

Abstract
p53 is a major sensor of cellular stresses, and its activation influences cell fate decisions. We identified SUV39H1, a histone code ‘writer’ responsible for the histone H3 Lys9 trimethylation (H3K9me3) mark for ‘closed’ chromatin conformation, as a target of p53 repression. SUV39H1 downregulation was mediated transcriptionally by p21 and post-translationally by MDM2. The H3K9me3 repression mark was found to be associated with promoters of representative p53 target genes and was decreased upon p53 activation. Overexpression of SUV39H1 maintained higher levels of the H3K9me3 mark on these promoters and was associated with decreased p53 promoter occupancy and decreased transcriptional induction in response to p53. Conversely, SUV39H1 pre-silencing decreased H3K9me3 levels on these promoters and enhanced the p53 apoptotic response. These findings uncover a new layer of p53-mediated chromatin regulation through modulation of histone methylation at p53 target promoters.
Chromatin conformation has a fundamental role in regulating gene transcription and silencing as well as DNA repair1. In response to stress, the p53 protein triggers cell cycle arrest and/or apoptosis2–4. p53 physically interacts with several transcriptional coactivators and co-repressors, which have intrinsic histone-modifying activities5,6, and also with histone deacetylase complexes that act specifically to remodel chromatin7,8. In addition, several subunits of the SWI/SNF ATP-dependent chromatin remodeling complex are either recruited onto p53 target promoters or interact with p53 itself9–11, suggesting their role in p53-mediated transactivation.
The N-terminal tails of histones undergo post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitination, sumoylation, biotinylation and ADP ribosylation12–15. p53 has also been shown to influence histone H3 acetylation at Lys9 (H3K9Ac) and Lys14 (H3K14Ac)16,17, although the mechanism for this is not known. Post-translational modifications such as acetylation alter chromatin structure by changing internucleosomal contacts, whereas others such as methylation serve to create docking sites for effector proteins, leading to distinct biological outcomes18. Five lysines on histone H3 (Lys4, Lys9, Lys27, Lys36 and Lys79) and one lysine on histone H4 (Lys20) can undergo methylation by specific histone methyltransferases (HMTases)13. Each of these lysine residues can be mono-, di- and trimethylated in vivo, adding further layers of combinatorial control. For example, H3K4me3 is usually associated with the transcriptional start sites of active genes, whereas mono- or dimethylated H3K4 is localized to the middle and ends of genes19. By contrast, H3K9me1 and H3K9me2 are mainly localized to silent domains within euchromatin, and H3K9me3 is generally localized to constitutive heterochromatin20.
In the present study, we demonstrate that SUV39H1, which writes the H3K9me3 chromatin mark, is a repression target of p53. We show that the promoters of several of the p53 established target genes are enriched with this mark in an uninduced state. SUV39H1 downregulation reduces H3K9me3 on these promoters, thereby enhancing p53 promoter occupancy and contributing to the activation of p53 target genes and the p53-induced apoptotic response. Thus, our findings uncover a new layer of regulation at the chromatin level, in which p53 modulates the histone methylation of its target promoters.
Results
p53 downregulates SUV39H1 expression
We hypothesized that p53 may influence chromatin-modifying enzymes that have crucial roles in altering the chromatin template to render it suitable for p53-dependent transcription. A tetracycline-regulated EJ-p53 expression system21 has been used to generate a cDNA expression array to analyze p53-modulated genes and to identify several new p53 targets22,23. By mining this same cDNA expression array (unpublished data), we observed a decrease in RNA levels of SUV39H1, an HMTase that catalyzes the H3K9me3 heterochromatin mark24,25, in response to p53 induction. Upregulation of p53 by various methods in several cell lines of different tissue origin led to decreased RNA and protein levels of SUV39H1 (Fig. 1 and Supplementary Fig. 1a). Known p53 transcriptional targets p21, PIG3 and MDM2 (refs. 26-28) served as positive controls (Fig. 1a,b,e and Supplementary Fig. 1a). To examine the specificity of SUV39H1 downregulation in response to p53 induction, we generated B5/589 cells that stably express short hairpin RNA (shRNA) directed against p53. Control cells, but not those expressing sh-p53, showed a reduction in SUV39H1 RNA level upon MI-219 treatment (Supplementary Fig. 1b). Further, this decrease in SUV39H1 RNA expression was observed in U87MG (WT p53) cells, but not in U373MG cells (R273H mutant p53), under the same conditions (Supplementary Fig. 1c).
Figure 1.
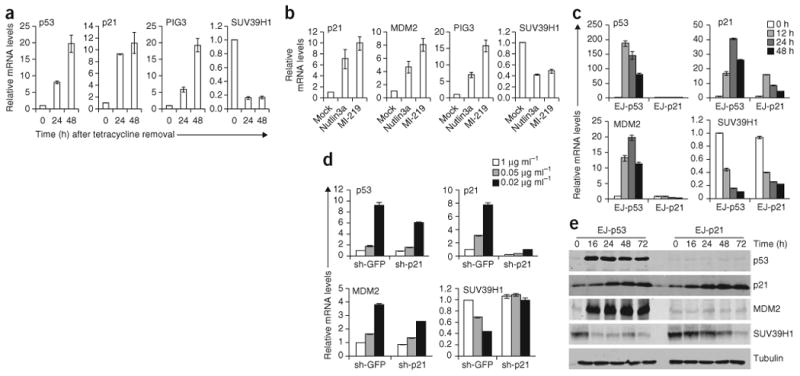
p53 downregulates SUV39H1 expression. (a) Real-time quantitative PCR (qrtPCR) of EJ-p53 cells induced for p53 for the indicated time points. (b) qrtPCR of B5/589 cells treated with either Nutlin3a or MI-219. (c) qrtPCR of EJ-p53 and EJ-p21 cells induced for p53 or p21, respectively, for the indicated time points. (d) qrtPCR of EJ-p53 cells stably expressing either sh-GFP or sh-p21 and induced for different levels of p53 for 24 h at the indicated tetracycline concentrations. (e) Western blot analysis of indicated proteins in EJ-p53 and EJ-p21 cells induced for either p53 or p21, respectively, for the indicated time points. All error bars represent s.e.m. of representative experiments done in triplicate.
