

Abstract
Background
Growth promoting signals from the epicardium are essential for driving myocardial proliferation during embryogenesis. In adults, these signals become reactivated following injury and promote angiogenesis and myocardial repair. Therefore, identification of such paracrine factors could lead to novel therapeutic strategies. The multi-functional peptide adrenomedullin (Adm = gene, AM = protein) is required for normal heart development. Moreover, elevated plasma AM following myocardial infarction offers beneficial cardioprotection and serves as a powerful diagnostic and prognostic indication of disease severity.
Results
Here, we developed a new model of Adm overexpression by stabilizing the Adm mRNA through gene-targeted replacement of the endogenous 3′ untranslated region. As expected, Admhi/hi mice express three-times more AM than controls in multiple tissues, including the heart. Despite normal blood pressures, Admhi/hi mice unexpectedly showed significantly enlarged hearts due to increased cardiac hyperplasia during development. The targeting vector was designed to allow for reversion to wild-type levels by means of Cre-mediated modification. Using this approach, we demonstrate that AM derived from the epicardium, but not the myocardium or cardiac fibroblast, is responsible for driving cardiomyocyte hyperplasia.
Conclusions
AM is produced by the epicardium and drives myocyte proliferation during development, thus representing a novel and clinically relevant factor potentially related to mechanisms of cardiac repair after injury.
Keywords: cardiac development, epicardium, myocardium, hyperplasia, mouse models
Introduction
Recently, many exciting connections between factors that regulate myocardial development and those that are elevated during myocardial injury have been appreciated (Degenhardt et al., 2013). Central to these connections are the unique roles of the epicardium during embryogenesis and the newly emerging concept that the epicardium becomes reactivated—re-expressing fetal genes—during disease processes (Gittenberger-de Groot et al., 2012). For example, studies in zebrafish and mice have shown that the epicardium is required for maintenance of myocardial proliferation during embryogenesis through several growth promoting signaling pathways including Raldh2, Wt-1, IGF, FGF, and β1 integrin (Chen et al., 2002; Lavine et al., 2005; Ieda et al., 2009; Guadix et al., 2011; Kikuchi et al., 2011). During cardiac injury, many of these same growth promoting factors become stimulated such that the potential of the epicardium to contribute to heart regeneration and repair is restored from its normally adult quiescent state (Huang et al., 2012; van Wijk et al., 2012). Several recent studies have begun to elucidate the pleiotropic roles for epicardial-derived cells (EPDCs) in the formation of numerous cardiac cell types, including coronary endothelial and smooth muscle cells, cardiac fibroblasts, heart valves and perhaps even cardiomyocytes (Gittenberger-de Groot et al., 2012; Degenhardt et al., 2013). However, it is also evident that an important function of the epicardium is to secrete paracrine factors that promote angiogenesis, myocardial regeneration and cardiac fibroblast proliferation (Smart et al., 2011; Duan et al., 2012). Therefore identifying new factors that exhibit these dual roles of supporting both embryonic and injured heart growth is a desirable and clinically promising area of research.
In this study, we find that adrenomedullin peptide, secreted from the epicardium, is a potent regulator of cardiomyocyte proliferation during embryogenesis. Since the discovery of adrenomedullin (protein = AM, gene = Adm) in 1993 (Kitamura et al., 1993), many diverse biological functions have been described for this small circulating peptide during development, normal physiology and disease (Caron and Smithies, 2002; Karpinich et al., 2011). We, and others, have previously shown that genetic deletion of the highly conserved Adm gene in mice causes embryonic lethality at mid-gestation (Caron and Smithies, 2001; Shindo et al., 2001; Shimosawa et al., 2002). The most prominent phenotype of Adm−/− embryos is their extreme hydrops fetalis, or interstitial edema, which we have attributed to a marked arrest of lymphatic endothelial cell proliferation and lymphangiogenesis at mid-gestation (Fritz-Six et al., 2008). However, the expression of Adm, although enriched in lymphatic endothelial cells, is not restricted. Indeed, the earliest and most robust expression of Adm can be observed in the heart at around E8.0 (Montuenga et al., 1997, 1998; Caron and Smithies, 2001). Consequently, we found that Adm−/− mice also have small hearts characterized by enhanced trabeculation and a thin compact zone (Caron and Smithies, 2001). Furthermore, loss of either the receptor for AM, calcitonin receptor-like receptor (Calcrl) (Dackor et al., 2006), or the receptor modifying protein Ramp2 (Fritz-Six et al., 2008) results in a phenocopy of the Adm−/− phenotype. Taken together, the highly conserved and robust cardiac phenotypes of mice with genetic deletion of AM signaling components demonstrates the essential function of AM in promoting embryonic heart development.
A multitude of clinical studies have also shown that circulating levels of AM are elevated two- to three-fold above basal levels during a variety of human disease conditions (Karpinich et al., 2011). Recently, the robust surge in plasma AM levels during cardiovascular disease is being used as a clinical diagnostic tool. The pro-hormone form of AM, mid regional pro-AM (MRpro-AM), is a highly effective clinical biomarker of patient prognosis following heart attack, providing greater sensitivity than traditional biomarkers such as atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) (Dhillon et al., 2010; Maisel et al., 2010, 2011). In addition, AM also offers protection from cardiovascular disease. Several studies using mice haploinsufficient for Adm, have clearly demonstrated an exacerbation of cardiovascular disease with a reduction in basal levels of AM (Shimosawa et al., 2002; Caron et al., 2007), highlighting the importance of circulating AM in controlling the extent of damage. Additionally, animal studies examining the impact of AM infusion on cardiovascular disease progression have revealed a protective effect of AM on disease progression (Rademaker et al., 1997; Nishikimi et al., 2003b). Furthermore, a limited number of studies performed in human patients have demonstrated that infusion of AM after a cardiovascular incident is associated with a reduction in the degree of damage (Nagaya et al., 2000; Nishikimi et al., 2009; Kataoka et al., 2010). However, because these studies have relied on the infusion of AM, it remains unclear whether the physiological three-fold increase of circulating AM during cardiovascular disease has any direct or protective implications.
Therefore, we sought to develop a mouse model that recapitulates the plasma elevation of AM observed during human disease as a useful tool for elucidating the precise functions of AM in cardioprotection. Herein, we describe the successful generation of a novel mouse model of Adm overexpression that was intentionally designed to conserve the endogenous promoter regulatory elements of the gene, with the added capability of reversing the overexpression by Cre-LoxP strategies. Surprisingly, we discovered that these animals have enlarged hearts due to cardiac hyperplasia during development, a phenotype that was reversed when high AM levels from the epicardium were genetically restored to basal levels.
Results
Transcriptional and Translational Up-Regulation of Adm Expression in Admhi/hi Mice
We recently described the generation of the Admhi/hi mice by gene targeting techniques which allowed for the stabilization of the Adm mRNA half-life by the bovine growth hormone 3′ untranslated region (UTR) (Li et al., 2013). Previous in vitro studies predicted that this modification would stabilize the mRNA transcript of Adm by approximately 3.5-fold compared with the native transcript, while still allowing for endogenous regulation of Adm gene expression by its native 5′UTR promoter region (Kakoki et al., 2004). Here we show that Adm mRNA transcript levels, as measured by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR), were statistically increased by approximately 3- to 15-fold in Admhi/hi mice compared with wild-type control animals in ten different tissues at 4 months of age (Fig. 1A). Moreover, a radioimmunoassay for total AM peptide levels revealed a statistically significant ∼three-fold increase in AM peptide levels from kidney and lung extracts (Fig. 1B). Analysis of circulating AM in the blood plasma of 4-month-old animals by a fluorescent EIA method also showed a significant two- to three-fold increase in Admhi/hi animals compared with wild-type littermates (Fig. 1C). Thus, the 3′UTR gene targeting approach successfully recapitulated the increases in Adm mRNA and peptide expression at a level that is comparable to that observed in human patients with cardiovascular disease and myocardial infarction.
Fig. 1.
