

Abstract
Purpose of review
This review discusses the current impact of pulmonary hypertension (PH) in the outcome and treatment of cystic fibrosis (CF).
Recent findings
Pulmonary hypertension is commonly encountered in advanced lung diseases such as CF. The prevalence of PH in CF patients varies based on disease severity and methodology used for diagnosis. Chronic alveolar hypoxia is the most likely etiology. The majority of recent studies have shown worse survival in CF patients who develop PH. The impact of PH-specific therapies on symptomatology and outcomes in CF patients has not been well studied.
Summary
Pulmonary hypertension is common in patients with CF and it occurs largely because of hypoxemia. The presence of PH in patients with CF is likely associated with worse outcome; however it remains unknown whether treatment with PH-specific therapies would be beneficial.
Keywords: Pulmonary hypertension, Cystic Fibrosis, Outcome Assessment, Treatment
Introduction
Pulmonary hypertension (PH) is a condition frequently found in patients with advanced parenchymal lung diseases of different etiologies [1, 2] and it is defined by a resting mean pulmonary artery pressure (PAP) of ≥ 25 mm Hg [3]. Cystic fibrosis (CF) is an autosomal recessive disease that typically affects the lungs eventually leading to progressive respiratory failure, pulmonary hypertension and right ventricular dysfunction [4, 5] (Figure 1). Indeed, respiratory failure continues to be the most common cause of morbidity and mortality in the disease [6].
Figure 1. Lung tissue of patients with CF and PH, stained with Hematoxylin and Eosin.
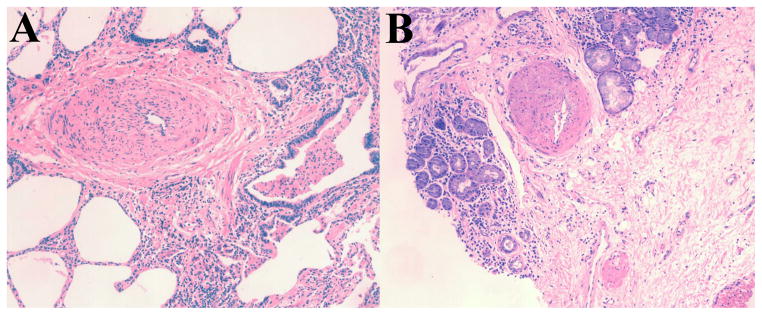
Tissue obtained from explanted lung at the time of transplantation. Note the intimal and medial hypertrophy with significant reduction of the arterial lumen. Patient A and patient B had mean PAP of 32 mm Hg and 30 mm Hg, respectively.
Cystic fibrosis associated PH has been incorporated in the group III, namely “PH due to lung diseases and/or hypoxia” of the latest clinical classification of PH (Dana Point, 2008) [7]. In particular, CF is part of a new subgroup called “Other pulmonary diseases with mixed restrictive and obstructive pattern” (Table 1). This subgroup also includes patients with chronic bronchiectasis and a syndrome characterized by the combination of pulmonary fibrosis and emphysema.
Table 1.
Updated Clinical Classification of Pulmonary Hypertension, Dana Point 2008
|
Adapted from reference [7]
The prevalence of PH in CF patients, its impact on outcome and potential therapeutic options has been infrequently studied; however in the last few years this association has gained notoriety. This article reviews the literature on PH in CF and summarizes the current evidence on prognosis and treatment.
Prevalence of PH in CF
Pulmonary hypertension is increasingly common in CF patients as greater numbers of individuals survive to adulthood. Right heart catheterization (RHC) continues to be the gold standard for the diagnosing of PH given that echocardiography is inaccurate, particularly in patients with advanced lung disease [1, 2, 8].
Table 2 shows the published studies in CF patients that reported the prevalence of PH by RHC [5, 9–16]. From these studies the prevalence of PH varies depending on the age of the patients (children and/or adults), severity of lung disease (variable or only severe), the methodology employed (echocardiography or RHC) and the criteria used to define patients (mean PAP ≥ 25 mm Hg or mean PAP ≥ 25 mm Hg with PAOP ≤ 15 mm Hg). Some studies included and other excluded patients with elevated pulmonary artery occlusion pressure (PAOP) (≥ 15 mmHg) since this finding suggests left ventricular diastolic dysfunction. This consideration is important because left ventricular diastolic function is frequently observed in CF patients with advanced disease [17].
Table 2.
Prevalence of PH by RHC in CF patients
| Author, Year | n | Age | Gender (%male) | Severity of lung disease | FEV1 (% of predicted) median (IQR) or mean ±SD |
PAP mmHg median (IQR) or mean ±SD |
Percentage of patients with PH |
|---|---|---|---|---|---|---|---|
| Goldring, 1964 [9] | 21 | Children and adults | 81 | Variable | N/A | 17 (13–28) |
|
| Siassi, 1971 [10] | 34 | Children | 26 | Variable | N/A | 21 (18–24) |
|
| Stern, 1980 [11] | 30 | Children and adults | 37 | Severe (all had corpulmonale) | Range (17–74) | 51.5 (41–60) |
|
| Burghuber, 1988 [12] | 16 | Adolescent and adults | 56 | Variable | 60 ± 8 | 20.8 ± 2 |
|
| Vizza, 2000 [13] | 146 | Adults | 55 | Severe |
Transplanted/Alive-waiting 22 ± 6 Died-waiting 22 ± 5 |
Transplanted/Alive-waiting 26 ± 5 Died-waiting 28 ± 7 |
N/A |
| Venuta, 2000 [14] | 45 | Children and adults | 33 | Severe |
Died-waiting 18 ± 4 Transplanted 21 ± 6 Alive-waiting 21± 4 |
Died-waiting 35 ± 12 Transplanted 22 ± 6 Alive-waiting 23 ± 6 |
Died-waiting
|
| Tonelli, 2010 [5] | 57 | Adults | 42 | Severe | 25 ± 8 | 26 (24–30) |
|
| Venuta, 2012 [15] | 179 | Children and adults | 54 | Severe | 23 ± 7 | 23 (19–28) |
|
| Belle-van Meerker, 2013 [16] | 93 | Adults | 51 | Severe |
No PH: 27 (22–31) PH: 22 (20–26) |
N/A |
|
Abbreviations: FEV1: forced expiratory volume in one second, IQR: interquartile range, mPAP: mean pulmonary artery pressure, N/A: not available, PH: pulmonary hypertension, SD: standard deviation.