p53 has been shown to repress the expression of several genes, including survivin, stathmin, CDC25C, cyclin A2 and cyclin B1 (refs. 29–34). Although these genes have all been found to have p53 binding sites on their promoters, subsequent studies have shown that in the absence of p21, p53 was unable to repress their expression. Moreover, ectopic expression of p21, in the absence of p53, was able to repress these same genes35. Although we observed p53 binding on the SUV39H1 promoter in EJ-p53 and B5/589 cells when induced for p53, we failed to observe such promoter occupancy in HCT116 WT cells (data not shown). Thus, we investigated the role of p21 in p53-mediated transcriptional repression of SUV39H1. We observed a similar decrease in SUV39H1 RNA expression in both EJ-p53 and EJ-p21 cells, when induced for p53 or p21, respectively (Fig. 1c). Further, p53 was unable to downregulate SUV39H1 transcript levels in EJ-p53 cells silenced for p21 (Fig. 1d). In EJ-p53 sh-GFP cells, an increase of two-fold and nine-fold in p53 level decreased SUV39H1 expression by 70% and 55%, respectively, whereas in EJ-p53 sh-p21 cells, a two-fold and six-fold increase in p53 level failed to reduce the expression of SUV39H1 at all. These results establish that p21 is both necessary and sufficient for p53-mediated SUV39H1 transcriptional downregulation, suggesting a potential involvement of an RB–E2F–dependent mechanism.
The p53 transcriptional target MDM2 has recently been reported to bind SUV39H1 and cause its proteosomal degradation36. Confirming these findings, we observed increased SUV39H1 ubiquitination in response to p53 activation (Supplementary Fig. 1d). Moreover, downregulation of SUV39H1 protein expression in response to p53 induction was inhibited when cells were treated with the proteosomal inhibitor MG-132 (Supplementary Fig. 1e). Further, exogenous expression of MDM2 resulted in a decreased protein level of exogenously expressed SUV39H1 in the absence of p53 (Supplementary Fig. 1f), and this decrease was rescued in the presence of MG-132 (Supplementary Fig. 1f). All of the above findings indicate that p53 also downregulates SUV39H1 expression at the protein level by MDM2-mediated proteosomal degradation. Further, p53 induction led to a rapid and sustained decrease in SUV39H1 protein level, whereas tet-regulated induction of p21 led to a comparable but less rapid decrease in the SUV39H1 protein level in the absence of any detectable MDM2 expression (Fig. 1e). Thus, p53 uses different mechanisms to downregulate SUV39H1 expression through two independent target genes, p21 and MDM2.
Induction of p53 abrogates the H3K9me3 heterochromatin mark
There is redundancy in the functions of remodeling factors that induce permissive alterations in chromatin accessibility37, whereas SUV39H1 is the only ubiquitously expressed HMTase that adds the repressive H3K9me3 mark38. Thus, we analyzed the effect of p53 activation on H3K9me3 levels and observed decreased levels of this mark in EJ-p53 (Fig. 2a), B5/589 (Fig. 2b) and HCT116 p53 WT cells (Fig. 2c) in response to p53 induction. Further, we did not observe a decrease in the H3K9me3 mark in either B5/589 cells stably transduced with sh-p53, when treated with MI-219 (Fig. 2b), or in HCT116 p53−/− cells treated with increasing doses of doxorubicin (Fig. 2c). Of note, the levels of H3K9me1 and H3K9me2 remained unaffected, confirming the specificity of the p53-induced decrease in H3K9me3 levels (data not shown).
Figure 2.
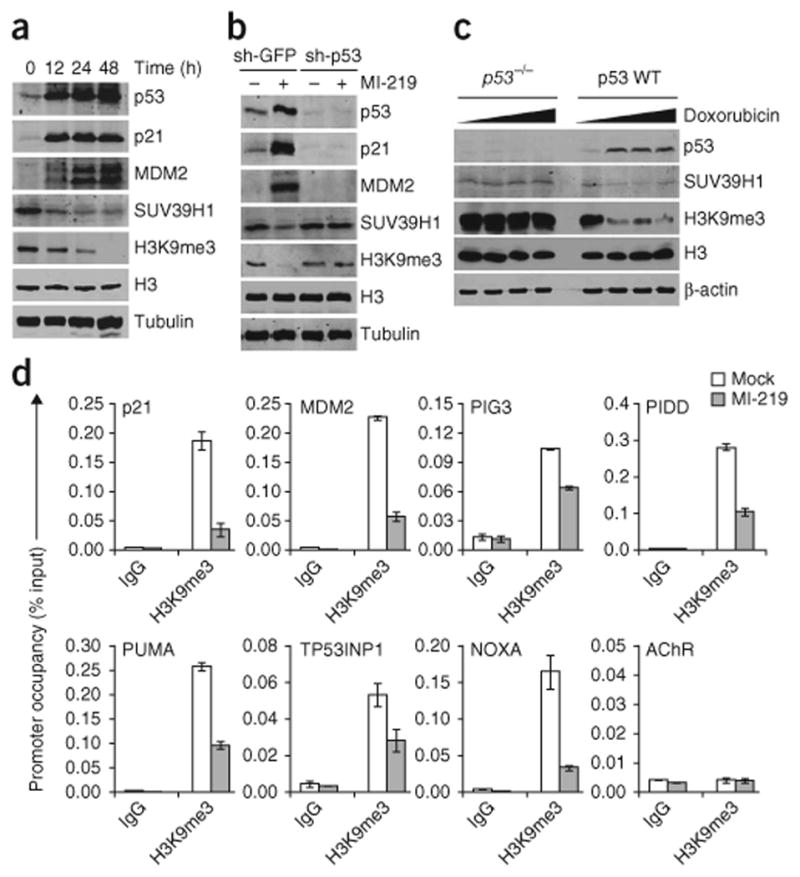
Induction of p53 abrogates the H3K9me3 heterochromatin mark. (a) Western blot analysis of EJ-p53 cells induced for p53 for the indicated time points by the complete removal of tetracycline from the medium. (b) Western blot analysis of B5/589 cells expressing either sh-GFP or sh-p53 and treated with MI-219. (c) Western blot analysis of HCT116 p53 WT and HCT116 p53−/− cells treated with 0, 0.05, 0.1 or 0.2 μg ml−1 of doxorubicin for 48 h. (d) ChIP analysis showing H3K9me3 occupancy on p53 target promoters in B5/589 cells treated with MI-219 for 24 h. The target sequences were detected by qrtPCR analysis of eluted DNA. The relative H3K9me3 occupancy over the input percentage is shown as a bar diagram. Acetylcholine receptor (AChR) is used as a negative control. All error bars represent s.e.m. of representative experiments done in triplicate.