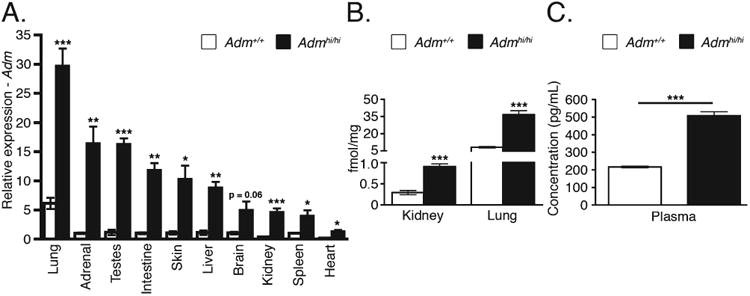
Transcriptional and translational up-regulation of Adm. A: Quantitative RT-PCR analysis of Adm from wild-type and Admhi/hi tissues (n = 3–10) at 4 months of age. B: Peptide levels of AM from 2- to 3-month-old wild-type and Admhi/hi lung and kidney as determined by a radioimmunoassay (n = 5–6). C: Plasma levels of AM from 4-month-old wild-type and Admhi/hi animals using a fluorescent immunoassay (n = 3). All values are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
Enhanced Adm Expression in Developing Heart of Admhi/hi Mice
Previous studies have shown the developing embryonic mouse heart expresses Adm as early as E8.0 and this expression persists throughout development and into adulthood (Montuenga et al., 1997; Caron and Smithies, 2001). Using immunohistochemistry, we find that endogenous levels of AM peptide expression are localized to the epicardium, with very low levels of expression in the compact zone and trabeculae at embryonic day (E) 13.5 (Fig. 2A,C). Similarly, Admhi/hi mice showed this same spatiotemporal pattern, but as expected, using semiquantitative imaging, we observed a robust and marked increase in AM expression from the epicardium and trabeculae compared with wild-type animals (Fig. 2B,D). By assessing the integrated density of AM at the epicardium, we confirmed that Admhi/hi embryos express higher levels of AM compared with wild-type controls (Fig. 2E). We also confirmed that AM is expressed in the epicardium by staining adjacent sections for AM and cytokeratin (Fig. 2F,G), which has been previously characterized as a marker of the epicardium (Viragh et al., 1993; Pennisi et al., 2003). This staining clearly demonstrates that AM is predominantly and robustly expressed in the epicardium, and to a much lesser extent in the myocardium. Another prominent feature of Admhi/hi hearts that can be appreciated from these images is the apparently larger size of the ventricle compared with Adm+/+ hearts viewed at the same magnification (compare Fig. 2A,B).
Fig. 2.
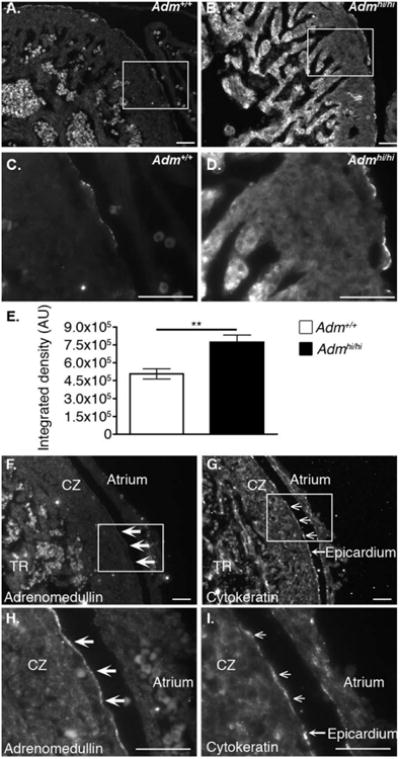
AM expression is localized to the developing epicardium and up-regulated in Admhi/hi mice. Immunofluorescence of AM peptide in heart sections of E13.5 embryos. A,C: Adm+/+ control mice. B,D: Admhi/hi mice. Images A,B were obtained with a 300 ms exposure, while images C and D were obtained with a 75 msec exposure. Boxes in A and B represent fields shown in panels C and D, respectively. E: The amount of AM expressed in the epicardium was assessed from panels C and D by measuring the integrated density of staining using Image J software. Data are expressed as arbitrary units (AU) of integrated density. Colocalization of AM peptide and the epicardial marker, cytokeratin. F,H: Adrenomedullin staining. G,I: Cytokeratin staining. The white boxes on panels F and G indicate the fields presented in panels H and I, respectively. Filled arrow heads on panels F and H indicate areas of AM staining, while open arrow heads in panels G and I highlight areas positive for cytokeratin staining. TR = trabecular zone; CZ = compact zone. Scale bars = 50 μM. All values are means ± SEM. **P < 0.01
Admhi/hi Mice Have Enlarged, Hyperplastic Hearts
Unexpectedly, we discovered that 2-month-old Admhi/hi mice had remarkably and visibly enlarged hearts compared with wild-type controls (Fig. 3A). Heart weight to body weight (HW:BW) ratios (Fig. 3B), as well as heart weight to tibia length (HW:TL) ratios (Fig. 3C) were significantly increased compared with wild-type control mice. Individual chamber dissections revealed that both ventricles and atria of Admhi/hi hearts were significantly enlarged compared with those of littermates (Fig. 3D).
Fig. 3.
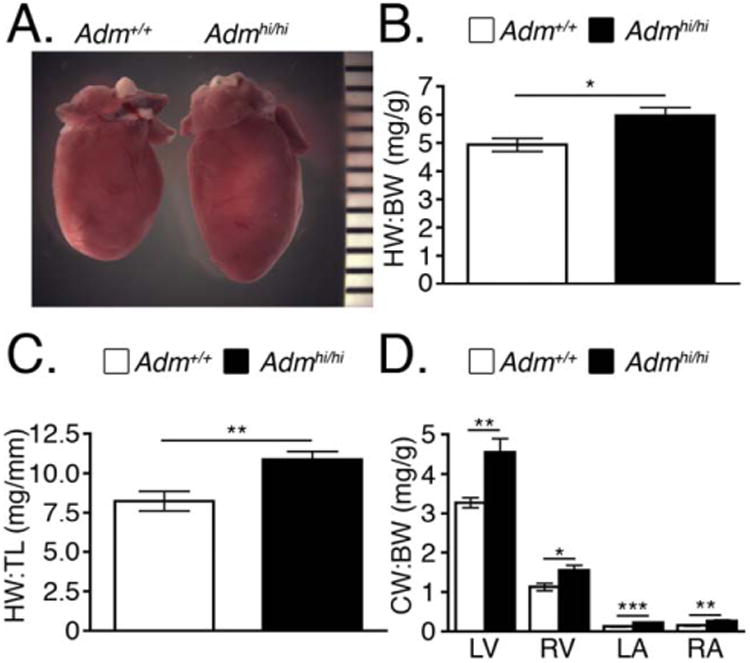
Admhi/hi mice have larger hearts that wild-type littermates. A: Gross appearance of hearts from 9-week-old male wild-type and Admhi/hi mice. B: Heart weight to body weight (HW:BW) ratios were assessed at 2 months of age (n = 6–12). C: Heart weight to tibia length (HW:TL) ratios were assessed at 2–4 months of age (n = 6). D: The ratio of left ventricle (LV), right ventricle (RV), left atria (LA), and right atria (RA) to body weight was assessed at 2 months of age to obtain chamber weight to body weight (CW:BW) ratios (n = 6–11). All values are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
Because either hypertrophic or hyperplastic mechanisms could contribute to enlargement of the heart, we used histological techniques to evaluate cardiomyocyte size and density. We found no difference in the cardiomyocyte cross-sectional area between wild-type and Admhi/hi animals, suggesting that the enlarged hearts of Admhi/hi animals were not hypertrophic (Fig. 4A). This conclusion was further supported by quantitation of cardiomyocyte cellular density from wheat germ agglutinin stained sections, which also revealed no difference in cellular myocyte density between Admhi/hi animals and wild-type controls (Fig. 4B,C). Additionally, qRT-PCR analysis of the pro-hypertrophic genes, Anp and Bnp, showed no significant differences between wild-type and Admhi/hi animals (Anp expression = 1.20 ± 0.3 vs. 1.87 ± 0.3; Bnp expression = 1.07 ± 0.2 vs. 1.83 ± 0.4 for wild-type and Admhi/hi, respectively), further confirming that Admhi/hi hearts are not hypertrophied. Finally, using isolectin staining, we also found no significant differences in vascular morphology or density between Admhi/hi animals and control animals (Fig. 4B,C). Based on these analyses, it was evident that the Admhi/hi hearts were enlarged due to hyperplasia, not hypertrophy, a conclusion that is further supported by the fact that Admhi/hi mice do not have elevated blood pressures (Fig. 4D).
Fig. 4.

Hearts of Admhi/hi mice are not hypertrophic. A: Representative heart sections were stained with hematoxylin and eosin (H&E). The cardiomyocyte cross-sectional area was determined from 15 individual myocytes per animal (n = 3). B: Representative heart sections were stained with isolectin (green), wheat germ agglutinin (WGA) (red), and DAPI (blue). We have also included single channel views of the WGA and isolectin staining to aid in the visualization of the vascular and cellular density. C: Total numbers of vessels and cardiomyocytes were quantitated from images at ×20 magnification (n = 3–8). Data shown here are representative of two independent experiments conducted by separate researchers. D: Mean arterial pressure (MAP) measurements assessed by tail cuff (n = 3–4). All values are means ± SEM.
Because Adm expression is prominently enriched in the heart during embryogenesis (Fig. 2), we performed bromodeoxyuridine (BrdU) incorporation studies to quantitate the number of proliferating cells in the hearts of wild-type and Admhi/hi embryos. We assessed proliferation at six different time points of embryonic development, and importantly found that Admhi/hi embryos always had more BrdU-positive cells per normalized field compared with wild-type, with significant differences in five out of six time points (Fig. 5A,B). From this analysis, it is also interesting to note that the shape of the proliferation curve for Admhi/hi animals recapitulates the shape of the curve exhibited by wild-type animals (see Fig. 5B), indicating that Admhi/hi hearts are still appropriately responding to signals responsible for regulating proliferation rates during development.