The exercise criteria (increase in mean PAP to > 30 mmHg) was removed from the latest definition of PH due to marked differences in the upper limit of normal depending on the level of activity and the age of the patients [3]. This is relevant, since adult CF patients with advanced lung disease may have significant increases in the mean PAP with exercise, a response that is probably common in these patients and may impair their functional capacity [18].
Pathophysiology
The exact mechanism by which PH develops in patients with CF has not been fully elucidated. It is allegedly due to chronic alveolar hypoxia, progressive destruction of pulmonary vascular bed and/or persistent increase in cardiac output. Nevertheless, in advanced stages of the disease, other factors may play a role such as left ventricular dysfunction (systolic and/or diastolic), portal hypertension, hypothyroidism and/or pulmonary embolism. A summary of the mechanisms involved in the development of PH in CF patients is shown in Figure 2.
Figure 2. Proposed algorithm for the development of PH in CF patients.

Abbreviations: LV: left ventricle, PAH: pulmonary arterial hypertension, PAOP: pulmonary artery occlusion pressure, PVH: pulmonary venous hypertension.
Hypoxemia is a powerful pulmonary vasoconstrictor [9, 19–21] and occurs in CF patients as a result of lung destruction, ventilation/perfusion (V/Q) mismatch and right-to-left shunt [22, 23]. When the pulmonary artery smooth muscle and endothelial cells sense low oxygen levels, pulmonary vasoconstriction ensues with the purpose of equilibrating ventilation and perfusion [24]. However, chronic alveolar hypoxia leads to intimal proliferation, subintimal fibrosis and muscularization of pulmonary arterioles. This remodeling of the pulmonary arteries can increase the mean PAP causing PH and right heart failure [25].
Chronic airway inflammation could also lead to pulmonary vascular remodeling and PH [26, 27]. In fact, the mean PAP was significantly higher in CF patients infected with Burkholderia cepacia than patients that were not. Interestingly, all but one isolate (that could not be subculture) belonged to the genomovar II, named Burkholderia multivorans [28]. Other mechanisms may contribute to the development of PH such as air trapping with vessel compression [29] and/or endothelial dysfunction caused by vessel inflammation or shear stress [29, 30]. Indeed, vascular endothelial dysfunction was observed in a cohort of young CF patients with fairly normal oxygen saturation and pulmonary function [31].
Henno et al. [32] investigated mechanisms potentially involved in the development of PH in CF patients. They assessed the vascular endothelial function in pulmonary arterial rings isolated from lungs of CF patients that underwent transplantation. A subset of CF patients (61 %) had pulmonary vascular endothelial dysfunction and of those a high proportion had PH (71%). Conversely, in patients with normal endothelial function, none had PH. In addition, the authors noted that in patients with endothelial dysfunction, endothelin-1 (a strong vasoconstrictor involved in vascular remodeling) was higher and the blockage of its receptor produced a marked vasodilator effect. Similarly, in another study of CF patients, plasma endothelin levels were noted to be directly associated with disease severity [33].
Recent investigations have studied the function of the right ventricle (RV) in CF patients. Using modern echocardiographic techniques, two prospective studies demonstrated the presence of RV dysfunction in these patients who had no or mild pulmonary disease [34, 35]. The reasons for the abnormalities in the structure and function of the RV at early stages of the CF disease are not clear, since traditionally the RV dysfunction was thought to be secondary to raised PAP in the setting of chronic hypoxia [36]. Potential factors that may affect the RV in CF include chronic inflammation, direct involvement of the heart by the disease and increased levels of neurohormones [34, 35]. Interestingly, a few studies demonstrated that the cystic fibrosis transmembrane conductance regulator (CFTR) intervenes in the regulation of the cardiomyocyte contraction by maintaining a resting membrane potential and preventing an overload of intracellular calcium that can lead to hypercontraction [37]. In addition, the activation of myocardial CFTR protects against necrotic myocardial injury induced by ischemia/reperfusion [38]. Thus the reduced myocardial activity of CFTR in CF patients may help explain the RV dysfunction in early stages of disease and the development of disproportionate elevation of pulmonary artery pressures in some individuals. More importantly, further investigations on the role of cardiac CFTR may lead to novel therapies that target this pathway.
There is growing evidence supporting an association between CF and hypothyroidism [39, 40] or pulmonary embolism, two conditions that can participate in the pathogenesis of PH. CF-related thyroid disorders have been reported by some authors [39, 40] possibly due to iodine deficiency [40] or mutations of the CFTR protein that affects the transport of ions across the epithelia and secretion of thyroid hormone [41]. A linked between hypothyroidism and PAH was initially suggested 30 years ago [42] and subsequently supported by other studies [43–45]. The mechanistic origin of this association is not clear but an underlying autoimmune process may contribute to both disorders. The risk of venous thromboembolism is increased in CF patients due to the presence of central venous catheter, immobility and acquired thrombophilia secondary to inflammation, vitamin K deficiency and/or liver dysfunction [46]. Studies in non-CF patients presenting with acute pulmonary embolism showed that a small percentage (0.6 – 4%) of subjects developed chronic thromboembolic pulmonary hypertension due to persistent fibrotic obstruction and vascular remodeling of the pulmonary arteries [47].
