

Abstract
Objective
Elevated serum phosphate has emerged as a major risk factor for vascular calcification. The sodium-dependent phosphate cotransporter, PiT-1, was previously shown to be required for phosphate-induced osteogenic differentiation and calcification of cultured human VSMCs, but its importance in vascular calcification in vivo, as well as the potential role of its homologue, PiT-2, have not been determined. We investigated the in vivo requirement for PiT-1 in vascular calcification using a mouse model of chronic kidney disease, and the potential compensatory role of PiT-2 using in vitro knockdown and over-expression strategies.
Approach and Results
Mice with targeted deletion of PiT-1 in VSMCs were generated (PiT-1Δsm). PiT-1 mRNA levels were undetectable whereas PiT-2 mRNA levels were increased 2 fold in the vascular aortic media of PiT-1Δsm compared to PiT-1flox/flox control. When arterial medial calcification was induced in PiT-1Δsm and PiT-1flox/flox by chronic kidney disease followed by dietary phosphate loading, the degree of aortic calcification was not different between genotypes, suggesting compensation by PiT-2. Consistent with this possibility, VSMCs isolated from PiT-1Δsm mice had no PiT-1 mRNA expression, increased PiT-2 mRNA levels, and no difference in sodium-dependent phosphate uptake or phosphate-induced matrix calcification compared to PiT-1flox/flox VSMCs. Knockdown of PiT-2 decreased phosphate uptake and phosphate-induced calcification of PiT-1Δsm VSMCs. Furthermore, over-expression of PiT-2 restored these parameters in human PiT-1-deficient VSMCs.
Conclusions
PiT-2 can mediate phosphate uptake and calcification of VSMCs in the absence of PiT-1. Mechanistically, PiT-1 and PiT-2 appear to serve redundant roles in phosphate-induced calcification of vascular smooth muscle cells.
Keywords: PiT-1, PiT-2, phosphate, vascular smooth muscle cell, vascular calcification
Introduction
Cardiovascular mortality remains the leading cause of death in patients with chronic kidney disease (CKD)1. The extent of arterial calcification independently predicts cardiovascular disease and mortality in CKD patients2, and this ectopic calcification occurs decades earlier in patients with CKD than in the general population3. The calcium phosphate mineral deposits occur in the form of hydroxyapatite or whitlockite4, and can contribute to disease progression in two main locations within arterial vessels: the intimal layer and the medial layer. Calcification associated with the intimal layer is a reflection of atherosclerotic burden, and may result in arterial occlusion. Arterial medial calcification (AMC) contributes to vascular stiffening without occlusion, and can lead to hypertension and heart failure. Studies have shown that AMC is a dominant form of vascular calcification in dialysis patients4-6.
Over the past decade, elevated serum phosphorus has emerged as a key risk factor for vascular calcification in CKD patients, as well as the general population 7-9. High extracellular phosphate has been widely established to induce vascular smooth muscle cell (VSMC) matrix calcification in vitro10-12. Indeed, elevated phosphate has been linked to several highly regulated processes in the vessel wall that promote AMC. These include VSMC osteogenic differentiation, VSMC apoptosis, and matrix degradation (reviewed in Lau et al13 and Shanahan et al14). The phenotype change is particularly striking, whereby the VSMCs cease to express smooth muscle markers (SM alpha actin, SM22) and instead express bone-forming genes (core-binding factor alpha-1 Runx2/Cbfa1, osterix, alkaline phosphatase, osteopontin)10, 15.
Active transport of phosphate into cells is primarily mediated by three classes of sodium-dependent phosphate (NaPi) cotransporters. Type I (SLC17 family) cotransporters are involved in organic anion transport, including phosphate16. The type II (SLC34 family) cotransporters, predominantly expressed in kidney and intestinal epithelium, are important for whole-body phosphate homeostasis17, 18. The type III (SLC20 family) cotransporters, PiT-1 and PiT-2, are more generally expressed19 and have been identified as the predominant phosphate transporters in rat20 and human21 VSMCs. PiT-1 and PiT-2 are 62% identical at the amino acid level and both contain 12 transmembrane spanning domains with a single large intracellular domain. They exhibit a high affinity for phosphate and have a preference to transport H2PO4- more than HPO42-, with a 2:1 ratio of sodium to phosphate in both acidic and alkaline pH19. The Michaelis-Menten affinity constant (Km) for sodium dependent phosphate uptake for PiT-1 and PiT-2 are similar and range from 25-300 μM phosphate depending on the species19, 20, 22-24.