Fig. 5.
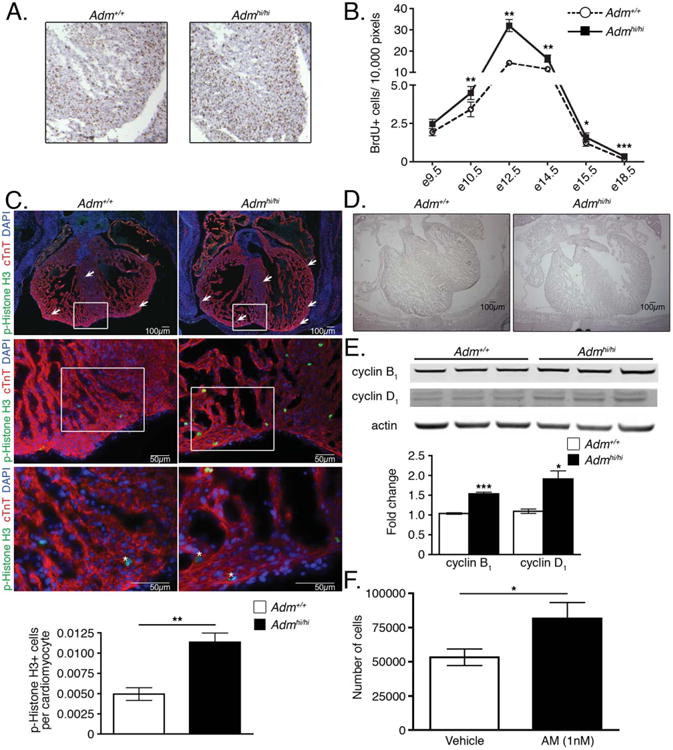
Enhanced proliferation of Admhi/hi hearts during embryonic development. A: Representative images of wild-type and Admhi/hi hearts at embryonic day (E) 12.5 stained for BrdU. B: Quantitation of the total number of BrdU positive cells from wild-type and Admhi/hi hearts throughout embryonic development normalized to the number of pixels in the image that were analyzed. C: Representative images of wild-type and Admhi/hi hearts at E12.5 stained with phospho-Histone H3 (p-Histone H3) (green), cardiac troponin T (cTnT) (red), and DAPI (blue). The white boxes on the ×4 (upper panels) and ×20 (middle panels) images represent the areas magnified in the ×20 and ×40 (lower panels) images, respectively. Asterisks on ×40 images indicate proliferating cardiomyocytes. The graph in the lower panel shows the number of phospho-Histone H3-positive cells per cardiomyocyte, which was determined by assessing three ×20 magnification fields per animal (n = 3). D: Low-power images of E15.5 wild-type and Admhi/hi hearts showing differences in heart size. Scale bar = 100 μm. E: Representative Western blot for cyclin B1 and cyclin D1 normalized to actin. Each lane on the blot is loaded with protein lysates from individual postnatal day 1 pups. Differences in lane loading were accounted for by normalizing each cyclin band to the corresponding actin band for each lane. These numbers were then normalized to a wild-type sample to compute the fold change. F: Proliferation of the HL-1 cardiomyocyte cell line with AM treatment. HL-1 cells were treated in serum-free media for 72 hours with either vehicle or 1 nM AM, before performing cell counts. Each condition was performed in triplicate. All values represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
To further confirm this developmental hyperplasia, we stained E12.5 hearts with phospho-Histone H3 (Fig. 5C), a less prevalent proliferation marker, and the cardiomyocyte marker, cardiac troponin T (cTnT). Consistent with the BrdU staining, we found a statistically significant increase in the level of phospho-Histone H3 labeling in Admhi/hi hearts compared with wild-type littermates (Fig. 5C). Moreover, by co-staining with cTnT, we could easily identify the phospho-Histone H3-positive cells as cardiomyocytes, by identifying phospho-H3-positive nuclei that were surrounded by cTnT signal, as indicated by the asterisks in the ×40 images in Figure 5C. Finally, we also compared the overall heart size of wild-type and Admhi/hi animals at the level of the ventricular valves at E15.5 and as expected, the enhanced levels of proliferation in Admhi/hi hearts resulted in a visible enlargement in the heart size of Admhi/hi embryos (Fig. 5D).
High AM Drives the Cardiomyocyte Cell Cycle
Cyclins and cyclin dependent kinases are critical for maintaining normal cardiomyocyte proliferation during development. Therefore, we further confirmed the accelerated degree of proliferation in Admhi/hi hearts by assessing protein levels of cyclin B1 and cyclin D1 by Western blotting of postnatal day 1 heart lysates. Our choice of postnatal day 1 tissue for these assays was based on the need to isolate sufficient amounts of protein for Western analysis but, furthermore, allowed us to interrogate whether the hyperplasia of Admhi/hi animals persists through the end of the proliferative phase of cardiac development. This analysis revealed a significant elevation of both cyclin B1 and cyclin D1 protein in the hearts of Admhi/hi pups compared with wild-type (Fig. 5E), even during the early postnatal period. As in the late stages of embryonic heart development, the proliferation rates in Admhi/hi hearts are modestly, but significantly elevated compared with wild-type animals, further highlighting the responsiveness of proliferating cardiomyocytes in Admhi/hi hearts to intrinsic regulators of the cell cycle.
To determine whether cells in Admhi/hi hearts do indeed exit the cell cycle in a manner comparable to wild-type animals, we also assessed proliferation in the hearts of 6-month-old adult wild-type and Admhi/hi animals by Ki67 staining. This analysis revealed that levels of proliferation are very low in the hearts of both wild-type and Admhi/hi groups, and we found no significant difference in the degree of proliferation (data not show), clearly demonstrating that the hyperplasia of Admhi/hi animals is limited to the developmental period.
We sought to further confirm our in vivo data demonstrating that AM can act directly on cardiomyocytes to drive the cell cycle by treating HL-1 cells, a cardiomyocyte cell line, with AM peptide. We chose to use the HL-1 cell line because previous studies have demonstrated that these cells retain many properties of adult cardiomyocytes in culture, including: cardiomyocyte-specific currents, gene expression profiles resembling adult cardiomyocytes, and the presence of cardiomyocyte-specific structures, such as intercalated discs (Claycomb et al., 1998). Additionally, HL-1 cells have the ability to proliferate in culture without losing these cardiomyocyte characteristics (Claycomb et al., 1998), unlike isolated neonatal cardiomyocytes. Thus, we treated the HL-1 cell line with 1nM AM or vehicle in serum-free media and measured cell counts. After 72-hr of treatment, there were significantly more HL-1 cells in the AM-treated conditions compared with vehicle-treated cells (Fig. 5F), indicating that AM can directly drive the proliferation of cardiomyocytes. These data, along with our in vivo data, clearly demonstrate first, that AM can act directly on cells to drive proliferation, and second, that elevated AM promotes cardiac hyperplasia.
Reversion of the Admhi Allele to Basal Levels Reverses the Cardiac Hyperplasia
The design of our targeting vector intentionally included loxP sites flanking the bGH 3′UTR stabilizing element to allow for tissue- and time-dependent reversion of high Adm levels back to wild-type levels (Fig. 6A). To confirm that this approach was effective, we crossed the Admhi/+ mice to the CMV-Cre line (Schwenk et al., 1995) to generate mice with a wild-type Adm allele, as well as a modified Admhi allele, referred to as Adm3′Δ. Adm3′Δ heterozygous mice were then intercrossed, at which point the CMV-Cre transgene was selected against to generate homozygous Adm3′Δ/3′Δ mice. By qRT-PCR, we found that Adm3′Δ/3′Δ mice express Adm at levels comparable to wild-type littermates (Fig. 6B), confirming that removal of the stabilizing bGH 3′UTR element results in a reversion of Adm levels to near basal levels.
Fig. 6.