Impact on outcome
Several studies have investigated whether the presence of PH carries prognostic significance in CF. Selimovic et al. retrospectively examined the prognostic value of cardiopulmonary hemodynamics in patients awaiting lung transplantation. The authors observed a direct association between higher pulmonary vascular resistance and mortality, along with a higher mean PAP and adverse outcomes [48]. Venuta et al. found a higher mean PAP (35 ± 12 mm Hg) in CF patients that died awaiting lung tranplantation (24 %) compared to subjects that received transplantation (22 ± 6 mm Hg) or were on the transplant list at the end of the study (23 ± 6 mm Hg, p=0.001) [14].
In a retrospective analysis of CF patients listed for lung transplantation, Vizza et al. reported that the mean PAP was not able to predict mortality. The mean PAP was 26 ± 5 mm Hg in patients that were alive at the end to the study versus 28 ± 7 mm Hg in those that died awaiting lung transplantation (p=0.15). However, a higher systolic PAP (in five mm Hg increments) was an independent risk factor for death after adjusting for confounders (hazard ratio: 1.41 (95% CI 1.11–1.8) [13]. Similarly, in CF patients evaluated for lung transplantation, Tonelli et al. found no difference in survival at 5 years in subjects with (40.8 %) or without PH (55.7 %, p=0.51); however the authors noted a suggestion that patients with mean PAP of ≥ 40 mm Hg had a trend towards worse survival than those with a mean PAP below that cut-off (1-year survival 50% versus 83%, p= 0.08) [5].
In the largest study to date, Venuta et al. demonstrated that the presence of PH increased the mortality in CF patients with advanced lung disease after adjusting the analysis by age, gender and forced expiratory volume in one second (FEV1). A receiver operating characteristic curve demonstrated that sensitivities and specificities, for predicting mortality at one year, varied depending on the mean PAP cut-offs. A cut-off of ≥ 25 mm Hg had a sensitivity and specificity of 70.2 % and 69.7 %; meanwhile a cut-off of ≥ 35 mm Hg had a sensitivity of 17.5% but a specificity of 97 % [15].
Belle-van Meerkerk et al. further investigated the impact of PH on the survival of CF patients evaluated for lung transplant. The authors found that the presence of PH in an unadjusted analysis did not affect their survival either before or after lung transplant; however, patients with PH had findings suggestive of more advanced CF disease as evidenced by the oxygen needs, spirometry and functional capacity [16].
It is possible that PH is a consequence of the advanced lung disease that develops in CF. As in COPD, the degree of PH in CF patients is usually mild; yet there is a group of patients that has higher pulmonary pressures for the degree of lung disease. In CF, a mean PAP ≥ 35 or 40 mm Hg could be considered a disproportionate elevation of PAP that may be associated with worse outcomes, as noted in patients with COPD [49, 50].
Echocardiography has limited accuracy in the diagnosis of PH, especially in the presence of parenchymal lung diseases [1]. However, a number of studies in CF patients have investigated the prognostic value of several echocardiographic parameters. Fraser et al. found that an elevated right ventricular systolic pressure (> 35 mm Hg) was associated with increased mortality in CF patients with severe lung disease (FEV1 < 40%) [36]. More recently, Bright-Thomas et al. found that adult CF patients, with different degrees of pulmonary involvement, had higher estimated right ventricular systolic pressures than age- and gender-matched healthy controls. The estimated right ventricular systolic pressure directly correlated with disease severity; however it did not predict survival when adjusted for FEV1 and arterial partial pressure of oxygen [51]. Manika et al. studied whether right ventricular systolic pressure has an impact on exercise capacity in adult CF patients with mild-to-moderate lung disease (FEV1 ≥ 40% of predicted). Within one minute postexercise, patients with CF (n=17) had higher estimated right ventricular systolic pressure than age- and height-matched healthy controls (n=10), and this echocardiographic measurement was associated with impaired exercise capacity [52].
Pulmonary flow acceleration time is the interval between the onset of ejection and the peak velocity of the pulmonary flow. This time is indirectly associated with mean PAP. Its main advantage is that it is measurable in most patients, unlike the right ventricular systolic pressure. Damy et al. prospectively studied the pulmonary flow acceleration time in patients with CF (FEV1 < 60%) and found that individuals in the lowest tertile had more advanced lung disease with a shorter time-to-lung transplantation than those in the other tertiles [53].
In PH, pulmonary arterial pressures are not good predictors of outcome because the pressures may decline when the RV fails and is no longer able to maintain an appropriate cardiac output. For this reason, the RV function may be a better predictor of outcomes. In fact, once CF patients develop right heart failure, the prognosis is usually poor. Stern et al. reported a median survival of four months after onset of cardiac dilation and fluid retention in CF patients [11]. Belkin et al. found that the presence of RV dysfunction was more common in CF patients that died (51%) compared to those awaiting lung transplantation (21%). On the other hand, the presence of PH was not different between survivors (21%) and non-survivors (29%) [54].
There is increasing recognition of the role of circulating biomarkers in PH. The brain natriuretic peptides (BNP) are secreted by the cardiac myocytes and in PH reflect the stress imposed on the right ventricle [55]. Both BNP and N-terminal-pro-BNP (NT-pro-BNP) have being associated with higher mortality in pulmonary arterial hypertension [56–58]. CF patients without heart failure had similar levels of NT-pro-BNP than the general population and more importantly this peptide was not associated with FEV1 or pulse oxygen saturation [59]. Consequently, a high level of NT-pro-BNP may suggest the presence of pulmonary hypertension or heart failure [59]; however further investigations are needed to confirm this hypothesis.