To investigate how alterations in the H3K9me3 mark might affect p53 function, we analyzed the relative enrichment of trimethylated H3K9 on p53 target promoters in response to p53 activation. Chromatin immunoprecipitation (ChIP) analysis with anti-H3K9me3 antibody showed the presence of this histone mark on each of the p53 target promoters analyzed in untreated B5/589 or uninduced EJ-p53 cells. H3K9me3 occupancy was reduced in each case upon p53 induction in both cell lines (Fig. 2d and data not shown). Furthermore, we observed a decrease in the H3K9me3 mark on these promoters as early as 6 h following p53 induction by MI-219 treatment in B5/589 cells (Supplementary Fig. 2a). This decrease correlated well with increased p53 occupancy on the same promoters (Supplementary Fig. 2a).
Overexpression of SUV39H1 inhibits p53-dependent apoptosis
To investigate the role of this repressive heterochromatin mark in p53-induced cell fate decisions, we exogenously overexpressed SUV39H1 in HCT116 WT as well as in B5/589 cells. B5/589 and HCT116 WT cells stably expressing SUV39H1 showed ∼3.2-fold and ∼4-fold greater SUV39H1 expression levels, respectively, than cells that stably express the vector. This abrogated the reduction in SUV39H1 and H3K9me3 expression levels that was induced by p53 activation in cells (Fig. 3a and data not shown) as well as the level of the H3K9me3 mark present on each of the p53 target promoters that were analyzed (Fig. 3b). p53 was less efficient in inducing its target genes in cells overexpressing SUV39H1, as observed at both RNA (Fig. 3c) and protein levels (Fig. 3a). The reduced ability of p53 to transactivate its target genes was consistent with reduced p53 occupancy on its target promoters under conditions of SUV39H1 overexpression (Fig. 3d), even though the extent of p53 activation was similar to that in control cells (Fig. 3a and Supplementary Fig. 2c). Finally, we observed a reduced ability of p53 to induce apoptosis, as determined by propidium iodide staining in SUV39H1-overexpressing cells (Fig. 3e). The relative reduction in apoptosis was decreased at higher etoposide concentrations, which is associated with a decreased ability to sustain overexpressed SUV39H1 protein levels at higher levels of p53–MDM2 induction (data not shown). We also observed reduced expression at both RNA and protein levels of the CDK inhibitor p21, which is both necessary and sufficient for p53-induced growth arrest26,39 (Fig. 3a,c), together with reduced p53 promoter occupancy (Fig. 3d). However, we did not observe any major decrease in p53-induced cell cycle arrest in cells overexpressing SUV39H1 (Supplementary Fig. 2b).
Figure 3.
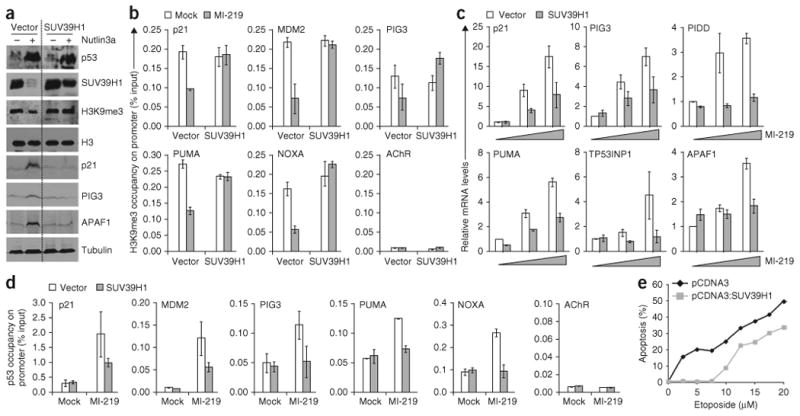
Overexpression of SUV39H1 inhibits p53-dependent apoptosis. (a) Western blot analysis of B5/589 cells overexpressing SUV39H1 and treated with Nutlin3a. The dotted line in the figure divides the vector from SUV39H1 bands. The discontinuity in the bands of the first three westerns is due to deletion of irrelevant lanes in this gel. (b) ChIP analysis showing H3K9me3 occupancy on p53 target promoters in B5/589 cells overexpressing SUV39H1 and treated with MI-219 for 24 h. The target sequences were detected by qrtPCR analysis of eluted DNA. The relative H3K9me3 occupancy over the percent input is shown as a bar diagram. Acetylcholine receptor (AChR) is used as a negative control. (c) qrtPCR of B5/589 cells overexpressing SUV39H1 and treated with 0 μM, 5 μM or 10 μM of MI-219. (d) ChIP analysis showing p53 occupancy on its target promoters in B5/589 cells overexpressing SUV39H1 and treated with MI-219 for 24 h. The target sequences were detected by qrtPCR analysis of eluted DNA. The relative p53 promoter occupancy over the percent input is shown as a bar diagram. (e) Propidium iodide staining of HCT116 cells overexpressing SUV39H1 and treated with increasing doses of etoposide. The percentage of cells undergoing apoptosis (less than 2N content of DNA) is shown as a line diagram. All error bars represent s.e.m. of representative experiments done in triplicate.