We have shown previously that PiT-1 was required for phosphate-induced osteogenic differentiation and calcification of human VSMCs in vitro21. When PiT-1 was knocked down in human aortic VSMCs using short hairpin RNA interference (shRNA), phosphate-induced mineralization was suppressed and VSMC phenotype was preserved. Subsequent over-expression of mouse PiT-1 restored phosphate transport, VSMC osteogenic phenotype change and calcification21.
Given the compelling in vitro evidence, we hypothesized that PiT-1 would be required for vascular calcification in vivo. To test this, a well-characterized mouse model of phosphate-driven uremic AMC was utilized 25-27. Calcification-prone DBA/2J mice28, when subjected to partial renal ablation, develop robust AMC upon dietary phosphate loading without atherosclerosis or inflammation25-27. As global deletion of PiT-1 is lethal during mouse embryogenesis24, 29, we generated VSMC-specific knockout of PiT-1 using a Sm22 promoter driven Cre/loxP system. This resulting PiT-1flox/flox;Sm22-Cre+/0 dual transgenic mouse line (abbreviated as PiT-1Δsm) lacked PiT-1 expression in VSMCs. Unexpectedly, sodium-dependent phosphate uptake and matrix mineralization in isolated VSMCs, as well as high phosphate-feeding induced uremic AMC did not differ between as PiT-1Δsm and PiT-1flox/flox control mice, prompting us to examine potential compensatory mechanisms. Our data indicate that PiT-2 compensates for PiT-1 in phosphate-uptake and matrix mineralization in vitro, and are the first to suggest redundant roles for PiT-1 and PiT-2 in phosphate-driven AMC.
Materials and Methods
An expanded materials and methods section is available in the supplementary data.
Results
PiT-1 and PiT-2 are the only NaPi cotransporters expressed by mouse VSMCs
Reverse-transcription PCR was used to determine the expression profile of NaPi cotransporters in cultured C57/Bl6 wild-type VSMCs. Using this technique, no specific bands were obtained using primers for type I transporters (SLC17A1, A4 and A7) or type II transporters (SLC34A1, A2 and A3) while both type III transporters were detected (supplemental Figure I).
VSMC-specific knockdown of PiT-1
In contrast to the global PiT-1 knockout that resulted in embryonic lethality due to anemia24, 29, mice with smooth muscle specific PiT-1 deletion were born at the expected Mendelian ratios, and showed no gross abnormalities in health, weight or lifespan (data not shown). Likewise, histological analyses of aortas from these mice were normal (data not shown). As shown in supplemental Figure II, PCR amplification of genomic DNA isolated from VSMCs from PiT-1Δsm mice showed a 500 bp band (compared with 950 bp band in the PiT-1flox/flox cells) consistent with deletion of exons 3 and 4. Western blotting confirmed the depletion of PiT-1 protein in whole aortic tissue in PiT-1Δsm mice (Figure 1A and B), and immunochemistry showed selective PiT-1 depletion in VSMCs but not endothelial cells (Figure 1C). Consistent with the immunostaining data, PiT-1 mRNA was undetectable in the aortic media isolated from healthy PiT-1Δsm mice (Supplemental Figure III). Since a trend for decreased expression of PiT-1 mRNA in cardiac tissue from PiT-1Δsm compared to PiT-1flox/flox was observed, and because Sm22α is transiently expressed in the heart during mouse embryogenesis30, there was a possibility that Sm22α promoter-driven PiT-1 deletion could have affected cardiogenesis. However, resting echocardiography under minimal sedation on healthy PiT-1Δsm mice showed no difference in ejection fraction, left ventricular wall thickness and left ventricular end-diastolic volume, compared to wild-type controls (supplemental Table II).
Figure 1. PiT-1 protein depletion in PiT-1Δsm aortic medial smooth muscle cells.