Treatment options for PH in CF patients
Although there are data supporting that PH is associated with worse outcomes in CF, no information is available to determine whether the treatment with PH-specific therapies is of benefit. Since PH may be a reflection of disease severity, its treatment might not necessarily improve outcomes. At this point, prevention of lung disease or its progression is the most important intervention. Therefore, adequate treatment of patients with CF is needed to delay the progression of the respiratory disease and consequently the development of PH [20].
In a small group of CF patients, oxygen supplementation demonstrated to decrease mean PAP and pulmonary vascular resistance [60, 61], as well as improve RV performance [62]. Goldring et al. performed RHC in CF patients (n=21) breathing different oxygen mixtures (fraction of inspired oxygen: 15% to 100%) and found that hypoxic mixtures increased and high-oxygen mixtures reduced the mean PAP [9]. However, no studies have assessed the hemodynamic effects of continuous long-term oxygen administration to CF patients with PH. Nocturnal supplemental oxygen over 3 years showed no effect on mortality, hospitalizations or progression of disease in a small study of CF patients [63]. The CF adult care consensus report expressed that one of the goals of oxygen therapy is the prevention of the development and/or progression of PH and recommends the screening of CF patients with moderate-to-severe respiratory disease for the presence of hypoxemia during exercise or sleep [64].
Diuretics have a place in the treatment of PH in the setting of hypervolemia and elevated PAOP. In advanced cases, care should be exercised to avoid a rapid reduction in the intravascular volume that may decrease the filling of the RV and hence impair cardiac output [20]. Digoxin does not seem to be beneficial in improving symptoms or survival in CF patients with right heart failure [11]. Anticoagulation is indicated in some patients with group I PH (pulmonary arterial hypertension) [65] but not in PH due to CF due to the lack of evidence to support its use and the possibility of increasing the risk of bleeding [66]. A small number of CF patients with PH have been treated with vasodilator agents such us nifedipine, diltiazem and hydralazine; nevertheless, no benefits were derived from these therapies, possible because these agents increased the flow to non-ventilated portions of the lung, accentuating right-to-left shunts and worsening hypoxemia [60, 61, 67].
PH-specific therapies have not been well studied in CF patients with PH. Phosphodiestearase-5 inhibitors like sildenafil may be useful in these patients; however this treatment continues to be controversial in PH due to parenchymal lung diseases such as COPD and interstitial lung disease [68–71]. Montgomery et al. showed that sildenafil improved exercise tolerance and reduced PAP in a patient with severe CF lung disease and PH [72]. Interestingly, phosphodiestearase-5 inhibitors are particularly attractive in CF patients with the most common disease allele (F508del), since they may promote the activity of the chloride channel (CFTR), the essential transepithelial ion transport abnormality in this condition [73].
Inhaled prostacyclin analogues may theoretically increase the pulmonary artery vascular flow in well-ventilated areas of the lung, potentially improving the match between ventilation and perfusion. Up to the time of this writing, there is only one case report showing that aerosolized iloprost lowered the pulmonary artery pressure, improved the functional status and facilitated lung transplantation in a patient with CF and PH [74]. Promising results have also been observed with the use of inhaled prostacyclin analogues in patients with PH due to other lung diseases [75–77]. Parenteral prostacyclin analogs are usually not given to patients with PH due to lung diseases as they may worsen the ventilation/perfusion match and oxygenation. Endothelin receptor antagonists such as bosentan and ambrisentan have not been tested in PH related to CF. Furthermore, in patients with COPD the utility of these medications is controversial [78] and in certain patients may worsen the functional status and oxygenation [79].
Lung transplantation is a valid treatment alternative for patients with a predicted survival shorter than the expected after transplantation, thus this intervention is indicated when the 2- to 3-year predicted survival from the lung disease is less than 50 % [80, 81]. Although lung transplantation showed no significant effect in the survival of CF children who were on the waiting list from 1992 through 2002 [82]; a recent study after the implementation of the lung allocation score (May 2005) showed that lung transplantation conferred a survival benefit in adult CF patients, directly related to the severity of disease [6]. In part this is due to better surgical techniques, organ preservation, critical care and post-transplant management [83]. At this point, lung transplantation remains the only option that improves the survival and quality of life of CF patients with end-stage lung disease [81]. In fact, after lung transplantation the median survival of CF patients is 7.5 years, better than other diagnosis like COPD or idiopathic pulmonary fibrosis [84].
The selection of the appropriate candidate for lung transplant is based on a comprehensive evaluation that incorporates several indicators of disease severity, including PH [80, 85]. In fact, the International Guidelines for the Selection of Lung Transplant Candidates includes PH as one of the factors to consider when deciding to perform lung transplant in CF patients [80]. The new allocation system of the United Network for Organ Sharing (UNOS) considers several determinants of disease severity in CF including systolic PAP [86]. Importantly, lung transplantation is not contraindicated in patients with PH and RV dysfunction as the RV function rapidly recovers after the intervention [81, 87].
Areas for further research
Areas of especial interest in CF that need further investigations include:
Study of the mechanisms for the development of PH in CF patients, with special emphasis in comprehending why some CF individuals have disproportionate PH in relation to the degree of lung disease.
Recognition of the prevalence, impact and prognostic implications of exercise-induced PH in CF.
Investigation of the origin of RV dysfunction in patients with mild CF.
Examination of the value of using PH-specific therapies to treat PH in CF patients.
