

Abstract
The binding between the HIV-1 trans-activator of transcription (Tat) and p300/(CREB-binding protein)-associated factor (PCAF) bromodomain is a crucial step in the HIV-1 life cycle. However, the structure of the full length acetylated Tat bound to PCAF has not been yet determined experimentally. Acetylation of Tat residues can play a critical role in enhancing HIV-1 transcriptional activation. Here, we have combined a fully flexible protein-protein docking approach with molecular dynamics simulations to predict the structural determinants of the complex for the common HIV-1BRU variant. This model reproduces all the crucial contacts between the Tat peptide 46SYGR(AcK)KRRQRC56 and the PCAF bromodomain previously reported by NMR spectroscopy. Additionally, inclusion of the entire Tat protein results in additional contact points at the protein-protein interface. The model is consistent with the available experimental data reported and adds novel information to our previous structural predictions of the PCAF bromodomain in complex with the rare HIVZ2 variant, which was obtained with a less accurate computational method. This improved characterization of Tat.PCAF bromodomain binding may help in defining the structural determinants of other protein interactions involving lysine acetylation.
Keywords: Tat, PCAF BRD, HIV-1, docking, protein-protein interaction
1. Introduction
The development of drugs against human immunodeficiency virus type 1 (HIV-1) was initiated about 30 years ago [1]. Since then, there have been 30 available antiretroviral drugs approved by the U.S. Food and Drug Administration (FDA). Current therapies against the AIDS pandemic are mainly based on inhibition of the three enzymes expressed by HIV-1 (protease, reverse transcriptase and integrase) [2,3,4,5]. Unfortunately, all of these approaches suffer from drug resistance due to the emergence of viral mutants [6]. In a rapid process of Darwinian evolution, the virus has developed functional enzyme variants not inhibited by current therapies/drugs. Several promising new strategies may, at least in part, counteract such viral hypervariability [7,8]. One of these strategies interferes with the HIV-1 transcriptional activity induced by the HIV-1 trans-activator of transcription (Tat) protein [9,10,11] (Figure 1a). Tat is a small and flexible protein, which acts as a molecular adaptor. Nuclear magnetic resonance (NMR) experiments reveal no secondary structure elements for free Tat in solution [12,13,14,15,16], although dramatic structural changes have been observed (both X-ray and NMR structures) when the protein is in complex with host cell partners [17,18]. The high plasticity of Tat is demonstrated by the fact that it can tolerate up to a 40% of sequence variation without loss of activity [19].
Figure 1.

Molecular representation of Tat.PCAF model and interactions. (a) HIV-1 Tat protein. The Tat protein lacks secondary structure elements [13]. Tat consists of six regions: Region I (in red) includes a proline-rich tract. It plays an important role in the entry of Tat into the cell [36]. Region II (in yellow) includes a cystein-rich tract. It is important for the transactivation ability of Tat [37]. Region III (in green) includes the motif RKGLGI. It is considered as the hydrophobic core of the protein and is conserved across HIV-1, HIV-2, and SIV Tat [38]. It does not have any known specific function [38]. Region IV (in orange) contains the basic domain (49RKKRRQRR56), arginine-rich motif, conserved across Tat proteins [39,40]. This region is required for binding to the viral RNA TAR (trans-activating response region) [41,42,43,44,45], as well as a variety of proteins, including PCAF BRD [18]. Region V (in violet) is a glutamine-rich region [46]. It is involved in microtubule polymerization and Tat-mediated apoptosis of T-cells [46]. Region VI (in blue, amino acid 73 to 86 or 101, depending on the virus strain) [10,11] includes the RGD motif that mediates Tat binding to cell surface integrins [47]. It is involved in cell adhesion [48]; (b) PCAF BRD. Cartoon representation of PCAF BRD NMR structure (PDBID 1JM4 [18]). It has a four-helix bundle (helices αZ, αA, αB, and αC); (c) Tat’s core domain and arginine-rich motif (ARM) interacting with PCAF BRD NMR structure [18]. PCAF BRD (orange) is represented in cartoon and traced with the residues in contact with Tat (green) in orange stick forms. Tat is represented in stick forms with oxygen and nitrogen atoms of AcK50 in red and blue, respectively.
Post-translational modifications—in particular acetylation of Tat residues can play a critical role in HIV replication, enhancing HIV-1 transcriptional activation [20,21,22].
Tat is acetylated at K50 by the p300/CPB co-activator and histone acetyltransferase (HAT) [10,11,21,23,24,25]. The p300/CPB protein contains several functional modules, including a nuclear receptor interaction domain (RID), a CREB and MYB interaction domain (KIX), cysteine/histidine regions (TAZ1/CH1 and TAZ2/CH3), an interferon response binding domain (IBiD), a histone acetyltransferase (PAT/HAT) domain [20,26,27,28,29], and a motif called the bromodomain (BRD). BRDs have recently been discovered to function as acetyl-lysine binding domains [30]. They feature a characteristic left-handed, four-helix bundle (helices αZ, αA, αB, and αC (Figure 1b). A large loop links helices αZ and αA (ZA loop), and a shorter loop links helices αB and αC (BC loop) [23]. Acetylated lysines bind between both loops by inserting the acetyl moiety along the longitudinal axis of the helices and between the four helices bundle [23,25,30]. In all the structures of BRDs in complex with cognate histone peptides so far determined [31], the acetyl moiety forms a hydrogen bond with a very conserved asparagine residue at the end of the cavity, serving as an anchor and providing specificity for acetylated versus non acetylated lysines [32]. Additional contacts with flanking amino acids in the target motif determine specificity/affinity for the large variety of BRD modules so far reported [31]. It is worth noticing that the binding regions belong to post-transcriptional modified histone tails, which are intrinsically disordered [33,34,35]. Therefore, it is not clear whether additional contacts may contribute to the recognition process.
Tat is also acetylated at K28 by the p300/CBP-associated factor (PCAF), thereby enhancing Tat binding to the protein P-TEFb [17]. This may in turn regulate functionally critical steps in transcription [20,21,22,49,50,51]. PCAF is a 90-kDa protein that contains the P300/SRC-1 domain (residues 1 to 352), the HAT domain (residues 518 to 649), which acetylates K28 [18,51] and a C-terminal BRD (residues 719-832) [18]. The BRD binds to the Tat protein, which is acetylated at lysine 50 (AcK50). Despite the fact that the structural determinants of full-length PCAF in complex with Tat have not yet been determined experimentally, the NMR structure of the complex between an acetylated Tat peptide (46SYGR(AcK)KRRQRC56) (Figure 1c) and the PCAF BRD has been solved by NMR spectroscopy [18].