A. Representative western blot showing whole aorta extract from PiT-1flox/flox (fl/fl) and PiT-1Δsm (ΔSM) mice using an anti-PiT-1 antibody (upper panel, arrow). Amido black stained loading control (lower panel). (B). Densitometric analysis of PiT-1 protein levels in pooled aortic media from PiT-1flox/flox (fl/fl, n=8) and PiT-1Δsm (ΔSM, n=8) mice normalized to the loading control and given in arbitrary units (AU). (C). Immunohistochemistry using anti-PiT-1 antibody performed on wildtype (WT) and PiT-1Δsm (ΔSM) paraffin embedded aorta sections. No primary: negative control that was not treated with the anti-PiT-1 antibody. Arrows indicate positive staining in medial VSMC (S) and endothelial cells (E) in WT, but only endothelial cells (E) in PiT-1Δsm. Scale bars=50 μm.
Deletion of PiT-1 from VSMCs had no effect on AMC induced by uremia and high phosphate feeding
We previously developed and characterized a CKD mouse model of AMC using calcification prone DBA/2J mice fed a high phosphate diet25-27. In this model, robust AMC in the absence of inflammation or atherosclerosis develops in CKD mice in response to high phosphate feeding, but does not occur in CKD mice fed a normal phosphate diet. Thus, this model is well suited to investigating the role of phosphate-dependent mechanisms in AMC. To determine if loss of PiT-1 expression in SMC would impact the extent of calcification in this animal model, partial renal ablation was used to generate CKD in PiT-1flox/flox and PiT-1Δsm mice (n=16 per group). After feeding with the high phosphate diet for four weeks, calcium accumulated in the aorta as expected, but the extent of calcification did not differ significantly between the two groups (Figure 2A). Consistent with previous studies25-27, no AMC was observed in CKD mice fed a normal phosphate diet (PiT-1flox/flox =0.9+/-0.1ug/mg; PiT-1Δsm =0.6 +/- 0.1ug/mg). Serum chemistries following renal ablation were not statistically different between groups fed high phosphate (Table 1), suggesting that differences in the degree of renal ablation or hyperphosphatemia achieved did not explain the lack of effect of PiT-1 deletion on AMC.
Figure 2. Aortic calcification in uremic, high phosphate-fed mice.
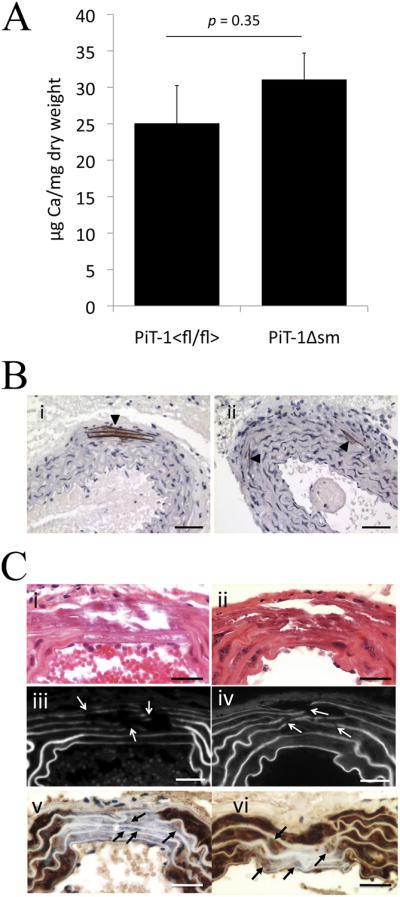
(A) Aortic calcium content was not different (p=.35) between the high phosphate fed CKD mice in the PiT-1Δsm group versus the PiT-1flox/flox group. Data are mean ± s.e. and n=16 per group. (B) Von Kossa staining (brown) with hematoxylin counterstain (blue) showing medial calcification in aortas from (i) PiT-1flox/flox mouse and, (ii) PiT-1Δsm mouse. Scale bars = 50 μm (C) Aortic sections from PiT-1flox/flox (i,iii,v) and PiT-1Δsm (ii,iv,vi) mice showing similar H&E staining (i,ii), eosin fluorescence (iii,iv) with arrowheads showing elastin strand breaks, and Sm22α staining with methyl green nuclear counterstain (v,vi), with arrows pointing to SMC nuclei within the calcified area that have lost or have much reduced Sm22α staining compared to adjacent non-calcified areas. Scale bars = 25 μm.