Conclusion
Pulmonary hypertension is common in patients with CF and advanced lung disease. Pulmonary hypertension may be associated with worse outcome and its identification may provide an opportunity for treatment, although no clinical trials using PH-specific therapies have been carried out to date.
Key points.
Pulmonary hypertension (PH) is frequently found in patients with cystic fibrosis (CF) and end-stage lung disease.
The degree of PH is usually mild; however there are cases of disproportional elevation of pulmonary pressures.
PH likely has a negative impact on survival.
There is no evidence that the use of specific therapies for PH is of benefit in CF patients.
Prevention of disease progression, supportive therapy and lung transplantation are accepted therapeutic modalities.
Acknowledgments
Funding sources: A.R.T is supported by the CTSA KL2 Grant # RR024990, from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Dr Tonelli is funded by CTSA KL2 [Grant # RR024990] (A.R.T.) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. I would to thank Dr Peter Drew from the Department of Pathology, Immunology and Laboratory Medicine at University of Florida for providing the pathology images shown in this manuscript.
Abbreviations
- BNP
brain natriuretic peptide
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive lung disease
- FEV1
forced expiratory volume in 1 second
- NT-pro-BNP
N-terminal-pro-brain natriuretic peptide
- PH
pulmonary hypertension
- PAOP
pulmonary artery occlusion pressure
- PAP
pulmonary artery pressure
- RHC
right heart catheterization
- RV
right ventricle
Footnotes
Disclosures: The author has no significant conflicts of interest with any companies or organization whose products or services may be discussed in this article.
References
- 1.Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735–740. doi: 10.1164/rccm.200210-1130OC. [DOI] [PubMed] [Google Scholar]
- 2.Homma A, Anzueto A, Peters JI, et al. Pulmonary artery systolic pressures estimated by echocardiogram vs cardiac catheterization in patients awaiting lung transplantation. J Heart Lung Transplant. 2001;20:833–839. doi: 10.1016/s1053-2498(01)00274-1. [DOI] [PubMed] [Google Scholar]
- 3.Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–66. doi: 10.1016/j.jacc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Ionescu AA, Payne N, Obieta-Fresnedo I, et al. Subclinical right ventricular dysfunction in cystic fibrosis. A study using tissue Doppler echocardiography. Am J Respir Crit Care Med. 2001;163:1212–1218. doi: 10.1164/ajrccm.163.5.9908005. [DOI] [PubMed] [Google Scholar]
- 5.Tonelli AR, Fernandez-Bussy S, Lodhi S, et al. Prevalence of pulmonary hypertension in end-stage cystic fibrosis and correlation with survival. J Heart Lung Transplant. 2010;29:865–872. doi: 10.1016/j.healun.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 6**.Thabut G, Christie JD, Mal H, et al. Survival Benefit of Lung Transplant for Cystic Fibrosis since Lung-Allocation-Score Implementation. Am J Respir Crit Care Med. 2013 doi: 10.1164/rccm.201303-0429OC. Using modern statistical methods the authors demonstrated a survival benefit from lung transplantation in adult patients with CF disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:615–621. doi: 10.1164/rccm.200811-1691OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldring RM, Fishman AP, Turino GM, et al. Pulmonary Hypertension and Cor Pulmonale in Cystic Fibrosis of the Pancreas. J Pediatr. 1964;65:501–524. doi: 10.1016/s0022-3476(64)80286-9. [DOI] [PubMed] [Google Scholar]
- 10.Siassi B, Moss AJ, Dooley RR. Clinical recognition of cor pulmonale in cystic fibrosis. J Pediatr. 1971;78:794–805. doi: 10.1016/s0022-3476(71)80350-5. [DOI] [PubMed] [Google Scholar]
- 11.Stern RC, Borkat G, Hirschfeld SS, et al. Heart failure in cystic fibrosis. Treatment and prognosis of cor pulmonale with failure of the right side of the heart. Am J Dis Child. 1980;134:267–272. doi: 10.1001/archpedi.1980.02130150025007. [DOI] [PubMed] [Google Scholar]
- 12.Burghuber OC, Salzer-Muhar U, Bergmann H, Gotz M. Right ventricular performance and pulmonary haemodynamics in adolescent and adult patients with cystic fibrosis. Eur J Pediatr. 1988;148:187–192. doi: 10.1007/BF00441398. [DOI] [PubMed] [Google Scholar]
- 13.Vizza CD, Yusen RD, Lynch JP, et al. Outcome of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2000;162:819–825. doi: 10.1164/ajrccm.162.3.9910102. [DOI] [PubMed] [Google Scholar]
- 14.Venuta F, Rendina EA, Rocca GD, et al. Pulmonary hemodynamics contribute to indicate priority for lung transplantation in patients with cystic fibrosis. J Thorac Cardiovasc Surg. 2000;119:682–689. doi: 10.1016/S0022-5223(00)70002-X. [DOI] [PubMed] [Google Scholar]
- 15**.Venuta F, Tonelli AR, Anile M, et al. Pulmonary hypertension is associated with higher mortality in cystic fibrosis patients awaiting lung transplantation. J Cardiovasc Surg (Torino) 2012;53:817–820. The presence of PH is a predictor of mortality when adjusted for age, gender and FEV1. [PubMed] [Google Scholar]