The structural determinants of the full-length Tat PCAF BRD complex were predicted based on molecular dynamics simulations, combined with biochemical and FRET experiments [52]. This model was built using the structure of the isolated Z2 variant of the HIV-1 Tat [15]. In contrast to the existing structures of BRDs in complex with histone peptides, that model pointed to the presence of contacts between the BRD and non-adjacent regions of the acetylated partner. Furthermore, Tat Regions III and IV made extensive contacts with the ZA loop of the PCAF BRD (Figure 1). Although these contacts were not present in the NMR structure, which only contained a Tat peptide, they were experimentally validated [52], supporting the idea that significant changes in the protein-protein interface may take place in the presence of the entire Tat molecule.
Here we construct an updated structural model of the Tat PCAF BRD complex using a more accurate docking procedure. This is the fully flexible protein-protein technique as implemented in the data-driven docking program HADDOCK version 2.1 [53,54] followed by molecular dynamics (MD) simulations. Importantly, we focused on the 86 amino acids Tat BRU variant, PDBID 1JFW [13], a representative of HIV-1 subtype B. This variant is actually the more common HIV subtype in Europe and North America [12]. The Z2 variant of the protein used in our previous study is evolutionally closer to early strains of the virus, but is nowadays much less abundant [12]. Docking studies involving Tat and its cellular partners can be expected to be feasible. Indeed, (i) HADDOCK (High Ambiguity Driven protein-protein DOCKing) [53,54] has been used to predict the protein-protein and protein-nucleic acid complexes based on available experimental data [55,56,57]; (ii) HADDOCK has also been applied to intrinsically disordered proteins for which NMR structural information is available, such as Tat [50,51,52,53]; (iii) HADDOCK has been successfully applied to a variety of NMR structures with a low number of NOE’s per atom [54,55], similar to the ones we found in the available Tat NMR structures. Moreover, HADDOCK complemented by MD also provides reasonable results in those cases where the protein undergoes conformational changes [56,57,58]. Therefore by using the docking procedure combined with the MD simulations, we believe that a reasonable model of Tat can be assured.
2. Results and Discussion
2.1. Intermolecular Contacts
In our full-length Tat.PCAF BRD model, the interface between the BRU Tat and PCAF BRD complex (BRU Tat.PCAF hereafter) consists of Tat residues 47–55 and PCAF BRD residues 722–781. We focus on the contacts at the protein/protein interface. We first describe the hydrophobic contacts (HCs). Then we move our attention to intermolecular hydrogen bonds (HBs) and salt-bridges (SBs). The residues belonging to PCAF BRD are indicated in the following text with italics, while those of Tat with normal text.
AcK50 forms HCs with F748, V752, K753, Y760, I764, N798, Y802 and Y809 (Table 1 and Figure 2a). These results are in agreement with in vitro mutagenesis experiments in which the mutants to alanine of these PCAF residues strongly dismissed the Tat.PCAF BRD binding [18]. The role of Y760 in binding has been confirmed also in vivo by fluorescence resonance energy transfer (FRET) experiments of full-length acetylated Tat with PCAF BRD [52]. The mutant Y760D indeed impaired Tat binding to PCAF BRD (Table 1) [52]. Q54 establishes HCs with PCAF E756. Both residues are located at the edge of a hydrophobic cavity of Tat (Figure 2b).
Table 1.
Effect of mutations in in vitro experiment for Tat46SYGR(AcK)KRRQRC56 (synthesis) complex with p300/(CREB-binding protein)-associated factor (PCAF) bromodomain (BRD) [18] or in vivo experiment for full-length acetylated Z2 Tat with PCAF BRD [52]. The corresponding contacts in both BRU Tat.PCAF docking and molecular dynamics (MD) models presented in this work are reported in the last columns (black = agreement; red = new contacts). Coverage in the last column is the percentage of the occurrence of hydrophobic contacts (HCs) over all frames in MD simulation.
| Effect on Tat.PCAF binding | Mutants | In contact with | BRU Tat.PCAF model (docking) | BRU Tat.PCAF model (MD) | Coverage (%) |
|---|---|---|---|---|---|
| inhibiting binding [18] | AcK50A | F748, V752, Y760, I764 | F748, V752, Y760, I764 | F748, V752, Y760 | 100% |
| Y802 | Y802 | Y802 | 80% | ||
| Y809 | Y809 | Absent | |||
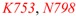 |
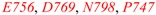 |
100% | |||
| inhibiting binding [18] | Y47A | V763 | Absent | Absent | |
| V763 | Y47 | P804, S807 | N803 | 80% | |
| E808 | E808 | 100% | |||
| Y809 | 80% | ||||
| Q54A | E756 | E756 | E756 | 80% | |
| E756A | Q54 | 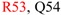 |
|
80% | |
| strongly diminishing binding [18] | R53E | E756 |  |
|
100% |
| F748A | Tat46SYGR(AcK)KRRQRC56 | AcK50 | AcK50 | 100% | |
| V752A | Tat46SYGR(AcK)KRRQRC56 | AcK50, R51 | AcK50, R51 | 100% | |
| Y802A | Tat46SYGR(AcK)KRRQRC56 | AcK50 | AcK50 | 100% | |
| Y809A | Tat46SYGR(AcK)KRRQRC56 | AcK50, S46 | S46 | 100% | |
| Diminishing binding [18] | R49A | PCAF BRD | P747, E750 | W746, P747, E750, Y802 | 100% |
| F748 | 80% | ||||
| K51A | PCAF BRD | E750, V752, K753 | F748, E750, V752, E756 | 100% | |
| K753 | 50% | ||||
| R52A | PCAF BRD | K753 | K753 | 25% | |
| E756 | E756 | 100% | |||
| Diminishing binding [52] | Y760D | AcK50 | AcK50 | AcK50 | 100% |
| Y761D | AcK50 | AcK50 | Absent | ||
| no effect [18] | W746A, D769A, C799A, N803A | R49 | 100% | ||
| E750A | R49 | R49 | 100% | ||
| T755A | R53 | R53 | 100% | ||
| I764A | AcK50 | AcK50 | 100% | ||
| N798A | AcK50 | AcK50 | 100% |
Figure 2.