SUV39H1 silencing causes p21-dependent cell cycle arrest
To assess the impact of SUV39H1 silencing on p53 target gene expression, we generated HCT116 WT and B5/589 cells stably expressing inducible SUV39H1 shRNA. Doxycycline induction (Supplementary Figs. 3a and 4a) led to a decrease in both SUV39H1 RNA (Fig. 4 and Supplementary Fig. 3d) and protein levels (Fig. 4c and Supplementary Fig. 4b). We observed a resulting reduction of H3K9me3 in total protein lysates (Fig. 4c and Supplementary Fig. 4b) as well as a reduction of the H3K9me3 mark on each of the p53 target promoters tested (Fig. 4b). We observed no detectable alteration in the expression level of any p53 pro-apoptotic transcriptional target analyzed or evidence of an apoptotic response under conditions of SUV39H1 silencing (Fig. 4a,e).
Figure 4.
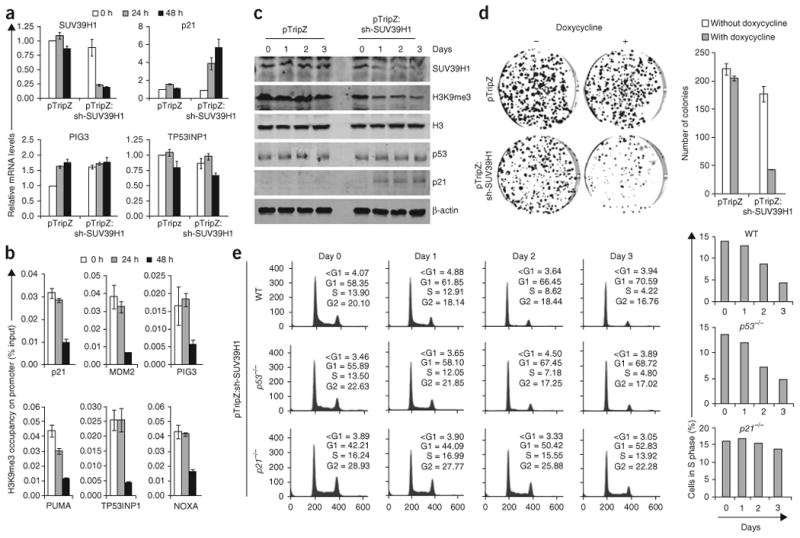
Silencing of SUV39H1 causes p21-dependent, but p53-independent, cell cycle arrest. (a) qrtPCR of HCT116 WT cells stably transduced with inducible sh-SUV39H1 and cultured in the presence of doxycycline for the indicated time points. (b) ChIP analysis showing H3K9me3 occupancy on p53 target promoters in HCT116 WT cells stably transduced with inducible sh-SUV39H1 and cultured in the presence of doxycycline for the indicated time points. The target sequences were detected by qrtPCR analysis of eluted DNA. The relative H3K9me3 occupancy over the percent input is shown as a bar diagram. (c) Western blot analysis of HCT116 WT cells stably transduced with inducible sh-SUV39H1. Day 0, 1, 2 and 3 represent time after doxycycline addition into the medium. (d) Colony formation assay in HCT116 WT cells stably transduced with sh-SUV39H1. The cells were grown in the absence or presence of doxycycline. The colonies formed after 9 d were counted and are shown as a bar diagram. (e) Propidium iodide staining in HCT116 p53 WT, HCT116 p53−/− and HCT116 p21−/− cells stably transduced with inducible sh-SUV39H1. Day 0, 1, 2 and 3 represent time after addition of doxycycline into the medium. A bar diagram for each cell line showing the percentage of cells in S phase is also shown on the right. All error bars represent s.e.m. of representative experiments done in triplicate.
A previous study indicated that SUV39H1 knockdown was associated with increased p21 RNA expression40. We confirmed increased expression of p21 at both RNA (Fig. 4a and Supplementary Fig. 3d) and protein levels (Fig. 4c and Supplementary Fig. 4b) in response to sh-SUV39H1 induction. Moreover, this response appeared to be independent of p53, as there was no detectable change in p53 RNA (Supplementary Fig. 3d) or protein levels (Fig. 4c). Of note, this modest increase in p21 was sufficient to cause cell cycle arrest, as determined by propidium iodide staining (Fig. 4e and Supplementary Fig. 4c) and colony-forming ability (Fig. 4d and Supplementary Figs. 4d and 5a) in both HCT116 WT and B5/589 cells. To rigorously exclude any p53-dependent contribution, we expressed inducible sh-SUV39H1 in HCT116 p53−/− cells (Supplementary Fig. 3b) and observed similar p21 induction (Supplementary Fig. 3e) associated with cell cycle arrest and colony suppression (Fig. 4e and Supplementary Fig. 5a). Thus, unlike the case with p53 pro-apoptotic target genes, loss of SUV39H1 expression removes the barrier to p21 expression, presumably allowing recruitment of transcription factors already present in the cell or induced by SUV39H1 silencing.