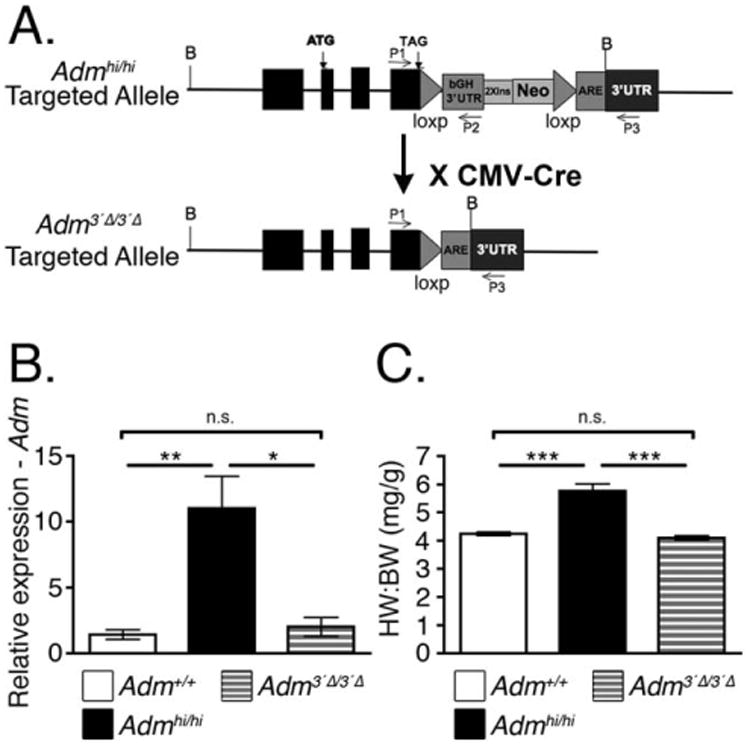
Mice with excised bGH 3′UTR have normal-sized hearts. A: Gene targeting design indicating the location of loxP sites flanking the bovine growth hormone (bGH) 3′UTR segment. B: Admhi/+ mice were bred to the CMV-Cre line to globally excise the bGH 3′UTR fragment from the Admhi allele. This new allele is designated as Adm3′Δ. After removing the Cre recombinase by means of breeding, Adm expression was assessed by qRT-PCR (n = 5–6). C: At 2–3 months of age, the heart weight to body weight (HW:BW) ratios of Adm3′Δ/3′Δ mice was compared with the HW:BW ratios of age-matched Admhi/hi and wild-type mice (n = 5–7). All values represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
Importantly, this genetic modification also reversed the cardiac hyperplasia phenotype of Admhi/hi mice. At 2 months of age, we assessed the HW:BW ratios of wild-type, Admhi/hi, and Adm3′Δ/3′Δ mice. We found that Adm3′Δ/3′Δ mice had HW:BW ratios that were significantly smaller than Admhi/hi mice, and indistinguishable from those of wild-type mice (Fig. 6C). Together, these data demonstrate the feasibility of genetically reversing the Admhi allele to normal levels, which is also associated with a reversal of the Admhi/hi cardiac hyperplasia phenotype.
AM Derived From Wt1-Positive Cells Promotes Enhanced Heart Size of Admhi/hi Mice
Our next goal was to use this genetic approach to identify the cellular source of AM that drives the observed cardiac hyperplasia during development. After confirming that we would be able to detect a decrease in HW:BW with a global reduction in Adm expression, we proceeded to cross the Admhi/hi mice to several different Cre mouse lines to assess the contribution of Adm from different cell types within the heart with respect to organ growth.
We began by assessing the contribution of cardiomyocyte-derived AM because the myocardium can release growth factors to regulate heart growth and development (Vega-Hernandez et al., 2011). Therefore, we crossed the Admhi/hi mice to two independent cardiomyocyte-specific Cre lines, the Nkx2.5-Cre (Moses et al., 2001) and αMHC-Cre (Agah et al., 1997) lines. At 2 months of age, we found there was no significant difference in the HW:BW ratio of Admhi/hi and Nkx2.5-Cre+: Adm3′Δ/3′Δ mice (Fig. 7A). Additionally, we found no difference in the HW:BW ratio of Admhi/hi and αMHC-Cre+: Adm3′Δ/3′Δ mice (data not shown). Together, these two independent cardiomyocyte-specific Cre lines clearly indicate that cardiomyocytes are not the cellular source of AM driving the heart growth during development.
Fig. 7.
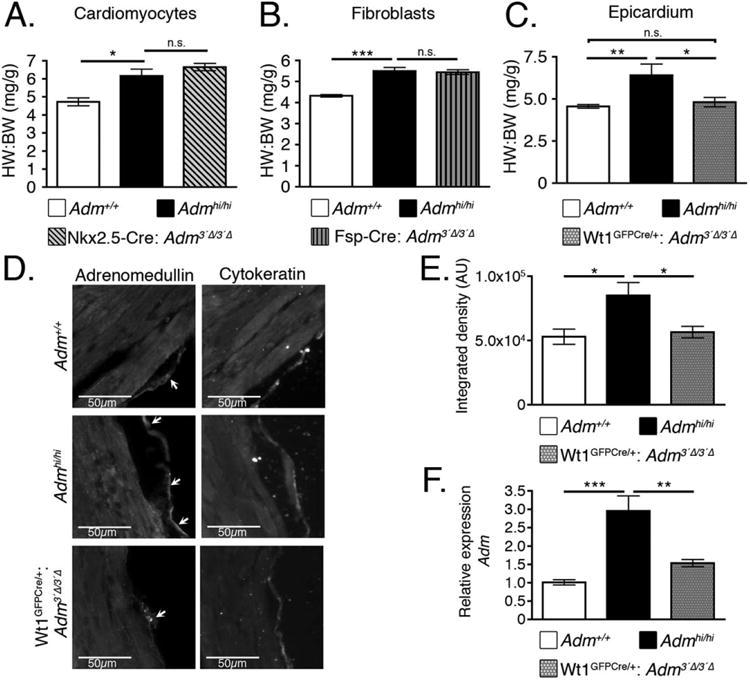
Epicardial-derived AM contributes to cardiac hyperplasia of Admhi/hi mice. A: Admhi/hi mice were crossed with the Nkx2.5-Cre mouse line to generate Admhi/hi animals with wild-type levels of Adm in cardiomyocytes. Heart weight to body weight (HW:BW) ratios were assessed at 2 months of age (n = 4–8). B: Admhi/hi mice were crossed to the Fsp-Cre mouse line to generate Admhi/hi mice with wild-type levels of Adm in fibroblasts. Heart weight to body weight (HW:BW) ratios were assessed at 2 months of age (n = 4–10). C: Admhi/hi mice were crossed with the Wt1GFPCre/+ mouse line to generate Admhi/hi mice with wild-type levels of Adm in the epicardium. At 2 months of age, heart weight to body weight (HW:BW) ratios were assessed (n = 3–6). D: Adjacent left ventricle sections from wild-type, Admhi/hi, and Wt1GFPCre/+:Adm3′Δ/3′Δ mice were stained with adrenomedullin (left panels) and the epicardial marker, cytokeratin (right panels). Adrenomedullin and cytokeratin images were acquired with a 130 ms exposure for all images. Images were acquired at ×20 magnification and the scale bar represents 50 μm. E: The amount of AM expressed in the epicardium was assessed from the images in panel D by measuring the integrated density using Image J software. The data are presented as arbitrary units (AU) of integrated density. F: RNA extracted from 2-month-old left ventricle samples was used to assess the relative expression levels of Adm by qRT-PCR. All values represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
Tomoda et al. found that the non-myocyte fraction of the rat heart expressed approximately 2.5 times more Adm than the myocyte fraction, suggesting that cardiac fibroblasts are a major source of AM in the heart (Tomoda et al., 2001). Therefore, we crossed the Admhi/hi mice to the Fsp-Cre (Bhowmick et al., 2004) mouse line to reduce Adm to wild-type levels in fibroblasts. Once again, we found there was no significant difference between Admhi/hi and Fsp-Cre+: Adm3′Δ/3′Δ mice at 2 months of age (Fig. 7B), suggesting that cardiac fibroblasts are not a major producer of AM during embryonic heart development.
Finally, we sought to determine whether AM derived from the epicardium contributed significantly to heart development because many studies have described the importance of the epicardium in secreting paracrine growth factors that regulate heart growth (Lavine et al., 2005; Lu et al., 2008; Li et al., 2011), and our data demonstrate that the epicardium is a major source of Adm expression during embryogenesis (Fig. 2). By crossing the Wt1GFPCre/+ (Zhou et al., 2008) mice to the Admhi/hi line, we were able to generate mice with reduced Adm in the epicardium (Fig. 7D). In this case, we discovered that Wt1GFPCre/+: Adm3′Δ/3′Δ mice had HW:BW ratios that were significantly lower that Admhi/hi animals, and not significantly different from wild-type animals (Fig. 7C). We also stained adjacent sections of the left ventricle from these animals for cytokeratin, to mark the epicardium, and AM (Fig. 7D). We found that AM colocalized with cytokeratin expression in the epicardium, further confirming the expression and contribution of epicardial-derived AM in heart development. These images also reveal that AM expression of Wt1GFPCre/+: Adm3′Δ/3′Δ mice is reduced in the epicardium compared with Admhi/hi animals (Fig. 7E) and is similar to wild-type littermates, thereby verifying that we successfully reduced AM expression in the epicardium of these animals, resulting in the normalization of heart size. Finally, to further corroborate these findings, we assessed Adm gene expression levels using RNA extracted from 2-month-old left ventricle samples from Admhi/hi and Wt1GFPCre/+:Adm3′Δ/3′Δ mice. This experiment revealed that Adm expression levels are significantly reduced to near baseline values in Wt1GFPCre/+:Adm3′Δ/3′Δ mice compared with Admhi/hi animals (Fig. 7F). Because Cre recombinase is expressed specifically in the pro-epicardium and epicardium in the Wt1GFPCre/+ line (Zhou et al., 2008), we concluded that a reduction in Adm expression level is due to a normalization of Adm expression levels specifically in the epicardium. Although Adm expression levels were still elevated over wild-type levels in Wt1GFPCre/+:Adm3′Δ/3′Δ animals, it is important to recognize that this difference can be attributed to the fact that other cell types in the heart, including cardiomyocytes and fibroblasts, are still overexpressing Adm. Because we have demonstrated that a reduction of Adm levels in cardiomyocytes and fibroblasts fails to affect the HW:BW ratios (Fig. 7A and 7B), these data indicate that the epicardium is a major source of Adm that promotes cardiac hyperplasia in the Admhi/hi line.