BRU Tat.PCAF BRD interface (a) HCs between R49, AcK50, R51 of BRU Tat (green) and PCAF BRD (orange) contacts at the core protein/protein interface are reported. Residues at the interaction interface are represented in stick forms; (b) The HCs between BRU Tat (green) and PCAF BRD (orange) contacts at the edge protein/protein interface are reported. Residues at the interaction interface are represented in stick forms; (c) The position of Y47 (shown with balls) and V763 (shown with sticks) represented in the complex of BRU Tat.PCAF model were superimposed with the same residues in Tat46SYGR(AcK)KRRQRC56.PCAF (black) NMR structure [18] (in green and orange, respectively); (d) A schematic view of the HCs between BRU Tat and PCAF BRD. The red dotted lines are the HCs newly found in BRU Tat.PCAF model; (e) The new HBs in BRU Tat.PCAF model (in green and orange, respectively) with oxygen and nitrogen atoms in red and blue, respectively; (f) salt-bridges (SBs) between BRU Tat (green) and PCAF (orange) between R53–E756, R49–E750, K51–E750.
R49 forms HCs with P747, E750; K51 with E750, V752, K753; R52 with K753, E756 (Figure 2a); and Y47 with P804, S807, E808 and Y809 (Figure 2b). These results are also consistent with in vitro mutagenesis experiments [18]; alanine substitution of residues R49, K51, R52, or R53 slightly weaken Tat’s binding to the BRD [18]. Similarly, the mutation of Y47 and Q54 to alanine hampers PCAF BRD binding to the Tat46SYGR(AcK)KRRQRC56 peptide. Most of these HCs are present in the Tat46SYGR(AcK)KRRQRC56.PCAF NMR structure [18] (Table 1). Additionally, our model predicts interactions between AcK50–K753, AcK50–N798 (Figure 2a) and S46–Y809 (Figure 2b); the last predicted interaction is possibly because residues flanking both sides of S46 contribute to the complex formation (Figure 2b). In addition, the V763–Y47 contact is present in the NMR structure only and absent in our model (Figure 2c). This is due to the fact that the formations of new HCs at the N-terminus affect the position of Y47 in the BRU Tat.PCAF model. The new HCs include Tat E2 to PCAF E756, A757, P758; P3 to P758; S46 to P804, Y809. As a result, the RMSD of Tat Y47 exhibits a difference of 4.8 Å with respect to the full length Tat.
A schematic view of the HCs between Tat and PCAF BRD is shown in Figure 2d.
Tat AcK50 forms HBs with V752, F748, N798, Y802 and Y809. Tat R53 and Tat Q54 make HBs with PCAF E756 (Figure 2e) (Table 2). These HBs are also present in the Tat46SYGR(AcK)KRRQRC56.PCAF NMR structure [18] (Figure 2e) and are consistent with our model of Z2 Tat.PCAF [52].
Table 2.
Selected intermolecular hydrogen bonds (HBs) in the BRU Tat.PCAF model by docking.
| Donors | Acceptors |
|---|---|
| AcK50 | V752 |
| AcK50 | F748 |
| AcK50 | Y809 |
| AcK50 | Y802 |
| N798 | AcK50 |
| R49 * | P747 * |
| R49 * | E750 * |
| R53 | E756 |
| R53 | K753 |
| R55 | E756 |
| Q54 | E756 |
| E2 * | P758 * |
| P3 * | P758 * |
* The residues form new HBs in the BRU Tat.PCAF model.
New HBs between R49 and E750, P747 were found in our model (Table 2) at the interface. In the N-terminal of Tat, E2 and P3 make a HB to PCAF P758.
Finally, the R53–E756, R49–E750, K51–E750 SBs are present (Figure 2f) in the BRU Tat.PCAF BRD model. These residues are all at the interface of Tat and PCAF with the high density of basic/acidic residues. Only the first SB was present in Tat46SYGR(AcK)KRRQRC56.PCAF NMR structure [18]. This SB was also present in our previous model [52].
The five representative clusters of Tat.PCAF complex from MD simulation are compared with the docked model and the NMR structure. The HCs are shown in Table 1. The structures obtained by docking and those obtained by MD are not too dissimilar (Table 1 and Table 2). In particular, some hydrophobic contacts (AcK50–E756, AcK50–D769, Ack50–P747, R49–W746, R49–F748, R49–Y802, K51–K753, K51–E756) are present in the MD structures but absent in the docked structures. None of the intermolecular HBs with a coverage of more than 50% over the frames is retained in MD. The models generated by data-driven docking and MD show that the hydrophobic interactions may play a leading role in Tat.PCAF BDB complex stabilization.
2.2. Structural Rearrangements of Tat and PCAF upon Binding
In the Tat46SYGR(AcK)KRRQRC56.PCAF BRD complex [18], the binding interface between Tat and PCAF BRD is composed of Tat residues 47–55 and PCAF BRD residues 722–781. The inclusion of the complete Tat protein in our model imposes structural constraints that result in conformational differences, which, however, do not significantly alter the identity of the contacts.
The structural changes mostly involve the Tat BRU variant: The RMSD of the backbone atoms between the Tat47YGR(AcK)KRRQR55 of our model and the NMR structure of Tat46SYGR(AcK)KRRQRC56.PCAF BRD [18] is as large as 4 Å. The RMSD of the 47YGRKKRRQR55segment relative to the Tat46SYGR(AcK)KRRQRC56.PCAF BRD NMR complex [18] ranges from 2.6 Å to 3.4 Å. A similar result is also found after MD simulations. Indeed, the binding regions (Tat47−55 and PCAF722−781 BRD) rearrange 3.1 Å and 2.6 Å, respectively, compared to the same residues on NMR structures, while the whole complex shows remarkable changes (RMSD 24.5 Å). This indicates that the binding region rearranges less than the rest of the protein.
Dramatic changes were instead observed in the N-terminal segment on Tat upon binding with the P-TEFb complex (see Supplementary Section S5 for more details [17]).