To directly establish that p21 is both necessary and sufficient for SUV39H1 knockdown–dependent cell cycle arrest, we generated inducible sh-SUV39H1 HCT116 p21−/− cells39 (Supplementary Fig. 3c) and observed that SUV39H1 silencing failed to induce cell cycle arrest and colony suppression in these cells (Fig. 4e and Supplementary Fig. 5a). We also generated double knockdown stable cells with sh-p21 and inducible sh-SUV39H1 in HCT116 WT cells (Supplementary Fig. 5b). Induction of sh-SUV39H1 led to cell cycle arrest in HCT116 WT cells but not in HCT116 cells silenced for p21 (Supplementary Fig. 5c,d). Furthermore, silencing of SUV39H1 cooperated with p53 in inducing p21 at both RNA (Fig. 5) and protein levels (Supplementary Fig. 6a) but did not result in enhanced cell cycle arrest (Supplementary Fig. 6b,c). Thus, SUV39H1 silencing, like overexpression, did not detectably influence p53-dependent cell cycle arrest (Supplementary Figs. 2b and 6), presumably because of the low threshold of p21 required to induce cell cycle arrest in the cells analyzed.
Figure 5.
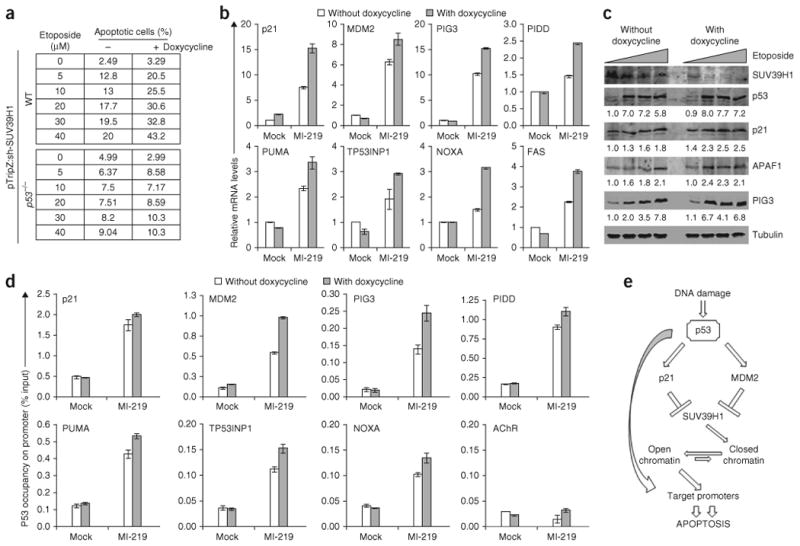
Pre-silencing of SUV39H1 cooperates with chemotherapy-induced apoptosis in a p53-dependent manner. (a) Propidium iodide (PI) staining in HCT116 p53 WT and HCT116 p53−/− cells stably transduced with sh-SUV39H1. The cells were pre-silenced for SUV39H1 by growing cells in the presence of doxycycline for two days followed by treatment with increasing doses of etoposide. The percentage of cells showing less than a 2N content of DNA (apoptosis) in each condition is shown in the table (see Supplementary Fig. 7 for actual FACS graphs). (b) Real-time analysis of HCT116 WT cells stably transduced with inducible sh-SUV39H1 and cultured in the presence of doxycycline for two days followed by treatment with MI-219. (c) Western blot analysis of HCT116 WT cells stably transduced with sh-SUV39H1 and treated with 0 μM, 5 μM, 10 μM or 20 μM of etoposide for 48 h. The cells were either untreated or pre-treated with doxycycline for two days before treating with etoposide. (d) ChIP analysis in HCT116 WT cells stably expressing sh-SUV39H1 showing p53 occupancy on its target promoters. The cells were either untreated or treated with doxycycline for two days, followed by MI-219 treatment for 24 h. The target sequences were detected by qrtPCR analysis of eluted DNA. The relative p53 promoter occupancy over the percent input is shown in the form of bar diagram. Acetylcholine receptor (AChR) was used as a negative control. Error bars represent s.e.m. of representative experiments done in triplicate. (e) Schematic diagram illustrating the role of SUV39H1 in p53-induced apoptosis. SUV39H1 is the HMTase that adds the H3K9me3 repressive chromatin mark on p53 target promoters, which keeps them in a closed chromatin conformation. Activation of p53 downregulates SUV39H1 expression, which in turn leads to a decrease in the H3K9me3 epigenetic mark on p53 target promoters. This results in a more open chromatin conformation that allows a higher level of p53 recruitment, leading to increased transcription of target genes and resulting in enhanced apoptosis.
SUV39H1 pre-silencing enhances p53-dependent apoptosis
To test the effects of a low basal level of SUV39H1 expression on p53-induced apoptosis, we first silenced SUV39H1 in WT p53-containing HCT116 cells. Both etoposide, a topoisomerase II inhibitor41, and paclitaxel, which interferes with the normal breakdown of microtubules during cell division42, induced considerably increased apoptosis under these conditions, as analyzed by propidium iodide staining (Fig. 5a, Supplementary Fig. 7a and data not shown) and Annexin-V staining (Supplementary Fig. 7b). This enhancement of chemotherapy-induced apoptosis was p53 dependent, as we did not observe any such cooperation in HCT116 p53−/− cells (Fig. 5a and Supplementary Fig. 7a). The increased apoptosis in HCT116 WT cells was associated with increased expression of p53 pro-apoptotic target genes, as measured at both RNA (Fig. 5b) and protein levels (Fig. 5c) in the absence of any noteworthy difference in p53 expression levels. In addition, increased p53 target gene expression was associated with increased p53 promoter occupancy (Fig. 5d). All of these findings argue that low basal SUV39H1 levels influence the chromatin conformation of p53 pro-apoptotic target genes, making their promoters more accessible and enhancing the p53 apoptotic response to chemotherapy.