Table 1.
Serum chemistries.
| Pre-CKD parameters | CKD parameters | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Treatment group | n | P (mg/dL) | Ca (mg/dL) | BUN (mg/dL) | P (mg/dL) | Ca (mg/dL) |
| PiT-1flox/flox | 16 | 8.5 ± 1.6 | 9.4 ± 0.2 | 45 ± 6.5 | 11 ± 0.9 | 9.6 ± 0.6 |
| PiT-1Δsm | 16 | 8.1 ± 1.3 | 9.3 ± 0.2 | 41 ± 5.1 | 13.3 ± 2.1 | 9.3 ± 0.9 |
BUN was measured in all mice. Pre-CKD Ca and P were measured from 6 mice per group, CKD Ca and P were measured in 3 mice per group. Data are mean ± s.e. BUN = blood urea nitrogen, Ca = calcium, n = number of mice in each treatment group, P = phosphate. Parameters were not significantly different between groups.
Aortas from PiT-1Δsm and PiT-1flox/flox mice showed similar histology and SMC gene expression
Aortas from high phosphate fed PiT-1Δsm and PiT-1flox/flox CKD mice showed similar calcified medial wall lesions by von Kossa staining, characterized by elastocalcinosis in the absence of inflammatory cells (Figure 2B). H&E evaluation showed no differences in vascular wall structure between PiT-1Δsm and PiT-1flox/flox CKD mice (Figure 2C.i and 2C.ii), and elastin degradation was evident in both experimental groups under eosin fluorescence imaging (Figure 2C.iii and 2C.iv). VSMC phenotype change, as characterized by loss of SM22α staining (Figure 2C.v and 2C.vi) was apparent in both groups. Furthermore, transcriptional profiling of aortic mRNA showed increased expression of osteochondrogenic genes Runx2, osteopontin, and osteoprotegerin in CKD versus healthy PiT-1flox/flox mice aortas, but no statistically significant differences in these genes between CKD PiT-1Δsm and PiT-1flox/flox mice fed a high phosphate diet (supplemental Table III). Finally, because recent studies have implicated PiT-1 in cell proliferation and survival31, 32, we counted the number of VSMCs per cross-sectional area of aorta to determine if there was a reduction in VSMCs within the medial layer. VSMC cell densities in PiT-1flox/flox and PiT-1Δsm were 3565 ± 378 cells/mm2 and 4158 ± 332 cells/mm2 (mean ± s.e.m.) respectively, showing no statistical difference between genotypes (p=0.27).
SMC specific deletion of PiT-1 did not alter phosphate-induced calcification of aortic rings and cultured VSMCs
To determine whether compensatory circulating factors might explain the lack of effect of PiT-1 deletion on AMC in uremic mice, in vitro calcification of aortic rings and isolated VSMCs was assessed. Aortas were harvested from PiT-1flox/flox and PiT-1Δsm mice to generate aortic rings and calcification was induced by incubation in high-phosphate media for 9 days. As shown in Figure 3A, there was no significant difference in calcification between the two groups. Similar to the results in aortic rings, we found no significant difference in the extent of calcification between PiT-1Δsm and PiT-1flox/flox in cultured VSMCs following treatment with high-phosphate media for 6-10 days (Figure 3B).
Figure 3. Aortic ring calcification and phosphate uptake in PiT-1Δsm (ΔSM) and PiT-1flox/flox (fl/fl) SMC.

(A) Calcification of mouse aortic rings in response to normal (1 mM; open bars) or elevated (2.6 mM; black bars) phosphate, n=5 per group. Data are given as mean ± sd.(B) Fold difference in calcification of PiT-1Δsm VSMC relative to PiT-1flox/flox VSMC following treatment with 2.6mmol/L phosphate. Data are the average of four separate experiments using cell passages 6-8 and were treated for 9-12 days in duration. Expressed as mean + s.e.m. (C) Radiolabeled phosphate transport assays in VSMCs from PiT-1flox/flox (circles) and PiT-1Δsm (squares) mice. Data are expressed as mean ± sd. (D) Lack of inhibition of phosphate uptake by the type II NaPi inhibitor phosphonoformic acid (PFA). Circles are PiT-1flox/flox, squares are PiT-1Δsm. Closed symbols are in the presence of sodium, open symbols are in the absence of sodium. Data are expressed as mean ± sd.