Notably, the RMSD between the present model (using the BRU variant) with that previously reported by us for the Z2 variant [15] (which has 79.1% sequence identity with BRU Tat, see Supplementary Figure S1) is as much as 9.5 Å. The main differences involve the N-termini, with a RMSD of 17 Å dissimilarity between the two structures, and the C-termini which differ by as much as 14 Å; the Tat46SYGR(AcK)KRRQRC56 is more conserved, with 3.3 Å dissimilarity. The AcK50 in BRU and Z2 Tat.PCAF BRD model was outside the Tat loop such as K50 in Tat full-length BRU variant in unbound conformers. In Z2 Tat full-length, the K50 instead turns inside the loop (See Supplementary Figure S1b). We further notice that in spite of their differences in sequence and overall structure (See Supplementary Figure S1b), the N-termini of Z2 and BRU Tat form similar contacts with PCAF BRD and residues in the N-terminal extremity of Tat. Different segments for the Z2 and BRU variants of Tat are conserved in terms of the intermolecular contacts with PCAF [12], as described in Section 2.1.
The PCAF BRD structure remains close to the protein in the free state in solution: The RMSD of the backbone relative to that of the NMR structure [18] is only 0.8 Å (Table 1).
We further notice that docking of the Tat46SYGR(AcK)KRRQRC56 peptide onto the PCAF BRD (see Methods Section and Supplementary Section S3) resulted in structural deviations differing by 0.9 Å and 1.3 Å for each of the components and 1.7 Å for the whole complex (Table 3). These values are well within the range of variation of RMSD reported for the NMR structure described in Table 3. This may suggest that our docking procedure is rather accurate.
Table 3.
Calculated backbone RMSD values between the BRU Tat.PCAF docking and MD models and the Tat47YGR(AcK)KRRQR55.PCAF NMR structure (conformer 8 [18], see Methods for details). The abbreviations min., max., avg., sd. stand for minimum, maximum, average, standard deviation of RMSD values, respectively.
| Systems | Backbone RMSD (Å):PCAF 722–781 | Backbone RMSD (Å): Tat47YGR(AcK)KRRQR55 | Backbone RMSD (Å): Full complex (same residues) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| min. | max. | avg. | min. | max. | avg. | min. | max. | avg. | |
| Tat47YGR(AcK)KRRQR55.PCAF*(1JM4) | 0.6 | 1.5 | 1.0 | 0.7 | 1.2 | 2.3 | 0.9 | 1.9 | 1.33 |
| sd. 0.2 | sd. 0.5 | sd. 0.26 | |||||||
| Tat47YGR(AcK)KRRQR55.PCAF (Prediction) | 1.3 | 0.9 | 1.7 | ||||||
| BRU Tat.PCAF (Prediction by docking) | 0.8 | 4.0 | 14.4 | ||||||
| BRU Tat.PCAF (Prediction by MD simulation, the last frame) | 2.9 3.3 3.1 sd. 0.2 | 2.8 3.4 2.6 sd. 0.4 | 24.7 25.8 24.5 sd. 1.2 | ||||||
2.3. Key Residues of PCAF BRD for Its Interaction with Tat
The sequence and structure conservation of Tat has been discussed by us in [12]. Here we focus on residues of PCAF human BRD important for Tat binding (See multiple sequence alignment of PCAF human BRD in Supplementary Figure S2). There are 53 PCAF human bromodomain (BRD) sequences as obtained from bromodomain PROSITE family, accession number PS50014 [59,60,61]. We start our discussion by listing the most conserved residues of this protein family (Table 4). In particular, P758, which makes HBs with E2 and P3 of Tat, is highly conserved among PCAF BRD sequences (71% identity, Table 4) and P747 (identity 53% respect to PCAF BRD sequences, Table 4 and Supplementary Figure S2). Notably, the N798, which makes an HB with AcK50, is one of the most conserved residues in PCAF BRD (96%, Table 4). P747 and N798 residues, which are involved in several HCs and HBs (Table 1 and Table 2 and Supplementary Tables S3 and S4), seem to play an important role for the Tat.PCAF interaction. Moreover, due to the overall structure similarities of PCAF BRD [62], the N798 may play an important role in the interaction of PCAF BRD with other cellular partners [63].
Table 4.
Conserved residues of the existing 53 PCAF human bromodomain (BRD) sequences as obtained from the bromodomain PROSITE family, accession number PS50014 [59,60,61].
| Residues | Identity (%) | Residues | Identity (%) |
|---|---|---|---|
| P747 | 53% | T772 | 69% |
| F748 | 91% | Y782 | 97% |
| P751 | 68% | F788 | 63% |
| V752 | 71% | D791 | 89% |
| P758 | 71% | F796 | 60% |
| I764 | 76% | N798 | 96% |
| P767 | 97% | Y802 | 77% |
| D769 | 85% | N803 | 88% |
| L770 | 66% |
3. Computational Methods Section
The structure of HIV-1 Tat (86 residues) has been solved by NMR (PDBID 1JFW [13]). Eleven conformers have been deposited. His13, His33, His65 are protonated in the Nε [13]. We superimposed residues 47 to 55 in each of these structures with the corresponding ones with the multiple conformers of Tat46SYGR(AcK)KRRQRC56 peptide in complex with human PCAF BRD, whose structure has been solved by NMR. His717 and His742 were protonated in Nδ as in the NMR structure (1JM4 [18]). The VMD 1.8 program [64] was used. The conformer n.8 of HIV-1 Tat and n.3 of the Tat46SYGR(AcK)KRRQRC56.PCAF complex exhibits the lowest pairwise root-mean-square deviation (RMSD) in respect Tat46SYGR(AcK)KRRQRC56 backbone (2.3 Å and 0.7 Å, respectively). All the RMSDs reported in this paper are calculated using only backbone atoms. Thus, these conformers were selected for all subsequent calculations. The full length Tat was fitted to Tat46SYGR(AcK)KRRQRC56 at the peptide then AcK50 was replaced with K50 in full length Tat using the tleap module in AMBER package [65] with acetylated lysine parameters adapted from Machado et al. [66]. The resulting structure was relaxed for 10 ns molecular dynamics simulation in water (See Supplementary Section S1; RMSF, RMSD of the system plotted as a function of simulated time in Supplementary Figure S3 and Figure S4 for further details). A clustering of the MD trajectory [67] identified seven clusters, 84 structures in total, which represented 98% of the conformations (Supplementary Section S2). These structures were docked on the 25 PCAF BRD conformers present in the NMR family of structures (1JM4 [18]) by a multiple conformations docking procedure using the HADDOCK 2.1 program [53,54,68]. Haddock is an information-driven flexible docking approach for the modeling of biomolecular complexes [40]. The entire protocol consists of four stages: (i) Topology and structure generation; (ii) Randomization of the starting orientation and rigid body energy minimization; (iii) Semi-flexible simulated annealing; (iv) Flexible refinement in explicit solvent (water).