SUV39H1 is known to influence global gene expression43. Thus, we carried out micrococcal nuclease (MNase) assays in EJ-p53 cells with and without p53 induction. p53 activation led to increased sensitivity to MNase treatment, as measured by increased DNA laddering, consistent with induction of a more relaxed chromatin structure (Supplementary Fig. 8a). We also analyzed the expression of several genes not known to be p53 transcriptional targets but reported to be targets of SUV39H1 (ref. 44). We confirmed that RNA expression levels of growth hormone receptor (GHR), myostatin (GDF8), scinderin (SCIN) and ets homologous factor (EHF) were upregulated in SUV39H1-silenced cells (Supplementary Fig. 8b). Similar gene expression changes were also observed in EJ-p53 cells upon p53 induction (Supplementary Fig. 8c). All of these results argue that SUV39H1 regulates the transcription of genes in addition to p53 direct targets. Further studies will be necessary to determine whether—and if so, how—these more global effects mediated by p53 repression of SUV39H1 influence p53 signaling.
Discussion
Our present study demonstrates the ability of the p53 tumor suppressor protein to influence its own transcriptional program by down-regulating the expression of SUV39H1, the histone code writer of the H3K9me3 mark. We identified the presence of the H3K9me3 repressive histone modification on several p53 target promoters. By inducing a decrease in this mark through downregulation of SUV39H1 expression, p53 causes a more ‘open’ chromatin conformation that allows increased p53 promoter occupancy and contributes to the activation of p53 target genes and the p53-induced apoptotic response.
Our results indicate that p53 regulates SUV39H1 expression at the RNA level by p21-mediated transcriptional downregulation and at the protein level by MDM2-mediated proteosomal degradation. p53 was unable to downregulate SUV39H1 transcript levels in cells silenced for p21. Furthermore, p21 was itself able to downregulate SUV39H1 RNA levels independently of p53. These data suggest that the cell cycle has a role in this repression and that the mechanism may involve the RB–E2F pathway. In fact, we observed putative E2F binding sites on the SUV39H1 promoter using MatInspector (http://www.genomatix.de/). Currently, we are analyzing the relative role of different E2F family members in p53-mediated downregulation of SUV39H1 transcription. The reduction in levels of SUV39H1 in HCT116 WT cells was modest when compared to that observed in EJ-p53 cells. However, there was a marked decrease in H3K9me3 levels in HCT116 WT cells compared to EJ-p53 cells (compareFig. 2a with Fig. 2c). Furthermore, SUV39H1 silencing alone was sufficient to reduce the levels of H3K9me3 on p53 target promoters, even without the activation of p53 (Fig. 4b). These results imply that there must be a dynamic equilibrium between the ‘erasers’ of this mark and SUV39H1, such that a change in the balance as a result of SUV39H1 silencing was sufficient to remove the H3K9me3 repressive chromatin mark.
The transforming adenovirus E1A and E1B proteins are well known to inactivate RB and p53 functions, respectively45. Recent studies have identified another adenoviral protein, E4-ORF3, which causes an increase in the H3K9me3 mark at p53 target promoters through a mechanism presumed to involve SUV39H1 function46. Evidence that E4-ORF3 can replace the need for E1B function in overcoming the host's p53 response strongly suggests that epigenetic regulation of the H3K9me3 mark has a major impact on p53 function. Our complementary findings establish that SUV39H1 is a p53 repression target that has a critical function in determining p53 cell fate decisions, through modulation of the H3K9me3 mark (Fig. 5e). Evidence that p53 exerts redundant mechanisms to silence SUV39H1 epigenetic function further argues (Fig. 5e) for the importance of SUV39H1 silencing in the p53 response.
Recently, increased binding of exogenously expressed SUV39H1 to the p21 promoter in the presence of exogenously expressed p53 and either exogenously expressed or amplified MDM2 has been reported47. Although this study observed increased levels of the H3K9me3 mark at the p21 promoter under these conditions of exogenous protein expression, we demonstrated decreased levels of this mark in response to p53 activation under physiological conditions. It was also reported that p53 induction in the presence of SUV39H1 knockdown resulted in increased p21 RNA levels, consistent with our findings and with what might be expected for loss rather than gain of the H3K9me3 repressive mark. This same group further found that SUV39H1 binding to the MDM2 acidic domain allows the p53–MDM2–SUV39H1 complex to bind DNA48. More recent evidence that MDM2 acts as a ubiquitin ligase that binds SUV39H1 as well as p53 and targets both proteins for proteosomal degradation36 could also explain the presence of all of these proteins at the p21 promoter.
Misregulation of various histone modifications has been observed in human cancers49–51. Loss of SUV39H1 function sensitizes mice to tumorigenesis43. Furthermore, reduced levels of H3K9me3 are associated with genomic instability43. By contrast, colorectal tumors show increased SUV39H1 expression levels52. Our present studies demonstrate that variations in basal levels of SUV39H1 have a major impact on p53 pro-apoptotic signaling by altering levels of the H3K9me3 mark, uncovering a new layer of regulation at the chromatin level by p53 modulation of histone methylation of its target promoters. Thus, normal cells with low basal levels may have an increased sensitivity to p53-induced apoptosis relative to otherwise comparable cells. Conversely, WT p53-containing cancer cells possessing high steady-state levels of SUV39H1 may be more resistant to chemotherapy and irradiation therapy than otherwise comparable tumor cells expressing low SUV39H1 levels.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
Online Methods
Plasmids, cell lines and treatments
The pLKO.1 puro vector expressing sh-p53 (ref. 53) was obtained from Addgene (#19119). The pLKO. 1 puro vector expressing sh-p21 was generated by cloning the sequence (CGCTCTACATCTTCTGCCTTA) into the pLKO.1 TRC Vector. The pLKO.1-sh-GFP vector was used as a control (Addgene; #30323). The pTripZ empty vector and pTripZ vector expressing a doxycycline-inducible shRNA against human SUV39H1 was purchased from Open Biosystems (#RHS4696-99356763). A full-length cDNA expressing human SUV39H1 was also procured from Open Biosystems (#MHS1011-61443) and cloned into pCDNA3 and VIRSP vectors using EcoRI and XhoI enzymes. The MDM2 expression construct, described previously54, was obtained from Addgene (#16233). B5/589 immortalized breast epithelial cells (p53 WT) were cultured in RPMI-1640 containing 10% FBS and 5 ng ml−1 of human EGF at 37 °C. EJ-p53 and EJ-p21 bladder carcinoma cells that express tetracycline-regulatable p53 or p21, respectively, were cultured in DMEM containing 10% FBS, 750 μg ml−1 G418, 100 μg ml−1 hygromycin and 1 μg ml−1 tetracycline21,55. HCT116 p53 WT, HCT116 p53−/−, HCT116 p21−/− and A549 lung carcinoma cells were cultured in DMEM with 10% FBS39,56. H1299 (p53 null) non–small cell lung carcinoma cells were cultured in RPMI-1640 with 5% FBS. Nutlin3a (Cayman chemicals #10004372) and MI-219 (ref. 57), were used for 16 h at a concentration of 20 μM and 10 μM, respectively57,58. Hydrogen peroxide and doxorubicin were purchased from Sigma (Cat #H1009; #D1515) and used at 1 mM and 0.2 μg ml−1, respectively, for 48 h, unless mentioned otherwise. The proteosomal inhibitor, MG-132, was purchased from Calbiochem (Cat #474790) and used at a concentration of 10 μM for 4 h. All transfections were carried out with Lipofectamine 2000 (Invitrogen), using the standard protocol.