Discussion
With this unique gene targeted mouse model, we have identified AM as a powerful regulator of cardiac growth during embryogenesis. Importantly, we have been able to demonstrate that AM secreted from epicardial cells is important for driving the cardiomyocyte cell cycle and promoting enlargement of the heart during embryogenesis. Considering the numerous and high profile clinical studies which link elevated plasma AM levels following myocardial infarction with both disease severity and cardioprotection, we consider that AM fulfills the characteristics of a newly recognized epicardial-derived growth factor that may play important roles in cardiac repair and regeneration.
Most importantly, the pharmacological targeting of AM signaling is readily achievable through small molecule agonists and antagonists capable of binding to the unique interface formed between the AM receptor, calcitonin receptor-like receptor and receptor activity modifying protein (RAMP) 2 (Archbold et al., 2011). Indeed this receptor- RAMP paradigm is already being exploited by the pharmaceutical industry for the treatment of migraine pain associated with CGRP signaling (Doods et al., 2007; Salvatore et al., 2010). Therefore, while the delivery of AM peptide has some limitations as a treatment strategy for myocardial infarction, activation of AM signaling through its unique G protein-coupled receptor paradigm is attainable.
In the literature, a limited number of mouse models have been reported that display cardiac hyperplasia (Chaudhry et al., 2004; Kerkela et al., 2008; Trivedi et al., 2008; Qiu et al., 2010; Heallen et al., 2011; Phillips et al., 2011). Many of these mutant animals die during the late embryonic development or the early postnatal period (Kerkela et al., 2008; Qiu et al., 2010; Heallen et al., 2011; Phillips et al., 2011), which contrasts with this Admhi/hi model, which survives into adulthood. This difference can, perhaps, be attributed to the presence of additional cardiac defects in some lines or differences in how the heart is enlarged. For example, many of these mouse lines display other cardiac abnormalities in addition to cardiomyocyte hyperplasia, including: ventricular septal defects (Kerkela et al., 2008; Heallen et al., 2011; Phillips et al., 2011), double outlet right ventricles (Kerkela et al., 2008), and dilated atria (Qiu et al., 2010; Phillips et al., 2011). The Admhi/hi mice do not display additional cardiac defects, which potentially explains why these mice are viable. Moreover, several of the other reported mouse lines displaying cardiac hyperplasia display a reduction in the luminal space of the ventricle due to a robust increase in the space occupied by the trabeculi as well as a drastic increase in the thickness of the compact layer (Kerkela et al., 2008; Qiu et al., 2010; Heallen et al., 2011; Phillips et al., 2011). Because the degree of trabeculation between wild-type and Admhi/hi animals appears similar, the lumens of the Admhi/hi mice remain unobstructed, allowing for normal heart function and viability of the mice.
We found that a reduction of Adm in the epicardium through the use of the Wt1GFPCre/+ mouse line resulted in a reversion of the HW:BW ratio back to wild-type levels, indicating that the cardiac hyperplasia phenotype of the Admhi/hi mice is caused by enhanced epicardial-derived AM. Some studies have indicated that Wt1 is expressed in a subset of cardiomyocytes during embryonic development (Rudat and Kispert, 2012), and therefore, the Wt1GFPCre/+ line may promote recombination directly in the cardiomyocytes. Additionally, other groups have suggested that epicardial cells may differentiate into cardiomyocytes (Cai et al., 2008; Zhou et al., 2008), and thus the recombined genes from the cross to the Wt1GFPCre/+ line may be transmitted to the cardiomyocyte lineage. Therefore, it is possible that additional recombination in cardiomyocytes through one of these two mechanisms is contributing to the reduction of the HW:BW ratio in Wt1GFPCre/+:Adm3′Δ/3′Δ mice. However, our data from two independent cardiomyocyte-specific Cre lines showing no reduction in HW:BW with reduced levels of AM expression in cardiomyocytes suggests that the contribution of cardiomyocyte-derived AM to the developmental hyperplasia observed in the Admhi/hi line is minimal.
Our conclusion that the epicardium is a major contributor of AM during heart development is further supported by the similarities between the heart phenotype of Adm−/− mice (Caron and Smithies, 2001) and animals with a disrupted epicardium (Kreidberg et al., 1993; Kwee et al., 1995; Pennisi et al., 2003; Li et al., 2011). Specifically, one characteristic of animals lacking an epicardium or with disrupted growth factor secretion from the epicardium is a thin compact zone (Kreidberg et al., 1993; Kwee et al., 1995; Pennisi et al., 2003; Li et al., 2011), which was also observed in the Adm−/− mice (Caron and Smithies, 2001). Although Adm−/− animals do not exhibit other cardiac defects that are sometimes described in other models with epicardial alterations, it is possible that AM acts independently or through different growth factors than those investigated to date. Additionally, it is possible that AM may play a role in the endocardium, as Adm−/− embryos also have abnormal trabeculation compared with wild-type littermates (Caron and Smithies, 2001), and the endocardium has been implicated in the regulation of trabecular development (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995; Stainier et al., 1995; Grego-Bessa et al., 2007). However, because the majority of the proliferation we observed in the hearts of Admhi/hi hearts was localized to the compact zone and septum (see Fig. 5C) and the development of these areas is largely regulated by the epicardium (Gittenberger-de Groot et al., 2000; Sucov et al., 2009), it seems likely that endocardial-derived AM contributes minimally to the observed cardiac hyperplasia.
Our current study does not excluded the possibility that AM may interact with other growth factors to promote cardiac hyperplasia in Admhi/hi animals. To date, several epicardial-derived factors critical to compact zone proliferation have been identified. These factors include several members of the fibroblast growth factor (FGF) family (Lavine et al., 2005), members of the insulin growth factor (IGF) signaling cascade (Li et al., 2011), as well as retinoic acid (RA) (Chen et al., 1998, 2002; Stuckmann et al., 2003; Merki et al., 2005) and erythropoietin (Epo) (Wu et al., 1999; Stuckmann et al., 2003). Presently, it remains unclear whether any interaction exists between AM and these factors in the heart to promote the cardiac hyperplasia observed in Admhi/hi animals. Similar to AM, the FGFs and their corresponding receptors are widely expressed throughout the body during development and play an important role in regulating cellular growth in many organ systems (Lea et al., 2009). Although there have been very few studies investigating any relationship between AM and the FGFs, one recent study found that transgenic overexpression of FGF18 in the lung during the early postnatal development period promoted an increase in Adm expression (Franco-Montoya et al., 2011), indicating the potential for crosstalk between these two signaling pathways; however, it is unknown whether a similar paradigm exists in the heart during embryonic development. Another potential mechanism by which the Adm signaling pathway may interact with other epicardial-derived signals is through the transactivation of FGF and IGF receptors. Guidolin et al. (2008) have reported that AM may transactivate VEGFR2 to promote angiogenesis in human vascular endothelial cells. Specifically, they found that the angiogenic properties of AM were dependent on the tyrosine kinase domain of the VEGFR2 receptor, (Guidolin et al., 2008) which suggests that other receptor tyrosine kinases, such as the FGF and IGF receptors, may be subject to transactivation by AM signaling. Furthermore, Cornish et al. (Cornish et al., 2004) found that AM-induced proliferation of osteoblasts was inhibited with the loss of IGF-1R, indicating a potentially broader paradigm for crosstalk between AM signaling and receptor tyrosine kinases. Finally, several studies have demonstrated that Epo and RA are essential regulators of epicardial-derived growth factors (Chen et al., 1998, 2002; Wu et al., 1999; Stuckmann et al., 2003; Merki et al., 2005). Although there are no data currently available that demonstrate a direct relationship between AM and Epo, both of these peptides are strongly induced by hypoxia through HIF-1alpha (Fandrey et al., 1994; Garayoa et al., 2000), which may indicate parallel function. Finally, a limited number of studies have examined the relationship between AM and RA (Minamino et al., 1995; Kubo et al., 1998). These studies have found that in macrophages (Kubo et al., 1998) and vascular smooth muscle cells (Minamino et al., 1995), RA induced Adm expression, indicating that Adm may be induced by RA in the epicardium during cardiac development to regulate cardiomyocyte proliferation.