- 16*.Belle-van Meerkerk G, Cramer MJ, Kwakkel-van Erp JM, et al. Pulmonary hypertension is a mild comorbidity in end-stage cystic fibrosis patients. J Heart Lung Transplant. 2013 doi: 10.1016/j.healun.2013.03.006. A quarter of adult CF patients with advanced lung disease had “precapillary PH”, a condition associated with lung disease severity. [DOI] [PubMed] [Google Scholar]
- 17.Koelling TM, Dec GW, Ginns LC, Semigran MJ. Left ventricular diastolic function in patients with advanced cystic fibrosis. Chest. 2003;123:1488–1494. doi: 10.1378/chest.123.5.1488. [DOI] [PubMed] [Google Scholar]
- 18.Hayes D, Kopp B, Mansour H, et al. Exercise-induced pulmonary arterial hypertension in pediatric and youg adult patietns with cystic fibrosis. Chest. 2012;124:773A. [Google Scholar]
- 19.Moss AJ. The cardiovascular system in cystic fibrosis. Pediatrics. 1982;70:728–741. [PubMed] [Google Scholar]
- 20.Eckles M, Anderson P. Cor pulmonale in cystic fibrosis. Semin Respir Crit Care Med. 2003;24:323–330. doi: 10.1055/s-2003-41093. [DOI] [PubMed] [Google Scholar]
- 21.Rovedder PM, Ziegler B, Pinotti AF, et al. Prevalence of pulmonary hypertension evaluated by Doppler echocardiography in a population of adolescent and adult patients with cystic fibrosis. J Bras Pneumol. 2008;34:83–90. doi: 10.1590/s1806-37132008000200004. [DOI] [PubMed] [Google Scholar]
- 22.Moss AJ, Desilets DT, Higashino SM, et al. Intrapulmonary shunts in cystic fibrosis. Pediatrics. 1968;41:438–445. [PubMed] [Google Scholar]
- 23.Rovedder PM, Ziegler B, Pasin LR, et al. Doppler echocardiogram, oxygen saturation and submaximum capacity of exercise in patients with cystic fibrosis. J Cyst Fibros. 2007;6:277–283. doi: 10.1016/j.jcf.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Evans AM, Hardie DG, Peers C, Mahmoud A. Hypoxic pulmonary vasoconstriction: mechanisms of oxygen-sensing. Curr Opin Anaesthesiol. 2011;24:13–20. doi: 10.1097/ACO.0b013e3283421201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryland D, Reid L. The pulmonary circulation in cystic fibrosis. Thorax. 1975;30:285–292. doi: 10.1136/thx.30.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat. 2002;201:335–348. doi: 10.1046/j.1469-7580.2002.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herget J, Palecek F, Preclik P, et al. Pulmonary hypertension induced by repeated pulmonary inflammation in the rat. J Appl Physiol. 1981;51:755–761. doi: 10.1152/jappl.1981.51.3.755. [DOI] [PubMed] [Google Scholar]
- 28.Fauroux B, Hart N, Belfar S, et al. Burkholderia cepacia is associated with pulmonary hypertension and increased mortality among cystic fibrosis patients. J Clin Microbiol. 2004;42:5537–5541. doi: 10.1128/JCM.42.12.5537-5541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. 2005;60:605–609. doi: 10.1136/thx.2005.042994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbera JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21:892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 31**.Poore S, Berry B, Eidson D, et al. Evidence of Vascular Endothelial Dysfunction in Young Patients with Cystic Fibrosis. Chest. 2012 doi: 10.1378/chest.12-1934. Systemic vascular endothelial dysfunction was observed in young and fairly healthy CF patients. [DOI] [PubMed] [Google Scholar]
- 32.Henno P, Maurey C, Danel C, et al. Pulmonary vascular dysfunction in end-stage cystic fibrosis: role of NF-kappaB and endothelin-1. Eur Respir J. 2009;34:1329–1337. doi: 10.1183/09031936.00186908. [DOI] [PubMed] [Google Scholar]
- 33.Siahanidou T, Nicolaidou P, Doudounakis S, et al. Plasma immunoreactive endothelin levels in children with cystic fibrosis. Acta Paediatr. 2000;89:915–920. doi: 10.1080/080352500750043332. [DOI] [PubMed] [Google Scholar]
- 34*.Ozcelik N, Shell R, Holtzlander M, Cua C. Decreased right ventricular function in healthy pediatric cystic fibrosis patients versus non-cystic fibrosis patients. Pediatr Cardiol. 2013;34:159–164. doi: 10.1007/s00246-012-0407-4. RV systolic and diastolic functions are reduced in children with CF and no lung disease. [DOI] [PubMed] [Google Scholar]
- 35*.Bano-Rodrigo A, Salcedo-Posadas A, Villa-Asensi JR, et al. Right ventricular dysfunction in adolescents with mild cystic fibrosis. J Cyst Fibros. 2012;11:274–280. doi: 10.1016/j.jcf.2012.03.002. Right ventricular abnormalities were observed in CF patients with mild disease. [DOI] [PubMed] [Google Scholar]
- 36.Fraser KL, Tullis DE, Sasson Z, et al. Pulmonary hypertension and cardiac function in adult cystic fibrosis: role of hypoxemia. Chest. 1999;115:1321–1328. doi: 10.1378/chest.115.5.1321. [DOI] [PubMed] [Google Scholar]
- 37.Sellers ZM, De Arcangelis V, Xiang Y, Best PM. Cardiomyocytes with disrupted CFTR function require CaMKII and Ca(2+)-activated Cl(−) channel activity to maintain contraction rate. J Physiol. 2010;588:2417–2429. doi: 10.1113/jphysiol.2010.188334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uramoto H, Okada T, Okada Y. Protective role of cardiac CFTR activation upon early reperfusion against myocardial infarction. Cell Physiol Biochem. 2012;30:1023–1038. doi: 10.1159/000341479. [DOI] [PubMed] [Google Scholar]
- 39.De Luca F, Trimarchi F, Sferlazzas C, et al. Thyroid function in children with cystic fibrosis. Eur J Pediatr. 1982;138:327–330. doi: 10.1007/BF00442510. [DOI] [PubMed] [Google Scholar]