RNA extraction and real-time PCR analysis
Total RNA was extracted from cultured cells by Trizol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA synthesis was done using Superscript II RT (Invitrogen), and SYBR Green (Roche; #04887352001)-based quantitative PCR was carried out using gene-specific primer sets. The sequences of the primers used for real-time PCR are shown in Supplementary Table 1.
Western blotting
Western blotting was done as described previously59, with the exception that Alexa Fluor secondary antibodies were used. All antibodies were used at a dilution of 1:1,000. All blots were developed using the Odyssey fluorescence image scanner, and the band intensities were quantified using LI-COR software.
Antibodies used
The following antibodies were used in the study: anti-p53 (SCBT; sc-6243), anti-p21 (BD; #556431), anti-MDM2 (Calbiochem; #OP46), anti-APAF1 (R and D; #MAB828), anti-PIG3 (abcam; #ab64798), anti-SUV39H1 (#05-615), anti-H3K9me3 (#07-442; #05-1242), anti-H3 (#05-928), all from Millipore; and anti-Tubulin (Sigma; #T5168), anti-β-actin (Sigma; #A5441), anti-ubiquitin (Covance; #PRB-268C), Alexa Fluor 680 goat anti-mouse IgG (Molecular Probes; #A21057) and Alexa Fluor goat anti-rabbit IgG (Molecular Probes; #A21076).
Flow cytometry and colony formation assay
Cell cycle analysis was conducted using propidium iodide staining according to the manufacturer's protocol (BD; #340242). Cells with less than 2N DNA content were considered apoptotic cells. Annexin-V staining was done according to the manufacturer's protocol (BD; #556547). All FACS data were analyzed using Flowjo 7.6. To determine colony formation ability, 1,500 cells were plated in a six-well plate and cultured for 9 d in DMEM with 10% FBS in the absence or presence of 1 μg ml−1 of doxycycline to induce shRNA expression. Fresh medium was replaced every third day. The cells were fixed in a 10% methanol and 10% acetic acid solution and stained with 1% crystal violet to visualize the colonies.
Micrococcal nuclease (MNase) assay
MNase digestion was carried out as described previously60. p53 induced or un-induced EJ-p53 cells were harvested and counted. Chromatin was isolated as described previously60 from each experimental condition consisting of 2.5 million cells. Each sample was treated with 0.025 units of MNase (Sigma; #N3755) for 2, 5 or 10 min at 37 °C, and the reaction was stopped with 1 mM EGTA. Samples were centrifuged for 10 min at 12,000g, and DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen; #69504). Equal amounts of DNA were resolved on a 1% agarose gel and visualized by ethidium bromide staining.
Quantitative chromatin immunoprecipitation assay
ChIP assay was done as described previously60, with the following modifications: Two and a half million cells were taken for ChIP assay. The whole cell extract was pre-cleared using 60 μl of Protein A/G PLUS-agarose beads (SCBT; sc-2003) and incubated overnight with anti-p53, anti-H3K9me3 or control IgG antibodies. One microgram of antibody was used for 100 μg of total lysate. Protein G agarose beads pre-blocked with salmon sperm DNA (to reduce nonspecific DNA binding) were purchased from Millipore (#16-2001), and 60 μl of this solution was used for each immunoprecipitation. Ten percent of chromatin from each sample was removed before immunoprecipitation and used for PCR amplification (input). Both immunoprecipitated and whole cell extract (input) were treated with RNaseA and proteinase K and DNA were purified using DNeasy Blood and Tissue kit (Qiagen; #69504). qrtPCR was carried out on this DNA to identify the amount of target sequence. The list of primers used is given in Supplementary Table 1.
Generation of stable cell lines
B5/589 cells expressing sh-p53 were generated by infecting cells with the pLKO.1 sh-p53 lentivirus53 followed by puromycin selection (2 μg ml−1). sh-GFP–expressing cells were used in all of the relevant experiments as control. B5/589, HCT116 p53 WT, HCT116 p53−/− or HCT116 p21−/− cells were infected with the pTripZ:sh-SUV39H1 lentivirus and selected for puromycin (2 μg ml−1) resistance. HCT116 p53 WT cells with sh-p21 and inducible sh-SUV39H1 were generated by infecting the cells together with lentiviruses expressing sh-p21 and sh-SUV39H1, followed by puromycin selection (2 μg ml−1). Stable puromycin-resistant clones were pooled to generate doxycycline-inducible shRNA cell lines. SUV39H1 overexpressing clones were generated in HCT116 WT and B5/589 cells by either transfecting cells with pCDNA3-SUV39H1 (under the control of the CMV promoter) or infecting them with VIRSP-SUV39H1 (under the control of the mPGK promoter) and selecting with G418 (800 μg ml−1) or puromycin (2 μg ml−1), respectively.