We first validated our docking procedure by redocking the Tat46SYGR(AcK)KRRQRC56 from PCAF BRD in the NMR structure of the complex (Supplementary Section S3) [18]. Then, the two binding partners were docked one onto the other by rigid body docking with different Ambiguous Interaction Restraints (AIRs) (Supplementary Section S3, Table S2, for the definition of AIRs, see [54]). The adduct exhibited a backbone RMSD of 1.3 Å for PCAF BRD and 1.7 Å for HIV-1 Tat46SYGR(AcK)KRRQRC56 with respect to the NMR structure (Supplementary Section S3, Figure S4) (Table 3). These results allow us to suggest that HADDOCK performs well for this system (Supplementary Section S3). Next, we performed rigid-body docking between PCAF BRD and Tat AcK50. The contacts F748–, V752–, Y809–, I764–, Y760–, Y802– to AcK50, E756– to R53, V763– to Y47, E756– to Q54, present in the NMR structure [18], were treated as AIRs. The rigid-body docking was followed by a rigid body energy minimization, a semi-flexible simulated annealing in torsion angle space and a refinement by MD at a temperature of 300K in explicit solvent [54,69]. Next we selected the 500 top structures in terms of the highest docking score and the lowest RMSD relative to the NMR structure [18]. In these 500 structures, a short MD simulation was performed to optimize the water positions while proteins were fixed (4,000 steps of MD consisting four times 1,000 steps at a temperature of 600, 500, 400 and 300 K, respectively [69]. This was followed by MD at 300K in water [54]. A clusterization procedure was performed with a cut-off at 3.0 Å from the final 500 structures (94% structures were clusterized). We finally selected the structure that had the lowest RMSD (0.8 Å for PCAF and 4 Å for Tat), which is representative for the most crowded cluster. This is the structure discussed in the Results and Discussion Section. Other representative structures are discussed in Supplementary Table S3.
The representative of the Tat.PCAF model from docking underwent MD simulations in explicit water. Tat.PCAF BRD was solvated in 104 × 70 × 66 Å3 box, containing 39.363 water molecules, 16 chlorine ions were added to neutralize the total charge of the system. The AMBER force field ff99SB [70] was used for the protein and the chlorine atoms; the TIP3P model [71] was used for water. The acetylated lysine topology parameter was taken from Machado et al. [66].
Periodic boundary conditions were used and a cutoff of 12 Å [72] was adopted for short-range non-bonded interactions. The particle mesh Ewald summation method [73] was used for long-range electrostatic interactions. A dielectric constant of one was assumed. All chemical bonds were constrained by using the SHAKE algorithm [74]. The equations of motion were integrated using a time step of 2 fs. Temperature and pressure were kept constant at 300 K and 1 atm by coupling the system to external baths [75] with a Langevin thermostat (coupling constants st = 0.05) and a Berendsen barostat (coupling constants sp = 0.5 ps), respectively.
After 5,000 steps of minimization (steepest descent algorithm for the first 1,500 steps before switching to the conjugate gradient algorithm for the remaining 3,500 steps), the system was equilibrated for 300 ps. Eighty nanoseconds of MD simulation were performed at 300 K and 2 ps for the time steps. We clusterized [67] the trajectory into five clusters. The backbone RMSD of the Tat.PCAF BRD structures over 80 ns is shown in Supplementary Figure S7. NAMD package was used for MD simulation [76].
Hydrophobic contacts at the protein/protein contact surface were defined as two non-polar carbon atoms at a distance of 4.0 Å or less. Hydrogen bonds were defined as a polar hydrogen at a distance of 2.5 Å or less from a polar atom (nitrogen or oxygen). Salt bridges were considered formed if the distance between any of the oxygen atoms of acidic residues and the nitrogen atoms of basic residues were less than 4.0 Å. Coverage was defined as the percentage of the occurrence of HCs over all frames in MD simulation or all representative structures in docking. The HADDOCK [53,54], LIGPLOT [77] and VMD [64] programs were used to calculate these quantities.
4. Conclusions
We have presented a model of the BRU Tat.PCAF complex produced by data-driven docking combined with MD simulations, which may add structural information to the intricate mechanism of Tat transcription and trans-activation, up to now not fully understood [10,11]. The structures obtained by docking and those obtained by MD are similar in the binding region. This model was obtained with a more accurate procedure than that used in [52] and it focuses on the more common BRU variant rather than the Z2 strain of HIV-1. The two models show conservation as far as the Tat46SYGR(AcK)KRRQRC56 and N-terminal contacts with PCAF BRD are concerned. In addition, the model suggests the presence, only for the Z2 strain of Tat, of hydrophobic contacts and hydrogen bonds between Tat and PCAF that are not present in the previous model of Z2 Tat.PCAF [52]. Specifically, the BRU Tat.PCAF model differs from the Z2 Tat.PCAF complex on HBs between R49 and P747, E750, between P758 and E2, P3 and on HCs between S46 and P804, Y809.
The model appears to be consistent with most of the experimental data obtained with Tat46SYGR(AcK)KRRQRC56.PCAF in vitro [18] (synthesized Tat peptide) and full length Tat.PCAF in vivo [52]. However, this comparison has to be interpreted with caution because of the assumption that most of the intermolecular interactions compared here do not vary when passing from the in vitro to the in vivo situation and/or from the Tat46SYGR(AcK)KRRQRC56 to the full length protein.
Several BRD amino acids have been identified as crucial for Tat binding (E750, V752, E756, A757, Y802, Y809) and as the targets for development of small molecules blocking Tat.PCAF association [78,79]. Our model allows us to suggest that the hydrophobic interactions seem to be a crucial factor for BRD binding to Tat.
Further, our model allows us to suggest that N798 in the BRD plays a key role for Tat binding, independently of whether this protein is expressed by the BRU or the ZZ variant of HIV-1. Indeed, this residue creates a HB with AcK50 in the X-ray structure of Tat and other cellular partners [41,42,49], in both the Z2 Tat.PCAF model [52] and in our model. Hence, interfering with N798-AcK50 interactions may be a valuable strategy for drug design [63,80]. Because this residue is highly conserved (Supplementary Figure S3 and Table 4), it may form key interactions also with other BRD cellular partners [18,31,32,62,81]. Moreover, some studies showed that specific small molecules binding at the acetylated lysine recognition pocket can antagonize the bromodomains and acetylate histone interactions, especially the HB between asparagine in αB loop and acetylated lysine [30,80,82,83,84,85,86,87,88,89]. Therefore, designing compounds, which bind to N798 may also lead to other potential therapeutic applications.
