step, the outcome has been the generation of a uniquely validated, cell type-specific transcriptome database.
Methods
Network visualization
A directed graphical model generated with Biotapestry has been used to document the network of regulatory gene interactions discussed within this study. BioTapestry (http://www.biotapestry.org) is an open source software package purpose built for molecular genetic cartography (Longabaugh 2012).
Genetically engineered chromosomes
Bacterial artificial chromosomes (BACs) have been genetically engineered to express GFP in lieu of an endogenous gene. The first exon of which, is replaced with a GFP cassette by way of homologous recombination (Lee et al. 2001). Each BAC harbors the entire locus of a gene that is exclusively expressed in the cell type of interest. The conceptual basis for utilizing BACs as a means to label individual cell types is the assumption that they are large enough to harbor the complete cis-regulatory apparatus that governs the expression of a marker gene. This is the principle that confers spatial and temporal precision of expression to a BAC reporter. In this study, we take advantage of two BACs that have been reported to recapitulate endogenous gene expression (Wahl et al. 2009; Damle and Davidson 2011). A library containing hundreds of genetically engineered BAC reporters may be accessed at http://www.spbase.org/SpBase/recomb_bac/recomb_bac_table.php. All BAC reporters shown are publically available upon request.
Developmental model organism
Adult sea urchins were sourced locally off the coast of Southern California. They were kept at Caltech’s Kerckhoff Marine Laboratory prior to being transferred to Caltech’s main campus for experimentation purposes.
Transgenesis
Adult sea urchins were spawned for their gametes. The eggs were briefly treated in filtered seawater (FSW) containing citric acid (0.5 M concentration) and aligned on protamine-coated Petri dishes. FSW containing para-aminobenzoic acid (300 µg/mL) was used in order to facilitate injection. Eggs were fertilized in situ, and the resulting zygotes were injected (1 pL/zygote) with one of two BACs (50 ng of DNA per µL of nuclease-free water). Injection needles were fabricated in-house from borosilicate glass capillary tubing (1 mm outer diameter × 0.75 mm inner diameter × 100 mm long) using a flaming/brown P-80 (Sutler Instruments) micropipette puller. The consecutive micromanipulation of thousands of embryos was achieved on an Axiovert 40 C (Zeiss) compound microscope equipped with a single-axis oil hydraulic MM0-220 (Narishige) micromanipulator and a picospritzer III (Parker) microinjection dispense system. Transgenic embryos were cultured at 15°C in FSW containing trace amounts of Penicillin and Streptomycin.
Embryo disaggregation
Transgenic embryos were harvested at mid-blastula stage and disaggregated en masse as delineated in the steps shown below.
Multiple blastulas were transferred into 2 mL of FSW.
One milliliter of FSW containing Pronase (2 mg/mL) was added to the above, and the resultant mixture incubated on ice for 2 min.
The blastulas were then collected using a tabletop centrifuge (800 times gravity for 1 min), and the supernatant was discarded.
These were then resuspended in 2 mL of calcium-free seawater containing bovine serum albumin (20 mg/mL), followed by an additional 2 mL of hyaline extraction medium. The resultant mixture was left to incubate on ice for 4 min.
As before, the blastulas were collected using a tabletop centrifuge (800g for 1 min) and the supernatant discarded.
At this point, the blastulas were resuspended in 2 mL of calcium-free seawater and disaggregated by force using a P1000 micropipette to repeatedly pipette the mixture up and down.
The disaggregated cells were collected using a tabletop centrifuge (1400g for 20 sec, 1100g for 30 sec, and 800g for 40 sec) and 1.7 mL of the supernatant discarded.
The cells were then resuspended in the remaining 300 µL of supernatant.
Finally, these cells were filtered through a nylon mesh (40 µm) directly into a polystyrene sample tube and immediately subjected to flow cytometry.
All of the above steps were performed on ice within a cold room kept at a constant temperature of 14°C. The contents of the buffers mentioned may be found within Supplemental Note 5.
nCounter analysis system
Embryos were harvested at mid-blastula stage either as intact whole embryos, or disaggregated en masse as described above. All samples were processed as delineated in the steps shown below.
Three hundred embryos (or the equivalent in cells collected via flow cytometer) were put into an empty 1.5 mL sample tube.
The embryos (or cells) were centrifuged and the supernatant discarded.
Fifteen microliters of RLT Plus buffer (Qiagen) containing 2-Mercaptoethanol (1:100) were added per tube.
Samples were vortexed for 1 min, then centrifuged and cooled to −70°C.
Once thawed, 5 µL lysate were processed following the manufacturer’s instructions (http://www.nanostring.com/lifesciences/).
Detailed information concerning the Nanostring Probe Set utilized in this study, the manner in which Raw Code Counts were normalized and threshold parameters derived, are listed within Supplemental File 1.
Flow cytometry
A FACSAria Flow Cytometer Cell Sorter (BD Biosciences) was used to isolate individual cells immediately after embryo disaggregation. The only distinction from the standard operating protocol was the utilization of twice-filtered seawater (0.2 µm) in lieu of the regular sample diluent. This operational alternative is of biological importance when assessing live cells derived from marine model organisms.