Deletion of PiT-1 in mouse VSMCs had no effect on phosphate uptake
Sodium dependent phosphate uptake kinetics were determined in cultured VSMCs from PiT-1flox/flox and PiT-1Δsm mice and revealed no significant differences between the two genotypes when non-linear curve fitting assuming Michaelis-Menten kinetics was performed (p=0.6) (Figure 3C). The Vmax was measured as 0.315 ± 0.042 and 0.285 ± 0.038 pmole/ug protein/min with a Km of 0.223 ± 0.061 mmol/Land 0.205 ± 0.057 mmol/Lf or the PiT-1flox/flox and PiT-1Δsm VSMCs respectively. Sodium dependent uptake was unchanged in the presence of 0-1mmol/Lphosphonoformic acid, an inhibitor of type II NaPi transport (Figure 3D). As the Ki of phosphonoformic acid for PiT-1 and PiT-2 is in the range of 2.5-5 mmol/L33, 34, lack of inhibition by phosphonoformic acid at sub-millimolar concentrations indicated that type II transporters were not contributing to phosphate uptake in the cultured VSMCs. This finding is consistent with the reverse-transcription PCR data that showed no expression of type II transporters in mouse VSMCs (supplemental Figure I).
VSMC-specific knockdown of PiT-1 was associated with PiT-2 upregulation
Because sodium-dependent phosphate uptake was unchanged in PiT-1 knockout cells compared to controls, we suspected that PiT-2 might be compensating for loss of PiT-1 in VSMC. Real-time qPCR showed that aortic media PiT-2 mRNA was 2-fold higher in PiT-1Δsm mice compared to PiT-1flox/flox controls. All other tissues examined showed no difference in PiT-2 expression between the genotypes (Figure 4A). Likewise, aortic VSMCs isolated from and PiT-1Δsm mice had 2-fold higher PiT-2 mRNA levels compared to PiT-1flox/flox (p<.002) (Figure 4B). Consistent with the mRNA data, PiT-2 protein levels were increased ∼1.6 fold in whole aorta extracts and ∼3.8 fold in VSMC extracts from PiT-1Δsm mice compared to PiT-1flox/flox (Figures 4C and 4D). Expression of type I (SLC17a1) and type II (SLC34a1, SLC34a2 or SLC34a3) sodium-dependent phosphate cotransporters was undetectable in the aortas of high phosphate fed PiT-1flox/flox and PiT-1Δsm CKD mice (data not shown).
Figure 4. PiT-2 expression in tissues and VSMC from PiT-1Δsm and PiT-1flox/flox mice.

(A) qPCR for PiT-2 mRNA expression in various tissues. mRNA levels were derived from standard curves and normalized to 18s, and expressed as ratio of mean mRNA levels in PiT-1Δsm tissue relative to mean levels in PiT-1flox/flox tissue. Error bars represent standard deviation. PiT-2 expression was 2-fold higher in the aortic media of PiT-1Δsm mice. PiT-2 levels were unchanged in the other tissues surveyed. (B) qPCR of PiT-1 and PiT-2 mRNA in cultured VSMCs, showing basal PiT-2 expression in PiT-1flox/flox (fl/fl) VSMCs and increased PiT-2 expression in PiT-1Δsm (ΔSM). (C) Western blot for PiT-2 (upper panel) and loading control (lower panel) in whole aorta extracts. (D) Western blot for PiT-2 (upper panel) and loading control (lower panel) in VSMC extracts.