Acknowledgments
This work was supported by the BANDO AIDS Grant from the Italian Government for “Targeting HIV transcription to control infection and to purge post-integrative latency”. SP is supported by ANII—Agencia Nacional de Investigación e Innovación, Programa de Apoyo Sectorial a la Estrategia Nacional de Innovación—INNOVA URUGUAY (Agreement n8 DCI–ALA/2007/19.040 between Uruguay and the European Commission).
Supplementary Files
PDF-Document (PDF, 4505 KB)
References
- 1.Flexner C. HIV drug development: The next 25 years. Nat. Rev. Drug Discov. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]
- 2.Wainberg M.A., Zaharatos G.J., Brenner B.G. Development of antiretroviral drug resistance. N. Engl. J. Med. 2011;365:637–646. doi: 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- 3.Detels R. The search for protection against HIV infection. Ann. Epidemiol. 2009;19:250–252. doi: 10.1016/j.annepidem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Mawsawi L.Q., Al-Safi R.I., Neamati N. Anti-infectives: Clinical progress of HIV-1 integrase inhibitors. Expert. Opin. Emerg. Drugs. 2008;13:213–225. doi: 10.1517/14728214.13.2.213. [DOI] [PubMed] [Google Scholar]
- 5.Menendez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: An update. Antivir. Res. 2010;85:210–231. doi: 10.1016/j.antiviral.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Menendez-Arias L. Targeting HIV: Antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002;23:381–388. doi: 10.1016/S0165-6147(02)02054-0. [DOI] [PubMed] [Google Scholar]
- 7.Al-Mawsawi L.Q., Neamati N. Blocking interactions between HIV-1 integrase and cellular cofactors: An emerging anti-retroviral strategy. Trends Pharmacol. Sci. 2007;28:526–535. doi: 10.1016/j.tips.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Adamson C.S., Freed E.O. Novel approaches to inhibiting HIV-1 replication. Antivir. Res. 2010;85:119–141. doi: 10.1016/j.antiviral.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ensoli B., Fiorelli V., Ensoli F., Cafaro A., Titti F., Butto S., Monini P., Magnani M., Caputo A., Garaci E. Candidate HIV-1 Tat vaccine development: From basic science to clinical trials. AIDS. 2006;20:2245–2261. doi: 10.1097/QAD.0b013e3280112cd1. [DOI] [PubMed] [Google Scholar]
- 10.Gatignol A. Transcription of HIV: Tat and cellular chromatin. Adv. Pharmacol. 2007;55:137–159. doi: 10.1016/S1054-3589(07)55004-0. [DOI] [PubMed] [Google Scholar]
- 11.Johri M.K., Mishra R., Chhatbar C., Unni S.K., Singh S.K. Tits and bits of HIV Tat protein. Expert. Opin. Biol. Ther. 2011;11:269–283. doi: 10.1517/14712598.2011.546339. [DOI] [PubMed] [Google Scholar]
- 12.Pantano S., Carloni P. Comparative analysis of HIV-1 Tat variants. Proteins. 2005;58:638–643. doi: 10.1002/prot.20323. [DOI] [PubMed] [Google Scholar]
- 13.Peloponese J.M., Jr., Gregoire C., Opi S., Esquieu D., Sturgis J., Lebrun E., Meurs E., Collette Y., Olive D., Aubertin A.M., et al. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C. R. Acad. Sci. III. 2000;323:883–894. doi: 10.1016/S0764-4469(00)01228-2. [DOI] [PubMed] [Google Scholar]
- 14.Gregoire C., Peloponese J.M., Jr., Esquieu D., Opi S., Campbell G., Solomiac M., Lebrun E., Lebreton J., Loret E.P. Homonuclear (1)H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers. 2001;62:324–335. doi: 10.1002/bip.10000. [DOI] [PubMed] [Google Scholar]
- 15.Bayer P., Kraft M., Ejchart A., Westendorp M., Frank R., Rosch P. Structural studies of HIV-1 Tat protein. J. Mol. Biol. 1995;247:529–535. doi: 10.1006/jmbi.1995.0158. [DOI] [PubMed] [Google Scholar]
- 16.Foucault M., Mayol K., Receveur-Brechot V., Bussat M.C., Klinguer-Hamour C., Verrier B., Beck A., Haser R., Gouet P., Guillon C. UV and X-ray structural studies of a 101-residue long Tat protein from a HIV-1 primary isolate and of its mutated, detoxified, vaccine candidate. Proteins. 2010;78:1441–1456. doi: 10.1002/prot.22661. [DOI] [PubMed] [Google Scholar]
- 17.Tahirov T.H., Babayeva N.D., Varzavand K., Cooper J.J., Sedore S.C., Price D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–751. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mujtaba S., He Y., Zeng L., Farooq A., Carlson J.E., Ott M., Verdin E., Zhou M.M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell. 2002;9:575–586. doi: 10.1016/S1097-2765(02)00483-5. [DOI] [PubMed] [Google Scholar]
- 19.Opi S., Peloponese J.M., Jr., Esquieu D., Campbell G., de Mareuil J., Walburger A., Solomiac M., Gregoire C., Bouveret E., Yirrell D.L., et al. Tat HIV-1 primary and tertiary structures critical to immune response against non-homologous variants. J. Biol. Chem. 2002;277:35915–35919. doi: 10.1074/jbc.M204393200. [DOI] [PubMed] [Google Scholar]
- 20.Ott M., Schnolzer M., Garnica J., Fischle W., Emiliani S., Rackwitz H.R., Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- 21.Kiernan R.E., Vanhulle C., Schiltz L., Adam E., Xiao H., Maudoux F., Calomme C., Burny A., Nakatani Y., Jeang K.T., et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakatani Y. HIV-1 transcription: Activation mediated by acetylation of Tat. Structure. 2002;10:443–444. doi: 10.1016/S0969-2126(02)00754-2. [DOI] [PubMed] [Google Scholar]
- 23.Dhalluin C., Carlson J.E., Zeng L., He C., Aggarwal A.K., Zhou M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 24.Shikama N., Chan H.M., Krstic-Demonacos M., Smith L., Lee C.W., Cairns W., La Thangue N.B. Functional interaction between nucleosome assembly proteins and p300/CREB-binding protein family coactivators. Mol. Cell. Biol. 2000;20:8933–8943. doi: 10.1128/MCB.20.23.8933-8943.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teufel D.P., Freund S.M., Bycroft M., Fersht A.R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7009–7014. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng L., de la Fuente C., Fu P., Wang L., Donnelly R., Wade J.D., Lambert P., Li H., Lee C.G., Kashanchi F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology. 2000;277:278–295. doi: 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- 27.Benkirane M., Chun R.F., Xiao H., Ogryzko V.V., Howard B.H., Nakatani Y., Jeang K.T. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998;273:24898–24905. doi: 10.1074/jbc.273.38.24898. [DOI] [PubMed] [Google Scholar]