- 40.Naehrlich L, Dorr HG, Bagheri-Behrouzi A, Rauh M. Iodine deficiency and subclinical hypothyroidism are common in cystic fibrosis patients. J Trace Elem Med Biol. 2013;27:122–125. doi: 10.1016/j.jtemb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Ganta S, Fong P. Altered ion transport by thyroid epithelia from CFTR(−/−) pigs suggests mechanisms for hypothyroidism in cystic fibrosis. Exp Physiol. 2010;95:1132–1144. doi: 10.1113/expphysiol.2010.054700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badesch DB, Wynne KM, Bonvallet S, et al. Hypothyroidism and primary pulmonary hypertension: an autoimmune pathogenetic link? Ann Intern Med. 1993;119:44–46. doi: 10.7326/0003-4819-119-1-199307010-00008. [DOI] [PubMed] [Google Scholar]
- 43.Curnock AL, Dweik RA, Higgins BH, et al. High prevalence of hypothyroidism in patients with primary pulmonary hypertension. Am J Med Sci. 1999;318:289–292. doi: 10.1097/00000441-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Arroliga AC, Dweik RA, Rafanan AL. Primary pulmonary hypertension and thyroid disease. Chest. 2000;118:1224–1225. doi: 10.1378/chest.118.4.1224. [DOI] [PubMed] [Google Scholar]
- 45.Ferris A, Jacobs T, Widlitz A, et al. Pulmonary arterial hypertension and thyroid disease. Chest. 2001;119:1980–1981. doi: 10.1378/chest.119.6.1980. [DOI] [PubMed] [Google Scholar]
- 46.Takemoto CM. Venous thromboembolism in cystic fibrosis. Pediatr Pulmonol. 2012;47:105–112. doi: 10.1002/ppul.21566. [DOI] [PubMed] [Google Scholar]
- 47.Kim NH, Lang IM. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2012;21:27–31. doi: 10.1183/09059180.00009111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selimovic N, Andersson B, Bergh CH, et al. Pulmonary hemodynamics as predictors of mortality in patients awaiting lung transplantation. Transpl Int. 2008;21:314–319. doi: 10.1111/j.1432-2277.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 49.Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:189–194. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 50.Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127:1531–1536. doi: 10.1378/chest.127.5.1531. [DOI] [PubMed] [Google Scholar]
- 51*.Bright-Thomas RJ, Ray SG, Webb AK. Pulmonary artery pressure in cystic fibrosis adults: characteristics, clinical correlates and long-term follow-up. J Cyst Fibros. 2012;11:532–538. doi: 10.1016/j.jcf.2012.04.012. Estimated PAP by echocardiography was associated with CF severity but not with survival when adjusted by FEV1 and PaO2. [DOI] [PubMed] [Google Scholar]
- 52*.Manika K, Pitsiou GG, Boutou AK, et al. The Impact of Pulmonary Arterial Pressure on Exercise Capacity in Mild-to-Moderate Cystic Fibrosis: A Case Control Study. Pulm Med. 2012;2012:252345. doi: 10.1155/2012/252345. Exercise induced elevation in estimated right ventricular systolic may be an important factor that could limit their exercise capacity in mild-to-moderated PH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53*.Damy T, Burgel PR, Pepin JL, et al. Pulmonary acceleration time to optimize the timing of lung transplant in cystic fibrosis. Pulm Circ. 2012;2:75–83. doi: 10.4103/2045-8932.94838. A short pulmonary acceleration time can identify patients with more advanced CF disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belkin RA, Henig NR, Singer LG, et al. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2006;173:659–666. doi: 10.1164/rccm.200410-1369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gan CT, McCann GP, Marcus JT, et al. NT-proBNP reflects right ventricular structure and function in pulmonary hypertension. Eur Respir J. 2006;28:1190–1194. doi: 10.1183/09031936.00016006. [DOI] [PubMed] [Google Scholar]
- 56.Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102:865–870. doi: 10.1161/01.cir.102.8.865. [DOI] [PubMed] [Google Scholar]
- 57.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 58.Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest. 2006;129:1313–1321. doi: 10.1378/chest.129.5.1313. [DOI] [PubMed] [Google Scholar]
- 59.Ben Tov A, Paret G, Sela BA, et al. N-terminal pro B-type natriuretic peptide (N-BNP) levels in cystic fibrosis patients. Pediatr Pulmonol. 2007;42:699–703. doi: 10.1002/ppul.20635. [DOI] [PubMed] [Google Scholar]
- 60.Geggel RL, Dozor AJ, Fyler DC, Reid LM. Effect of vasodilators at rest and during exercise in young adults with cystic fibrosis and chronic cor pulmonale. Am Rev Respir Dis. 1985;131:531–536. doi: 10.1164/arrd.1985.131.4.531. [DOI] [PubMed] [Google Scholar]
- 61.Davidson A, Bossuyt A, Dab I. Acute effects of oxygen, nifedipine, and diltiazem in patients with cystic fibrosis and mild pulmonary hypertension. Pediatr Pulmonol. 1989;6:53–59. doi: 10.1002/ppul.1950060113. [DOI] [PubMed] [Google Scholar]
- 62.Alpert BE, Gainey MA, Schidlow DV, Capitanio MA. Effect of oxygen on right ventricular performance evaluated by radionuclide angiography in two young patients with chronic lung disease. Pediatr Pulmonol. 1987;3:149–152. doi: 10.1002/ppul.1950030307. [DOI] [PubMed] [Google Scholar]
- 63.Adler FR, Aurora P, Barker DH, et al. Lung transplantation for cystic fibrosis. Proc Am Thorac Soc. 2009;6:619–633. doi: 10.1513/pats.2009008-088TL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yankaskas JR, Marshall BC, Sufian B, et al. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125:1S–39S. doi: 10.1378/chest.125.1_suppl.1s. [DOI] [PubMed] [Google Scholar]