Future studies to elucidate these interactions are underway. For example, this Adm overexpression model will be useful for understanding the importance of AM elevation during disease, and with Cre-recombinase technology, we can begin to address in what tissues and at what times AM elevation is advantageous. Additionally, the role of AM during developmental processes of many organs, including the heart, lymphatic system, and other organ systems, can be studied more thoroughly due to the viability of these animals.
Experimental Procedures
Animals
Admhi/hi mice were generated using standard gene targeting techniques, as described in detail in (Li et al., 2013). Briefly, the targeting vector was designed to replace the endogenous 3′UTR with a stabilizing cassette that increases the Adm mRNA half-life. Additionally, loxP sites allow for the deletion of the stabilizing element and its replacement by an 80 bp of AU/U-rich element of the mouse c-fos gene (ARE) (Fig. 6A). For genotyping, a 3-primer, PCR-based strategy was used: primer 1: 5′-AACCTTAC ACCTTGCTGAGACATTC-3′; primer 2: 5′-TTTATTAGGAAAGGACAGTGG GAGTG-3′; primer 3: 5′CCCACATT CGTGTCAAACGCTAC-3′. Primers 1 and 3 amplify a 760-bp wild-type allele, while primers 2 and 3 amplify a 600-bp targeted allele. Mice used in these studies were either from a mixed genetic background or backcrossed to the C57Bl6 strain for over nine generations. For all experiments, littermate animals were used as controls.
To generate mice with reduced Adm expression either globally or in specific cells types, Admhi/hi mice were crossed with the following previously characterized Cre mouse lines: CMV-Cre (Schwenk et al., 1995), αMHC-Cre (Agah et al., 1997), Nkx2.5-Cre (Moses et al., 2001), Fsp-Cre (Bhowmick et al., 2004), or Wt1GFPCre/+ (Zhou et al., 2008). The recombined Admhi allele was designated Adm3′Δ. Heart weight to body weight (HW:BW) ratios were assessed at 2 months of age. All studies were approved by the Institutional Animal Care and Use Committee of UNC-CH.
Gene Expression Analysis
Adm gene expression was analyzed by qRT-PCR with the Mx3000P Real Time PCR System from Stratagene. Primers for Adm amplification were 5′-GAGCGAAGCCCACATTCGT-3′ and 5′-GAAGCGGCATCCATTGCT-3′ and the probe sequence was 5′-FAM-CTACCGCCAGAGCATGAACCAGGG-TAMRA-3′. Primers for Bnp amplification were 5′-CTGCTGGAGCTGATA AGAGA-3′ and 5′ -TGCCCAAAGCAGC TTGAGAT-3′ and the probe sequence was 5′-FAM-CTCAAGGCAGCACCCT CCGGG-TAMRA-3′. Taqman Gene Expression Assays for Anp and GAPDH were purchased from Applied Biosystems. Embryonic lung RNA from Ambion was used as calibrator. RNA was isolated from mouse tissues with Trizol and subsequently DNaseI treated. The ΔΔCt method was used to determine the relative levels of gene expression. Assays were repeated 3 times, each time with triplicates.
Measurement of AM Peptide
Mouse AM was measured using a specific immunoradiometric assay kit (AM RIA, Shionogi) with some modifications, as previously reported (Nishikimi et al., 2002, 2003a). Plasma levels of AM were measured by a fluorescent immunoassay (Phoenix Pharmaceuticals, FEK-010-08) following the manufacturer's protocol.
Adrenomedullin Staining
Paraffin embedded left ventricle sections were blocked for 1 hr in 5% normal donkey serum at room temperature. Slides were then incubated with adrenomedullin antibody (Novus, NBP1-19731) overnight at room temperature. Slides were washed with phosphate buffered saline (PBS) before incubation with secondary antibodies for 2 hr at room temperature.
Cytokeratin Staining
Paraffin embedded tissues were subjected to three, 5-min rounds of antigen retrieval in 10 mM citric acid buffer/0.05% Tween 20. Slides were then blocked in 5% normal donkey serum/0.1% Triton X at room temperature for 1 hr. Slides were incubated with diluted cytokeratin primary antibody (Dako, Z0622) overnight at room temperature. After washing slides with PBS, secondary antibody was applied for 2 hr at room temperature in the dark.
Capillary Density and Cardiomyocyte Density Assay
Paraffin embedded heart sections were blocked in 1%BSA/0.2% triton X-100 in PBS for 1 hr, then incubated with Isolectin B4 fluorescein isothio-cyanate conjugate (Sigma L2895, 1:200), Rhodamine wheat germ agglutinin (Vector Laboratories, RL-1022, 1:1,000), and 1:1,000 DAPI in 1%BSA in PBS for 2.5 hr at room temperature, then washed in PBS and rinsed with ddH2O.
Measurement of Mean Arterial Pressure
The mean arterial pressure (MAP) was assessed by the tail cuff method as previously described (Krege et al., 1995). Briefly, mice were acclimated to the restraint apparatus for 2 days before data collection. For 3 days following the acclimation period, blood pressure data were collected for each mouse. The data from the 3 days were then averaged together to generate the MAP value.
BrdU Incorporation Assay
Timed matings were performed between Admhi/+ males and females. Two hours before the dissection of the embryos, the pregnant female was injected with BrdU (0.1 mg/g of BW) by means of tail vein injection. Embryos were carefully dissected and genotypes were determined from DNA extracted from either the yolk sac or the tail. Frozen sections of the heart were stained for BrdU using a diaminobenzidine (DAB)-based protocol.
Phospho-Histone H3 (Ser10) and Cardiac Troponin T Co-staining
Frozen E12.5 embryo heart sections were thawed overnight before antigen retrieval. Slides were blocked in 10% normal goat serum in 0.05 M Tris buffer for 1 hr before applying primary phospho-Histone H3 antibody (Cell Signaling, 9701) and cardiac troponin T antibody (Thermo, MS-295-P) slides overnight at 4°C. Slides were washed with three changes of 0.05 M Tris buffer before incubating with fluorescent secondary antibody for 2 hr at room temperature in the dark.
Cyclin B1 and Cyclin D1 Western blot
Hearts were dissected from postnatal day 1 pups and flash-frozen in liquid nitrogen. Protein was isolated from wild-type and Admhi/hi hearts using standard procedures. Samples were denatured and run on NuPage Bis-Tris gels and proteins were transferred to PVDF membranes. Membranes were probed with cyclin B1 (Rockland, 100-401-152), cyclin D1 (Rockland, 100-401-153) and actin (Sigma, A4700) antibodies overnight at 4°C. Membranes were incubated with secondary antibodies for 45minutes and imaged using a Li-Cor Odyssey CLx. Integrated density was assessed using ImageJ software.
Proliferation of HL-1 Cells With AM Treatment
HL-1 cells are a cardiomyocyte cell line first developed and characterized by Claycomb et al. in 1998 (Claycomb et al., 1998). When cultured in Claycomb media (Sigma, 51800C), supplemented with norepinephrine and L-glutamine, these cells retain many properties of cardiomyocytes, but, unlike isolated cardiomyocytes, will proliferate in culture (Claycomb et al., 1998). For this experiment, HL-1 cells were plated in fibronectin/gelatin-coated 24-well dishes at a density of 50,000 cells per well in serum-free media. Cells were allowed to adhere to the plates for 4 hr before treatment. At the time of treatment, the media was removed from each well and replaced with serum-free media containing either tissue culture-grade water as a vehicle control or 1nM AM (Phoenix Pharmaceuticals, 010-31). Total cell numbers in each well were counted using a Countess Automated Cell Counter. Each condition was performed in triplicate to allow for statistical analysis.
Statistics
Statistical analyses were performed with JMP software (SAS). One-way analysis of variance was used to determine significance at P < 0.05. In all figures, error bars represent SEM.
Key Findings.
Adrenomedullin is a secreted peptide with important functions during embryonic development and cardiovascular disease.
Adrenomedullin overexpression in mice was accomplished by gene targeted insertion of a stabilizing element within the endogenous 3′ UTR.
Admhi/hi mice show enlarged heart size due to cardiomyocyte hyperplasia during development.
Reversal of the cardiac hyperplasia is accomplished by Cre-mediated excision of the genetic stabilizing element in epicardial cells.
Epicardial-derived adrenomedullin drives myocyte proliferation during development and thus represents a novel mitogenic factor potentially related to mechanisms of cardiac repair after injury.
Acknowledgments
We thank current and past members of the Caron Lab, including Scott T. Espenschied, Helen Willcockson and Kimberly Fritz-Six, Kirk McNaughton of the Histopathology Core and Dr. Mauricio Rojas of the Mouse Cardiovascular Core for technical assistance and helpful discussions. We would also like to thank Dr. Monte Willis for generously providing us with HL-1 cells and Dr. Joan Taylor for providing us with cardiac troponin antibody. K.M.C. was funded by the Burroughs Wellcome Fund and S.E.W-S. was funded by the AHA.