- 28.Deng L., Wang D., de la Fuente C., Wang L., Li H., Lee C.G., Donnelly R., Wade J.D., Lambert P., Kashanchi F. Enhancement of the p300 HAT activity by HIV-1 Tat on chromatin DNA. Virology. 2001;289:312–326. doi: 10.1006/viro.2001.1129. [DOI] [PubMed] [Google Scholar]
- 29.Lusic M., Marcello A., Cereseto A., Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22:6550–6561. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng L., Zhou M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/S0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 31.Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Muller S., Pawson T., et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeng L., Zhang Q., Gerona-Navarro G., Moshkina N., Zhou M.M. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure. 2008;16:643–652. doi: 10.1016/j.str.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 34.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 35.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 36.Kuppuswamy M., Subramanian T., Srinivasan A., Chinnadurai G. Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 1989;17:3551–3561. doi: 10.1093/nar/17.9.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia J.A., Harrich D., Pearson L., Mitsuyasu R., Gaynor R.B. Functional domains required for tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J. 1988;7:3143–3147. doi: 10.1002/j.1460-2075.1988.tb03181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeang K.T., Xiao H., Rich E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 39.Dorn P., DaSilva L., Martarano L., Derse D. Equine infectious anemia virus tat: Insights into the structure, function, and evolution of lentivirus trans-activator proteins. J. Virol. 1990;64:1616–1624. doi: 10.1128/jvi.64.4.1616-1624.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noiman S., Yaniv A., Tsach T., Miki T., Tronick S.R., Gazit A. The Tat protein of equine infectious anemia virus is encoded by at least three types of transcripts. Virology. 1991;184:521–530. doi: 10.1016/0042-6822(91)90422-8. [DOI] [PubMed] [Google Scholar]
- 41.Roy S., Katze M.G., Parkin N.T., Edery I., Hovanessian A.G., Sonenberg N. Control of the interferon-induced 68-kilodalton protein kinase by the HIV-1 tat gene product. Science. 1990;247:1216–1219. doi: 10.1126/science.2180064. [DOI] [PubMed] [Google Scholar]
- 42.Churcher M.J., Lamont C., Hamy F., Dingwall C., Green S.M., Lowe A.D., Butler J.G., Gait M.J., Karn J. High affinity binding of TAR RNA by the human immunodeficiency virus type-1 tat protein requires base-pairs in the RNA stem and amino acid residues flanking the basic region. J. Mol. Biol. 1993;230:90–110. doi: 10.1006/jmbi.1993.1128. [DOI] [PubMed] [Google Scholar]
- 43.Dingwall C., Ernberg I., Gait M.J., Green S.M., Heaphy S., Karn J., Lowe A.D., Singh M., Skinner M.A., Valerio R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 1989;86:6925–6929. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weeks K.M., Crothers D.M. RNA recognition by Tat-derived peptides: Interaction in the major groove? Cell. 1991;66:577–588. doi: 10.1016/0092-8674(81)90020-9. [DOI] [PubMed] [Google Scholar]
- 45.Chang Y.N., Jeang K.T. The basic RNA-binding domain of HIV-2 Tat contributes to preferential trans-activation of a TAR2-containing LTR. Nucleic Acids Res. 1992;20:5465–5472. doi: 10.1093/nar/20.20.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Mareuil J., Carre M., Barbier P., Campbell G.R., Lancelot S., Opi S., Esquieu D., Watkins J.D., Prevot C., Braguer D., et al. HIV-1 Tat protein enhances microtubule polymerization. Retrovirology. 2005;2:5. doi: 10.1186/1742-4690-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiorelli V., Barillari G., Toschi E., Sgadari C., Monini P., Sturzl M., Ensoli B. IFN-gamma induces endothelial cells to proliferate and to invade the extracellular matrix in response to the HIV-1 Tat protein: Implications for AIDS-Kaposi's sarcoma pathogenesis. J. Immunol. 1999;162:1165–1170. [PubMed] [Google Scholar]
- 48.Barillari G., Gendelman R., Gallo R.C., Ensoli B. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7941–7945. doi: 10.1073/pnas.90.17.7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaehlcke K., Dorr A., Hetzer-Egger C., Kiermer V., Henklein P., Schnoelzer M., Loret E., Cole P.A., Verdin E., Ott M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol. Cell. 2003;12:167–176. doi: 10.1016/s1097-2765(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 50.Bres V., Tagami H., Peloponese J.M., Loret E., Jeang K.T., Nakatani Y., Emiliani S., Benkirane M., Kiernan R.E. Differential acetylation of Tat coordinates its interaction with the co-activators cyclin T1 and PCAF. EMBO J. 2002;21:6811–6819. doi: 10.1093/emboj/cdf669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorr A., Kiermer V., Pedal A., Rackwitz H.R., Henklein P., Schubert U., Zhou M.M., Verdin E., Ott M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002;21:2715–2723. doi: 10.1093/emboj/21.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pantano S., Marcello A., Ferrari A., Gaudiosi D., Sabo A., Pellegrini V., Beltram F., Giacca M., Carloni P. Insights on HIV-1 Tat:P/CAF bromodomain molecular recognition from in vivo experiments and molecular dynamics simulations. Proteins. 2006;62:1062–1073. doi: 10.1002/prot.20805. [DOI] [PubMed] [Google Scholar]
- 53.de Vries S.J., van Dijk A.D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 54.Dominguez C., Boelens R., Bonvin A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 55.Lowry J.A., Gamsjaeger R., Thong S.Y., Hung W., Kwan A.H., Broitman-Maduro G., Matthews J.M., Maduro M., Mackay J.P. Structural analysis of MED-1 reveals unexpected diversity in the mechanism of DNA recognition by GATA-type zinc finger domains. J. Biol. Chem. 2009;284:5827–5835. doi: 10.1074/jbc.M808712200. [DOI] [PubMed] [Google Scholar]