- 65.Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J. 2006;28:999–1004. doi: 10.1183/09031936.06.00015206. [DOI] [PubMed] [Google Scholar]
- 66.Flume PA, Mogayzel PJ, Jr, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: pulmonary complications: hemoptysis and pneumothorax. Am J Respir Crit Care Med. 2010;182:298–306. doi: 10.1164/rccm.201002-0157OC. [DOI] [PubMed] [Google Scholar]
- 67.Moskowitz WB, Gewitz MH, Heyman S, et al. Cardiac involvement in cystic fibrosis: early noninvasive detection and vasodilator therapy. Pediatr Pharmacol (New York) 1985;5:139–148. [PubMed] [Google Scholar]
- 68.Blanco I, Santos S, Gea J, et al. Sildenafil to improve respiratory rehabilitation outcomes in COPD: a controlled trial. Eur Respir J. 2013 doi: 10.1183/09031936.00176312. [DOI] [PubMed] [Google Scholar]
- 69.Lederer DJ, Bartels MN, Schluger NW, et al. Sildenafil for chronic obstructive pulmonary disease: a randomized crossover trial. COPD. 2012;9:268–275. doi: 10.3109/15412555.2011.651180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blanco I, Gimeno E, Munoz PA, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. 2010;181:270–278. doi: 10.1164/rccm.200907-0988OC. [DOI] [PubMed] [Google Scholar]
- 71.Corte TJ, Gatzoulis MA, Parfitt L, et al. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010;15:1226–1232. doi: 10.1111/j.1440-1843.2010.01860.x. [DOI] [PubMed] [Google Scholar]
- 72.Montgomery GS, Sagel SD, Taylor AL, Abman SH. Effects of sildenafil on pulmonary hypertension and exercise tolerance in severe cystic fibrosis-related lung disease. Pediatr Pulmonol. 2006;41:383–385. doi: 10.1002/ppul.20393. [DOI] [PubMed] [Google Scholar]
- 73**.Noel S, Dhooghe B, Leal T. PDE5 Inhibitors as Potential Tools in the Treatment of Cystic Fibrosis. Front Pharmacol. 2012;3:167. doi: 10.3389/fphar.2012.00167. Detailed description on the potential use of phosphodiestearase-5 inhibitors in patients with CF and F508 deletion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tissieres P, Nicod L, Barazzone-Argiroffo C, et al. Aerosolized iloprost as a bridge to lung transplantation in a patient with cystic fibrosis and pulmonary hypertension. Ann Thorac Surg. 2004;78:e48–50. doi: 10.1016/j.athoracsur.2003.12.096. [DOI] [PubMed] [Google Scholar]
- 75.Olschewski H, Ghofrani HA, Walmrath D, et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160:600–607. doi: 10.1164/ajrccm.160.2.9810008. [DOI] [PubMed] [Google Scholar]
- 76.Dernaika TA, Beavin M, Kinasewitz GT. Iloprost improves gas exchange and exercise tolerance in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Respiration. 2010;79:377–382. doi: 10.1159/000242498. [DOI] [PubMed] [Google Scholar]
- 77.Hegewald MJ, Elliott CG. Sustained improvement with iloprost in a COPD patient with severe pulmonary hypertension. Chest. 2009;135:536–537. doi: 10.1378/chest.08-1515. [DOI] [PubMed] [Google Scholar]
- 78.Valerio G, Bracciale P, Grazia D’Agostino A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2009;3:15–21. doi: 10.1177/1753465808103499. [DOI] [PubMed] [Google Scholar]
- 79.Stolz D, Rasch H, Linka A, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. 2008;32:619–628. doi: 10.1183/09031936.00011308. [DOI] [PubMed] [Google Scholar]
- 80.Orens JB, Estenne M, Arcasoy S, et al. International guidelines for the selection of lung transplant candidates: 2006 update--a consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2006;25:745–755. doi: 10.1016/j.healun.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 81.Spahr JE, Love RB, Francois M, et al. Lung transplantation for cystic fibrosis: current concepts and one center’s experience. J Cyst Fibros. 2007;6:334–350. doi: 10.1016/j.jcf.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 82.Liou TG, Adler FR, Cox DR, Cahill BC. Lung transplantation and survival in children with cystic fibrosis. N Engl J Med. 2007;357:2143–2152. doi: 10.1056/NEJMoa066359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Inci I, Stanimirov O, Benden C, et al. Lung transplantation for cystic fibrosis: a single center experience of 100 consecutive cases. Eur J Cardiothorac Surg. 2012;41:435–440. doi: 10.1016/j.ejcts.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 84**.Christie JD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report-2012. J Heart Lung Transplant. 2012;31:1073–1086. doi: 10.1016/j.healun.2012.08.004. Transplant report of the ISHLT for the year 2012. [DOI] [PubMed] [Google Scholar]
- 85.Braun AT, Merlo CA. Cystic fibrosis lung transplantation. Curr Opin Pulm Med. 2011;17:467–472. doi: 10.1097/MCP.0b013e32834b8bdb. [DOI] [PubMed] [Google Scholar]
- 86.Christie JD, Edwards LB, Aurora P, et al. The Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth Official Adult Lung and Heart-Lung Transplantation Report-2009. J Heart Lung Transplant. 2009;28:1031–1049. doi: 10.1016/j.healun.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 87.Vizza CD, Lynch JP, Ochoa LL, et al. Right and left ventricular dysfunction in patients with severe pulmonary disease. Chest. 1998;113:576–583. doi: 10.1378/chest.113.3.576. [DOI] [PubMed] [Google Scholar]