RNA processing
Total RNA was extracted from each of the various cell populations isolated by FACS utilizing an RNeasy Plus Mini Kit (Qiagen). The only distinction from the manufacturer’s recommended protocol was a twofold increase in the DNase incubation time. Amplified cDNA was prepared from each sample of total RNA utilizing the Ovation RNA-seq System V2 (NuGEN).
Illumina sequencing
Amplified cDNA samples were purified, fragmented, and ligated into a library specifically designed for multiplexed paired-end sequencing. Quality control was monitored using the 2100 Bioanalyzer (Agilent) and a P330 Nano Photometer (Implen). Sequencing was performed on the HiSeq system (Illumina).
Mapping Illumina sequencing reads
Raw sequencing reads were mapped to the Sp genome using STAR (version 2.1) (Dobin et al. 2013). The number of reads mapped to each gene locus was counted using the HTSeq Python package (version 0.5.3), described in detail at http://www-huber.embl.de/users/anders/HTSeq/. The genomic coordinates used to identify each locus were in turn defined by our laboratory in a previous RNA-seq study (Tu et al. 2012).
Differential gene expression analysis
Gene counts (as defined by the number of sequencing reads that map to a gene’s locus) were normalized to the total count observed for each sample. A pseudocount was then added in order to be able to compute a ratio for genes in which no reads were observed in one of the two samples. Normalized gene counts were then used for differential gene expression analysis as calculated by DESeq (version 1.8.3) (Anders and Huber 2010). The dispersion was estimated using fitted values, as suggested by the authors for instances in which the number of replicates is not large. Lastly, the P-values have not been adjusted for multiple comparison testing.
Data visualization
The results were visualized predominantly in the form of scatterplots made possible by R software (R Development Core Team 2013) (version 3.0.1; http://www.r-project.org) and the ggplot2 package (Wickham 2009) (version 0.9.3; http://ggplot2.org). The online resource was generated using Shiny software (RStudio Inc 2013) (server version v1.0.0.42, R package version 0.8.0; http://www.rstudio.com/shiny/).
RNA in situ hybridization
Whole mount RNA in situ hybridization was performed on Sp blastula following a published method optimized by our laboratory (Ransick 2004). The sequence of the primers used and that of the mRNA targeted are listed within Supplemental File 2.
Microscopy
Live transgenic embryos were monitored for accurate reporter expression prior to disaggregation, using an Axioskop 2 plus (Zeiss) compound microscope equipped for fluorescence and differential interference contrast microscopy. Fixed embryos were imaged on an alternate Axioskop (Zeiss) compound microscope optimized for capturing color microphotographs. All digital images were taken using an Axiocam MRm (Zeiss) camera. Embryos shown were visualized through a 20× objective lens, whereas individual cells were imaged using a 40× objective lens.
Gene expression profile dynamics
Gene expression level time-course data pertaining to PMC enriched biomineralization and calcium toolkit cohorts were obtained during a developmental transcriptome study carried out by our laboratory (Tu et al. 2014). Detailed time-course quantification for all of the genes cataloged in this study can be found at http://www.spbase.org:3838/quantdev/.
Data access
All raw data supporting the findings communicated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRA105057. Processed data and the corresponding query/visualization tools are available via SpBase, the public sea urchin genome database (http://www.spbase.org:3838/cellspecific/).
Acknowledgments
Research was supported by NIH grant HD067454 to E.H.D. We thank Dina Malounda and Erika Vielmas for exceptional technical assistance in acquiring RNA in situ hybridization data sets; Diana Perez and Pat Koen for their effort to adapt the flow cytometer toward filtered seawater; Igor Antoshechkin for general guidance pertaining to Illumina sequencing; Michael Collins and Sagar Damle for critical reading of the manuscript; Jongmin Nam for practical discussions concerning the procedure described herein; Klara Stefflova for standardizing vector graphics across figures; and finally, above all, Rochelle Diamond for her expertise in all FACS-related matters.
Author contributions: J.C.B. and E.H.D. designed the research; J.C.B. performed the research; J.C.B. and Q.T. analyzed the data; and J.C.B. and E.H.D. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.167668.113.