Grant sponsor: AHA; Grant numbers: EIA 0940097N; PRE11710002; Grant sponsor: NIH; Grant numbers: HL091973; HD060860; Grant sponsor: Burroughs Wellcome Fund.
Abbreviations
- AM
adrenomedullin protein
- Adm
adrenomedullin gene
- ANP
atrial natriuretic peptide
- BNP
B-type natriuretic peptide
- bGH
bovine growth hormone
- MAP
mean arterial pressure
- DAB
diaminobenzidine
- qRT-PCR
quantitative RT-PCR
- EIA
enzyme immunoassay
- HW:BW
heart weight to body weight
- HW:TL
heart weight to tibia length
- CW:BW
chamber weight to body weight
References
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archbold JK, Flanagan JU, Watkins HA, Gingell JJ, Hay DL. Structural insights into RAMP modification of secretin family G protein-coupled receptors: implications for drug development. Trends Pharmacol Sci. 2011;32:591–600. doi: 10.1016/j.tips.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104–108. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron K, Hagaman J, Nishikimi T, Kim HS, Smithies O. Adrenomedullin gene expression differences in mice do not affect blood pressure but modulate hypertension-induced pathology in males. Proc Natl Acad Sci U S A. 2007;104:3420–3425. doi: 10.1073/pnas.0611365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci U S A. 2001;98:615–619. doi: 10.1073/pnas.021548898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron KM, Smithies O. Multiple roles of adrenomedullin revealed by animal models. Microsc Res Tech. 2002;57:55–59. doi: 10.1002/jemt.10046. [DOI] [PubMed] [Google Scholar]
- Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, Wolgemuth DJ. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem. 2004;279:35858–35866. doi: 10.1074/jbc.M404975200. [DOI] [PubMed] [Google Scholar]
- Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- Chen T, Chang TC, Kang JO, Choudhary B, Makita T, Tran CM, Burch JB, Eid H, Sucov HM. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev Biol. 2002;250:198–207. doi: 10.1006/dbio.2002.0796. [DOI] [PubMed] [Google Scholar]
- Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish J, Grey A, Callon KE, Naot D, Hill BL, Lin CQ, Balchin LM, Reid IR. Shared pathways of osteoblast mitogenesis induced by amylin, adrenomedullin, and IGF-1. Biochem Biophys Res Commun. 2004;318:240–246. doi: 10.1016/j.bbrc.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Dackor RT, Fritz-Six K, Dunworth WP, Gibbons CL, Smithies O, Caron KM. Hydrops fetalis, cardiovascular defects, and embryonic lethality in mice lacking the calcitonin receptor-like receptor gene. Mol Cell Biol. 2006;26:2511–2518. doi: 10.1128/MCB.26.7.2511-2518.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Singh MK, Epstein JA. New approaches under development: cardiovascular embryology applied to heart disease. J Clin Invest. 2013;123:71–74. doi: 10.1172/JCI62884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon OS, Khan SQ, Narayan HK, Ng KH, Struck J, Quinn PA, Morgenthaler NG, Squire IB, Davies JE, Bergmann A, Ng LL. Prognostic value of mid-regional pro-adrenomedullin levels taken on admission and discharge in non-ST-elevation myocardial infarction: the LAMP (Leicester Acute Myocardial Infarction Peptide) II study. J Am Coll Cardiol. 2010;56:125–133. doi: 10.1016/j.jacc.2010.01.060. [DOI] [PubMed] [Google Scholar]
- Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28:580–587. doi: 10.1016/j.tips.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, Majesky M, Deb A. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31:429–442. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrey J, Pagel H, Frede S, Wolff M, Jelkmann W. Thyroid hormones enhance hypoxia-induced erythropoietin production in vitro. Exp Hematol. 1994;22:272–277. [PubMed] [Google Scholar]
- Franco-Montoya ML, Boucherat O, Thibault C, Chailley-Heu B, Incitti R, Delacourt C, Bourbon JR. Profiling target genes of FGF18 in the postnatal mouse lung: possible relevance for alveolar development. Physiol Genomics. 2011;43:1226–1240. doi: 10.1152/physiolgenomics.00034.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Invest. 2008;118:40–50. doi: 10.1172/JCI33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, Trepel J, Montuenga LM, Ryan H, Johnson R, Gassmann M, Cuttitta F. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Mol Endocrinol. 2000;14:848–862. doi: 10.1210/mend.14.6.0473. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Vrancken Peeters MP, Bergwerff M, Mentink MM, Poelmann RE. Epicardial outgrowth inhibition leads to compensatory mesothelial outflow tract collar and abnormal cardiac septation and coronary formation. Circ Res. 2000;87:969–971. doi: 10.1161/01.res.87.11.969. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Winter EM, Bartelings MM, Goumans MJ, DeRuiter MC, Poelmann RE. The arterial and cardiac epicardium in development, disease and repair. Differentiation. 2012;84:41–53. doi: 10.1016/j.diff.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Perez-Pomares JM, de la Pompa JL. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadix JA, Ruiz-Villalba A, Lettice L, Velecela V, Munoz-Chapuli R, Hastie ND, Perez-Pomares JM, Martinez-Estrada OM. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development. 2011;138:1093–1097. doi: 10.1242/dev.044594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidolin D, Albertin G, Spinazzi R, Sorato E, Mascarin A, Cavallo D, Antonello M, Ribatti D. Adrenomedullin stimulates angiogenic response in cultured human vascular endothelial cells: Involvement of the vascular endothelial growth factor receptor 2. Peptides. 2008;29:2013–2023. doi: 10.1016/j.peptides.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GN, Thatcher JE, McAnally J, Kong Y, Qi X, Tan W, DiMaio JM, Amatruda JF, Gerard RD, Hill JA, Bassel-Duby R, Olson EN. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science. 2012;338:1599–1603. doi: 10.1126/science.1229765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakoki M, Tsai YS, Kim HS, Hatada S, Ciavatta DJ, Takahashi N, Arnold LW, Maeda N, Smithies O. Altering the expression in mice of genes by modifying their 3′ regions. Dev Cell. 2004;6:597–606. doi: 10.1016/s1534-5807(04)00094-2. [DOI] [PubMed] [Google Scholar]
- Karpinich NO, Hoopes SL, Kechele DO, Lenhart PM, Caron KM. Adrenomedullin function in vascular endothelial cells: insights from genetic mouse models. Curr Hypertens Rev. 2011;7:228–239. doi: 10.2174/157340211799304761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka Y, Miyazaki S, Yasuda S, Nagaya N, Noguchi T, Yamada N, Morii I, Kawamura A, Doi K, Miyatake K, Tomoike H, Kangawa K. The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J Cardiovasc Pharmacol. 2010;56:413–419. doi: 10.1097/FJC.0b013e3181f15b45. [DOI] [PubMed] [Google Scholar]
- Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118:3609–3618. doi: 10.1172/JCI36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 2011;20:397–404. doi: 10.1016/j.devcel.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, Eto T. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192:553–560. doi: 10.1006/bbrc.1993.1451. [DOI] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- Kubo A, Minamino N, Isumi Y, Katafuchi T, Kangawa K, Dohi K, Matsuo H. Production of adrenomedullin in macrophage cell line and peritoneal macrophage. J Biol Chem. 1998;273:16730–16738. doi: 10.1074/jbc.273.27.16730. [DOI] [PubMed] [Google Scholar]