- 56.Gelis I., Bonvin A.M., Keramisanou D., Koukaki M., Gouridis G., Karamanou S., Economou A., Kalodimos C.G. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J., Hu W., Cai S., Lee B., Song J., Chen Y. The intrinsic affinity between E2 and the Cys domain of E1 in ubiquitin-like modifications. Mol. Cell. 2007;27:228–237. doi: 10.1016/j.molcel.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lowry J.A., Gamsjaeger R., Thong S.Y., Hung W., Kwan A.H., Broitman-Maduro G., Matthews J.M., Maduro M., Mackay J.P. Structural analysis of MED-1 reveals unexpected diversity in the mechanism of DNA recognition by GATA-type zinc finger domains. J. Biol. Chem. 2009;284:5827–5835. doi: 10.1074/jbc.M808712200. [DOI] [PubMed] [Google Scholar]
- 59.Haynes S.R., Dollard C., Winston F., Beck S., Trowsdale J., Dawid I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992;20:2603. doi: 10.1093/nar/20.10.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tamkun J.W. The role of brahma and related proteins in transcription and development. Curr. Opin. Genet. Dev. 1995;5:473–477. doi: 10.1016/0959-437X(95)90051-H. [DOI] [PubMed] [Google Scholar]
- 61.Tamkun J.W., Deuring R., Scott M.P., Kissinger M., Pattatucci A.M., Kaufman T.C., Kennison J.A. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-E. [DOI] [PubMed] [Google Scholar]
- 62.Umehara T., Nakamura Y., Jang M.K., Nakano K., Tanaka A., Ozato K., Padmanabhan B., Yokoyama S. Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. J. Biol. Chem. 2010;285:7610–7618. doi: 10.1074/jbc.M109.062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hewings D.S., Wang M., Philpott M., Fedorov O., Uttarkar S., Filippakopoulos P., Picaud S., Vuppusetty C., Marsden B., Knapp S., et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011;54:6761–6770. doi: 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 65.Case D.A., Darden T.A., Cheatham T.E., Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M., et al. Amber 11. University of California; San Francisco, CA, USA: 2010. [Google Scholar]
- 66.Machado M.R., Dans P.D., Pantano S. Isoform-specific determinants in the HP1 binding to histone 3: Insights from molecular simulations. Amino Acids. 2010;38:1571–1581. doi: 10.1007/s00726-009-0371-3. [DOI] [PubMed] [Google Scholar]
- 67.De Mori G.M., Colombo G., Micheletti C. Study of the Villin headpiece folding dynamics by combining coarse-grained Monte Carlo evolution and all-atom molecular dynamics. Proteins. 2005;58:459–471. doi: 10.1002/prot.20313. [DOI] [PubMed] [Google Scholar]
- 68.HADDOCK, version 2.1 program; free of charge for non commercial users. [(accessed on 31 July 2012)]. Available online: http://www.nmr.chem.uu.nl/haddock/download.html.
- 69.van Dijk A.D., Bonvin A.M. Solvated docking: Introducing water into the modelling of biomolecular complexes. Bioinformatics. 2006;22:2340–2347. doi: 10.1093/bioinformatics/btl395. [DOI] [PubMed] [Google Scholar]
- 70.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 72.Makov G., Payne M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B Condens Matter. 1995;51:4014–4022. doi: 10.1103/PhysRevB.51.4014. [DOI] [PubMed] [Google Scholar]
- 73.Darden T., York D., Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 74.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 75.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 76.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 78.Zeng L., Li J., Muller M., Yan S., Mujtaba S., Pan C., Wang Z., Zhou M.M. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 2005;127:2376–2377. doi: 10.1021/ja044885g. [DOI] [PubMed] [Google Scholar]
- 79.Pan C., Mezei M., Mujtaba S., Muller M., Zeng L., Li J., Wang Z., Zhou M.M. Structure-guided optimization of small molecules inhibiting human immunodeficiency virus 1 Tat association with the human coactivator p300/CREB binding protein-associated factor. J. Med. Chem. 2007;50:2285–2288. doi: 10.1021/jm070014g. [DOI] [PubMed] [Google Scholar]
- 80.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Umehara T., Nakamura Y., Wakamori M., Ozato K., Yokoyama S., Padmanabhan B. Structural implications for K5/K12-di-acetylated histone H4 recognition by the second bromodomain of BRD2. FEBS Lett. 2010;584:3901–3908. doi: 10.1016/j.febslet.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mujtaba S., He Y., Zeng L., Yan S., Plotnikova O., Sachchidanand, Sanchez R., Zeleznik-Le N.J., Ronai Z., Zhou M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell. 2004;13:251–263. doi: 10.1016/S1097-2765(03)00528-8. [DOI] [PubMed] [Google Scholar]
- 83.Chandy M., Gutierrez J.L., Prochasson P., Workman J.L. SWI/SNF displaces SAGA-acetylated nucleosomes. Eukaryot. Cell. 2006;5:1738–1747. doi: 10.1128/EC.00165-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh M., D'Silva L., Holak T.A. DNA-binding properties of the recombinant high-mobility-group-like AT-hook-containing region from human BRG1 protein. Biol. Chem. 2006;387:1469–1478. doi: 10.1515/BC.2006.184. [DOI] [PubMed] [Google Scholar]
- 85.Huang H., Zhang J., Shen W., Wang X., Wu J., Shi Y. Solution structure of the second bromodomain of Brd2 and its specific interaction with acetylated histone tails. BMC Struct. Biol. 2007;7:57. doi: 10.1186/1472-6807-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chung C.W., Coste H., White J.H., Mirguet O., Wilde J., Gosmini R.L., Delves C., Magny S.M., Woodward R., Hughes S.A., et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 87.Chung C.W., Witherington J. Progress in the discovery of small-molecule inhibitors of bromodomain—Histone interactions. J. Biomol. Screen. 2011;16:1170–1185. doi: 10.1177/1087057111421372. [DOI] [PubMed] [Google Scholar]
- 88.Chung C.W., Dean A.W., Woolven J.M., Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: Inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012;55:576–586. doi: 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- 89.Filippakopoulos P., Picaud S., Fedorov O., Keller M., Wrobel M., Morgenstern O., Bracher F., Knapp S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012;20:1878–1886. doi: 10.1016/j.bmc.2011.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDF-Document (PDF, 4505 KB)