References
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tabou de-Leon S, Su Y-H, Lin K-T, Li E, Davidson EH 2013. Gene regulatory control in the sea urchin aboral ectoderm: spatial initiation, signaling inputs, and cell fate lockdown. Dev Biol 374: 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JE, Place ES, Eve AMJ, Bradshaw CR, Sesay A, Morrell NW, Smith JC 2013. Global analysis of the haematopoietic and endothelial transcriptome during zebrafish development. Mech Dev 130: 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damle S, Davidson EH 2011. Precise cis-regulatory control of spatial and temporal expression of the alx-1 gene in the skeletogenic lineage of s. purpuratus. Dev Biol 357: 505–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH. 2006. The regulatory genome. Academic, Burlington, MA. [Google Scholar]
- de-Leon SB, Davidson EH 2010. Information processing at the foxa node of the sea urchin endomesoderm specification network. Proc Natl Acad Sci 107: 10103–10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duloquin L, Lhomond G, Gache C 2007. Localized VEGF signaling from ectoderm to mesenchyme cells controls morphogenesis of the sea urchin embryo skeleton. Development 134: 2293–2302 [DOI] [PubMed] [Google Scholar]
- Estrada B, Choe SE, Gisselbrecht SS, Michaud S, Raj L, Busser BW, Halfon MS, Church GM, Michelson AM 2006. An integrated strategy for analyzing the unique developmental programs of different myoblast subtypes. PLoS Genet 2: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. 2008. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26: 317–325 [DOI] [PubMed] [Google Scholar]
- Haenni S, Ji Z, Hoque M, Rust N, Sharpe H, Eberhard R, Browne C, Hengartner MO, Mellor J, Tian B, et al. 2012. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res 40: 6304–6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp RT, Wu C-H, Mobilia KC, Joester D 2012. Recombinant sea urchin vascular endothelial growth factor directs single-crystal growth and branching in vitro. J Am Chem Soc 134: 17908–17911 [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73: 56–65 [DOI] [PubMed] [Google Scholar]
- Li E, Materna SC, Davidson EH 2012. Direct and indirect control of oral ectoderm regulatory gene expression by Nodal signaling in the sea urchin embryo. Dev Biol 369: 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Materna SC, Davidson EH 2013. New regulatory circuit controlling spatial and temporal gene expression in the sea urchin embryo oral ectoderm GRN. Dev Biol 382: 268–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Cui M, Peter IS, Davidson EH 2014. Encoding regulatory state boundaries in the pregastrular oral ectoderm of the sea urchin embryo. Proc Natl Acad Sci 111: E906–E913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston BT, Killian CE, Wilt F, Cameron A, Landrum MJ, Ermolaeva O, Sapojnikov V, Maglott DR, Buchanan AM, Ettensohn CA 2006. A genome-wide analysis of biomineralization-related proteins in the sea urchin Strongylocentrotus purpuratus. Dev Biol 300: 335–348 [DOI] [PubMed] [Google Scholar]
- Longabaugh WJR 2012. BioTapestry: a tool to visualize the dynamic properties of gene regulatory networks. Methods Mol Biol 786: 359–394 [DOI] [PubMed] [Google Scholar]
- Materna SC, Ransick A, Li E, Davidson EH 2013. Diversification of oral and aboral mesodermal regulatory states in pregastrular sea urchin embryos. Dev Biol 375: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveri P, Tu Q, Davidson EH 2008. Global regulatory logic for specification of an embryonic cell lineage. Proc Natl Acad Sci 105: 5955–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Davidson EH 2009. Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS Lett 583: 3948–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Davidson EH 2011. A gene regulatory network controlling the embryonic specification of endoderm. Nature 474: 635–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Faure E, Davidson EH 2012. Predictive computation of genomic logic processing functions in embryonic development. Proc Natl Acad Sci 109: 16434–16442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poustka AJ, Kühn A, Radosavljevic V, Wellenreuther R, Lehrach H, Panopoulou G 2004. On the origin of the chordate central nervous system: expression of onecut in the sea urchin embryo. Evol Dev 6: 227–236 [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2013. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- R Studio, Inc. 2013. shiny: Web Application Framework for R. http://www.rstudio.com/shiny/.
- Rafiq K, Cheers MS, Ettensohn CA 2012. The genomic regulatory control of skeletal morphogenesis in the sea urchin. Development 139: 579–590 [DOI] [PubMed] [Google Scholar]
- Ransick AJ 2004. Detection of mRNA by in situ hybridization and RT-PCR. Methods Cell Biol 74: 601–620 [DOI] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, et al. 2011. A spatial and temporal map of C. elegans gene expression. Genome Res 21: 325–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Q, Cameron RA, Worley KC, Gibbs RA, Davidson EH 2012. Gene structure in the sea urchin Strongylocentrotus purpuratus based on transcriptome analysis. Genome Res 22: 2079–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Q, Cameron RA, Davidson EH 2014. Quantitative developmental transcriptomes of the sea urchin Strongylocentrotus purpuratus. Dev Biol 385: 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl ME, Hahn J, Gora K, Davidson EH, Oliveri P 2009. The cis-regulatory system of the tbrain gene: alternative use of multiple modules to promote skeletogenic expression in the sea urchin embryo. Dev Biol 335: 428–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer, New York. [Google Scholar]
- Wu SY, Ferkowicz M, McClay DR 2007. Ingression of primary mesenchyme cells of the sea urchin embryo: a precisely timed epithelial mesenchymal transition. Birth Defects Res C Embryo Today 81: 241–252 [DOI] [PubMed] [Google Scholar]
- Zhu X, Mahairas G, Illies M, Cameron RA, Davidson EH, Ettensohn CA 2001. A large-scale analysis of mRNAs expressed by primary mesenchyme cells of the sea urchin embryo. Development 128: 2615–2627 [DOI] [PubMed] [Google Scholar]