- Kwee L, Baldwin HS, Shen HM, Stewart CL, Buck C, Buck CA, Labow MA. Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development. 1995;121:489–503. doi: 10.1242/dev.121.2.489. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lea R, Papalopulu N, Amaya E, Dorey K. Temporal and spatial expression of FGF ligands and receptors during Xenopus development. Dev Dyn. 2009;238:1467–1479. doi: 10.1002/dvdy.21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- Li M, Schwerbrock NM, Lenhart PM, Fritz-Six KL, Kadmiel M, Christine KS, Kraus DM, Espenschied ST, Willcockson HH, Mack CP, Caron KM. Fetal-derived adrenomedullin mediates the innate immune milieu of the placenta. J Clin Invest. 2013;123:2408–2420. doi: 10.1172/JCI67039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Bruning JC, Pashmforoush M, Sucov HM. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development. 2011;138:1795–1805. doi: 10.1242/dev.054338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SY, Sheikh F, Sheppard PC, Fresnoza A, Duckworth ML, Detillieux KA, Cattini PA. FGF-16 is required for embryonic heart development. Biochem Biophys Res Commun. 2008;373:270–274. doi: 10.1016/j.bbrc.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisel A, Mueller C, Nowak R, Peacock WF, Landsberg JW, Ponikowski P, Mockel M, Hogan C, Wu AH, Richards M, Clopton P, Filippatos GS, Di Somma S, Anand I, Ng L, Daniels LB, Neath SX, Christenson R, Potocki M, McCord J, Terracciano G, Kremastinos D, Hartmann O, von Haehling S, Bergmann A, Morgenthaler NG, Anker SD. Mid-region pro-hormone markers for diagnosis and prognosis in acute dyspnea: results from the BACH (Biomarkers in Acute Heart Failure) trial. J Am Coll Cardiol. 2010;55:2062–2076. doi: 10.1016/j.jacc.2010.02.025. [DOI] [PubMed] [Google Scholar]
- Maisel A, Mueller C, Nowak RM, Peacock WF, Ponikowski P, Mockel M, Hogan C, Wu AH, Richards M, Clopton P, Filippatos GS, Di Somma S, Anand I, Ng LL, Daniels LB, Neath SX, Christenson R, Potocki M, McCord J, Hartmann O, Morgenthaler NG, Anker SD. Midregion prohormone adrenomedullin and prognosis in patients presenting with acute dyspnea: results from the BACH (Biomarkers in Acute Heart Failure) trial. J Am Coll Cardiol. 2011;58:1057–1067. doi: 10.1016/j.jacc.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- Minamino N, Shoji H, Sugo S, Kangawa K, Matsuo H. Adrenocortical steroids, thyroid hormones and retinoic acid augment the production of adrenomedullin in vascular smooth muscle cells. Biochem Biophys Res Commun. 1995;211:686–693. doi: 10.1006/bbrc.1995.1866. [DOI] [PubMed] [Google Scholar]
- Montuenga LM, Mariano JM, Prentice MA, Cuttitta F, Jakowlew SB. Coordinate expression of transforming growth factor-beta1 and adrenomedullin in rodent embryogenesis. Endocrinology. 1998;139:3946–3957. doi: 10.1210/endo.139.9.6205. [DOI] [PubMed] [Google Scholar]
- Montuenga LM, Martinez A, Miller MJ, Unsworth EJ, Cuttitta F. Expression of adrenomedullin and its receptor during embryogenesis suggests autocrine or paracrine modes of action. Endocrinology. 1997;138:440–451. doi: 10.1210/endo.138.1.4881. [DOI] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2–5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Nagaya N, Satoh T, Nishikimi T, Uematsu M, Furuichi S, Sakamaki F, Oya H, Kyotani S, Nakanishi N, Goto Y, Masuda Y, Miyatake K, Kangawa K. Hemodynamic, renal, and hormonal effects of adrenomedullin infusion in patients with congestive heart failure. Circulation. 2000;101:498–503. doi: 10.1161/01.cir.101.5.498. [DOI] [PubMed] [Google Scholar]
- Nishikimi T, Mori Y, Kobayashi N, Tadokoro K, Wang X, Akimoto K, Yoshihara F, Kangawa K, Matsuoka H. Renoprotective effect of chronic adrenomedullin infusion in Dahl salt-sensitive rats. Hypertension. 2002;39:1077–1082. doi: 10.1161/01.hyp.0000018910.74377.93. [DOI] [PubMed] [Google Scholar]
- Nishikimi T, Tadokoro K, Mori Y, Wang X, Akimoto K, Yoshihara F, Minamino N, Kangawa K, Matsuoka H. Ventricular adrenomedullin system in the transition from LVH to heart failure in rats. Hypertension. 2003a;41:512–518. doi: 10.1161/01.HYP.0000053447.64213.C4. [DOI] [PubMed] [Google Scholar]
- Nishikimi T, Yoshihara F, Horinaka S, Kobayashi N, Mori Y, Tadokoro K, Akimoto K, Minamino N, Kangawa K, Matsuoka H. Chronic administration of adrenomedullin attenuates transition from left ventricular hypertrophy to heart failure in rats. Hypertension. 2003b;42:1034–1041. doi: 10.1161/01.HYP.0000097604.64716.D2. [DOI] [PubMed] [Google Scholar]
- Nishikimi T, Karasawa T, Inaba C, Ishimura K, Tadokoro K, Koshikawa S, Yoshihara F, Nagaya N, Sakio H, Kangawa K, Matsuoka H. Effects of long-term intravenous administration of adrenomedullin (AM) plus hANP therapy in acute decompensated heart failure: a pilot study. Circ J. 2009;73:892–898. doi: 10.1253/circj.cj-08-0487. [DOI] [PubMed] [Google Scholar]
- Pennisi DJ, Ballard VL, Mikawa T. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dyn. 2003;228:161–172. doi: 10.1002/dvdy.10360. [DOI] [PubMed] [Google Scholar]
- Phillips MD, Mukhopadhyay M, Poscablo C, Westphal H. Dkk1 and Dkk2 regulate epicardial specification during mouse heart development. Int J Cardiol. 2011;150:186–192. doi: 10.1016/j.ijcard.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Cang Y, Goff SP. c-Abl tyrosine kinase regulates cardiac growth and development. Proc Natl Acad Sci U S A. 2010;107:1136–1141. doi: 10.1073/pnas.0913131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademaker MT, Charles CJ, Lewis LK, Yandle TG, Cooper GJ, Coy DH, Richards AM, Nicholls MG. Beneficial hemodynamic and renal effects of adrenomedullin in an ovine model of heart failure. Circulation. 1997;96:1983–1990. doi: 10.1161/01.cir.96.6.1983. [DOI] [PubMed] [Google Scholar]
- Rudat C, Kispert A. Wt1 and epicardial fate mapping. Circ Res. 2012;111:165–169. doi: 10.1161/CIRCRESAHA.112.273946. [DOI] [PubMed] [Google Scholar]
- Salvatore CA, Moore EL, Calamari A, Cook JJ, Michener MS, O'Malley S, Miller PJ, Sur C, Williams DL, Jr, Zeng Z, Danziger A, Lynch JJ, Regan CP, Fay JF, Tang YS, Li CC, Pudvah NT, White RB, Bell IM, Gallicchio SN, Graham SL, Selnick HG, Vacca JP, Kane SA. Pharmacological properties of MK-3207, a potent and orally active calcitonin gene-related peptide receptor antagonist. J Pharmacol Exp Ther. 2010;333:152–160. doi: 10.1124/jpet.109.163816. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosawa T, Shibagaki Y, Ishibashi K, Kitamura K, Kangawa K, Kato S, Ando K, Fujita T. Adrenomedullin, an endogenous peptide, counteracts cardiovascular damage. Circulation. 2002;105:106–111. doi: 10.1161/hc0102.101399. [DOI] [PubMed] [Google Scholar]
- Shindo T, Kurihara Y, Nishimatsu H, Moriyama N, Kakoki M, Wang Y, Imai Y, Ebihara A, Kuwaki T, Ju KH, Minamino N, Kangawa K, Ishikawa T, Fukuda M, Akimoto Y, Kawakami H, Imai T, Morita H, Yazaki Y, Nagai R, Hirata Y, Kurihara H. Vascular abnormalities and elevated blood pressure in mice lacking adrenomedullin gene. Circulation. 2001;104:1964–1971. doi: 10.1161/hc4101.097111. [DOI] [PubMed] [Google Scholar]
- Smart N, Bollini S, Dube KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, Pu WT, Riley PR. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier DY, Weinstein BM, Detrich HW, III, Zon LI, Fishman MC. Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development. 1995;121:3141–3150. doi: 10.1242/dev.121.10.3141. [DOI] [PubMed] [Google Scholar]
- Stuckmann I, Evans S, Lassar AB. Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev Biol. 2003;255:334–349. doi: 10.1016/s0012-1606(02)00078-7. [DOI] [PubMed] [Google Scholar]
- Sucov HM, Gu Y, Thomas S, Li P, Pashmforoush M. Epicardial control of myocardial proliferation and morphogenesis. Pediatr Cardiol. 2009;30:617–625. doi: 10.1007/s00246-009-9391-8. [DOI] [PubMed] [Google Scholar]
- Tomoda Y, Kikumoto K, Isumi Y, Katafuchi T, Tanaka A, Kangawa K, Dohi K, Minamino N. Cardiac fibroblasts are major production and target cells of adrenomedullin in the heart in vitro. Cardiovasc Res. 2001;49:721–730. doi: 10.1016/s0008-6363(00)00291-1. [DOI] [PubMed] [Google Scholar]
- Trivedi CM, Lu MM, Wang Q, Epstein JA. Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J Biol Chem. 2008;283:26484–26489. doi: 10.1074/jbc.M803686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PLoS One. 2012;7:e44692. doi: 10.1371/journal.pone.0044692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Hernandez M, Kovacs A, De Langhe S, Ornitz DM. FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development. 2011;138:3331–3340. doi: 10.1242/dev.064410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viragh S, Gittenberger-de Groot AC, Poelmann RE, Kalman F. Early development of quail heart epicardium and associated vascular and glandular structures. Anat Embryol (Berl) 1993;188:381–393. doi: 10.1007/BF00185947. [DOI] [PubMed] [Google Scholar]
- Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML. Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development. 1999;126:3597–3605. doi: 10.1242/dev.126.16.3597. [DOI] [PubMed] [Google Scholar]
- Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]