Supplementary Material
Acknowledgments
The authors would like to thank E. Bernstein and her lab members as well as C. Munoz-Fontela, M. Kracikova and W. Murk for helpful discussions. The project was supported by grant P01CA080058 from the National Cancer Institute (to S.A.A. and S.W.L.). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the US National Institutes of Health.
Footnotes
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Author Contributions: S.K.M. and S.A.A. planned the project. S.K.M. conducted all the experiments, with participation by E.K.B. S.W. provided the MI-219 MDM2 inhibitor. S.A.A. supervised the study, along with S.W.L. and W.G. S.K.M. and S.A.A. wrote the paper.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med. 2011;3:64rv1. doi: 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 4.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 7.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2011;2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drost J, et al. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol. 2010;12:380–389. doi: 10.1038/ncb2038. [DOI] [PubMed] [Google Scholar]
- 10.Lee D, et al. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem. 2002;277:22330–22337. doi: 10.1074/jbc.M111987200. [DOI] [PubMed] [Google Scholar]
- 11.Naidu SR, Love IM, Imbalzano AN, Grossman SR, Androphy EJ. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene. 2009;28:2492–2501. doi: 10.1038/onc.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 13.Jaskelioff M, Peterson CL. Chromatin and transcription: histones continue to make their marks. Nat Cell Biol. 2003;5:395–399. doi: 10.1038/ncb0503-395. [DOI] [PubMed] [Google Scholar]
- 14.Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–272. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 15.Schneider R, Grosschedl R. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev. 2007;21:3027–3043. doi: 10.1101/gad.1604607. [DOI] [PubMed] [Google Scholar]
- 16.Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allison SJ, Milner J. Loss of p53 has site-specific effects on histone H3 modification, including serine 10 phosphorylation important for maintenance of ploidy. Cancer Res. 2003;63:6674–6679. [PubMed] [Google Scholar]
- 18.Jacobs SA, et al. specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001;20:5232–5241. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sims RJ, III, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Rice JC, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 21.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA. 1997;94:9648–9653. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ide T, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36:379–392. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Muñoz-Fontela C, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205:1929–1938. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fodor BD, Shukeir N, Reuter G, Jenuwein T. Mammalian Su(var) genes in chromatin control. Annu Rev Cell Dev Biol. 2010;26:471–501. doi: 10.1146/annurev.cellbio.042308.113225. [DOI] [PubMed] [Google Scholar]
- 25.Lehnertz B, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 26.El-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 27.Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315–320. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 28.Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010;17:93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 29.Mirza A, et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613–2622. doi: 10.1038/sj.onc.1205353. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 31.Johnsen JI, et al. p53-mediated negative regulation of stathmin/Op18 expression is associated with G2/M cell-cycle arrest. Int J Cancer. 2000;88:685–691. doi: 10.1002/1097-0215(20001201)88:5<685::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 32.Krause K, et al. Expression of the cell cycle phosphatase cdc25C is down-regulated by the tumor suppressor protein p53 but not by p73. Biochem Biophys Res Commun. 2001;284:743–750. doi: 10.1006/bbrc.2001.5040. [DOI] [PubMed] [Google Scholar]
- 33.Badie C, Itzhaki JE, Sullivan MJ, Carpenter AJ, Porter AC. Repression of CDK1 and other genes with CDE and CHR promoter elements during DNA damage-induced G2/M arrest in human cells. Mol Cell Biol. 2000;20:2358–2366. doi: 10.1128/mcb.20.7.2358-2366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Innocente SA, Abrahamson JL, Cogswell JP, Lee JM. p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci USA. 1999;96:2147–2152. doi: 10.1073/pnas.96.5.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Löhr K, Moritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem. 2003;278:32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 36.Bosch-Presegué L, et al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol Cell. 2011;42:210–223. doi: 10.1016/j.molcel.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 37.Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 38.O'Carroll D, et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol. 2000;20:9423–9433. doi: 10.1128/mcb.20.24.9423-9433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381:713–716. doi: 10.1038/381713a0. [DOI] [PubMed] [Google Scholar]
- 40.Cherrier T, et al. p21WAF1gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene. 2009;28:3380–3389. doi: 10.1038/onc.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y, et al. Medicinal chemistry of paclitaxel and its analogues. Curr Med Chem. 2009;16:3966–3985. doi: 10.2174/092986709789352277. [DOI] [PubMed] [Google Scholar]
- 43.Peters AH, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 44.Kondo Y, et al. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 46.Soria C, Estermann FE, Espantman KC, O'Shea CC. Heterochromatin silencing of p53 target genes by a small viral protein. Nature. 2010;466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, et al. MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO J. 2010;29:2538–2552. doi: 10.1038/emboj.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cross B, et al. Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J Biol Chem. 2011;286:16018–16029. doi: 10.1074/jbc.M111.228981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chi P, Allis CD, Wang GG. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 51.Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br J Cancer. 2004;90:761–769. doi: 10.1038/sj.bjc.6601575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozdağ H, et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 2006;7:90. doi: 10.1186/1471-2164-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Godar S, et al. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou BP, et al. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 55.Fang L, et al. p21Waf1/Cip1/Sdi1induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999;18:2789–2797. doi: 10.1038/sj.onc.1202615. [DOI] [PubMed] [Google Scholar]
- 56.Bunz F, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 57.Shangary S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 59.Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66:4715–4724. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- 60.Schaniel C, et al. Smarcc1/Baf155 couples self-renewal gene repression with changes in chromatin structure in mouse embryonic stem cells. Stem Cells. 2009;27:2979–2991. doi: 10.1002/stem.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.