Overexpression of PiT-2 in PiT-1 knockdown VSMCs restored phosphate uptake and phosphate-induced calcification
We previously reported that knockdown of PiT-1 expression in human VSMCs using shRNA decreased phosphate uptake, phosphate-induced calcification and osteogenic differentiation without compensation by PiT-221. Thus, to determine if PiT-2 could functionally compensate for loss of PiT-1 in VSMCs, we overexpressed PiT-2 by retroviral transduction into PiT-1 knockdown human VSMCs that were previously shown to express ∼80% less PiT-1 than controls 21. Overexpression of PiT-2 was confirmed by qPCR (Figure 5A, graph), and western blotting (Figure 5A, inset). Overexpression of PiT-2 restored phosphate uptake (Figure 5B) as well as calcification (Figure 5C) in PiT-1 shRNA cells. Overexpression of PiT-2 also significantly promoted osteogenic differentiation of VSMCs as measured by a 30% increase in osteogenic index (ratio of OPN/SM22α mRNA levels) compared with vector control following elevated phosphate treatment (data not shown). These data indicated that PiT-2 could functionally compensate for loss of PiT-1 in terms of phosphate uptake, phosphate-induced osteogenic differentiation and calcification.
Figure 5. Overexpression of PiT-2 in PiT-1 knockdown human aortic VSMCs.
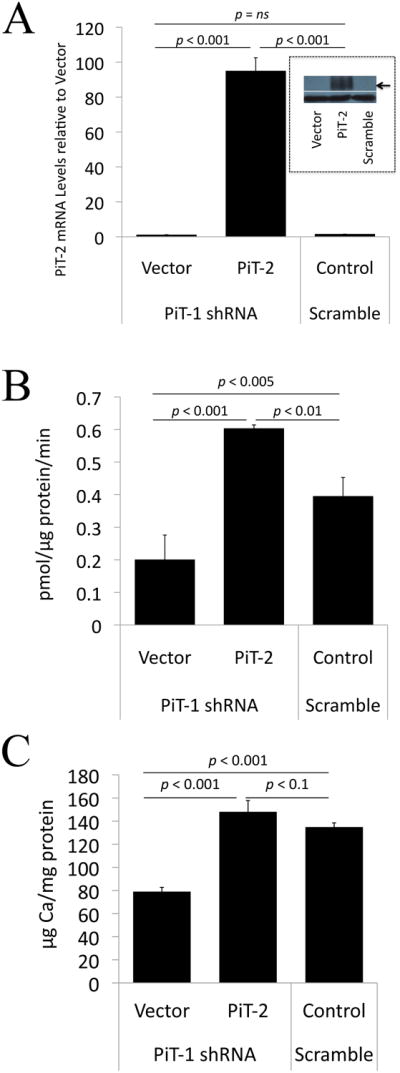
(A) PiT-2 levels in PiT-1 knockdown (PiT-1 shRNA-treated) human VSMCs transduced with vector alone (Vector) or PiT-2 cDNA (PiT-2), or control VSMCs treated with scrambled shRNA (Scramble). Graph shows PiT-2 mRNA levels as determined by qPCR, expressed as PiT-2 mRNA levels relative to Vector. Inset shows PiT-2 protein levels (upper panel; arrow) and b-tubulin loading control (lower panel) as determined by Western blotting (B) Restoration of phosphate (Pi) uptake by PiT-2 overexpression in PiT-1-knockdown cells. Phosphate uptake was measured in 0.1mmol/L phosphate in a 30 min period. (C) Restoration of phosphate-induced calcification by PiT-2 overexpression in PiT-1-knockdown cells. Data are mean ± s.d., n=3 per group.
Knockdown of PiT-2 in PiT-1 deficient mouse VSMCs decreased phosphate transport and phosphate-induced calcification
To determine if PiT-2 was functionally compensating for PiT-1 in the PiT-1Δsm VSMCs, we transduced these cells with scrambled control or PiT-2 specific shRNA to create PiT-1/PiT-2 double knockdown VSMCs. In these cells, PiT-2 mRNA expression was reduced by ∼80% compared to the control cells (Figure 6A). Subsequently, sodium-dependent phosphate uptake was significantly decreased by ∼35% (Figure 6B) and phosphate-induced calcification was significantly decreased by ∼20% in PiT-1 knockdown VSMCs compared to the scrambled control (Figure 6C).
Figure 6. Knockdown of PiT-2 in PiT-1Δsm mouse VSMCs.
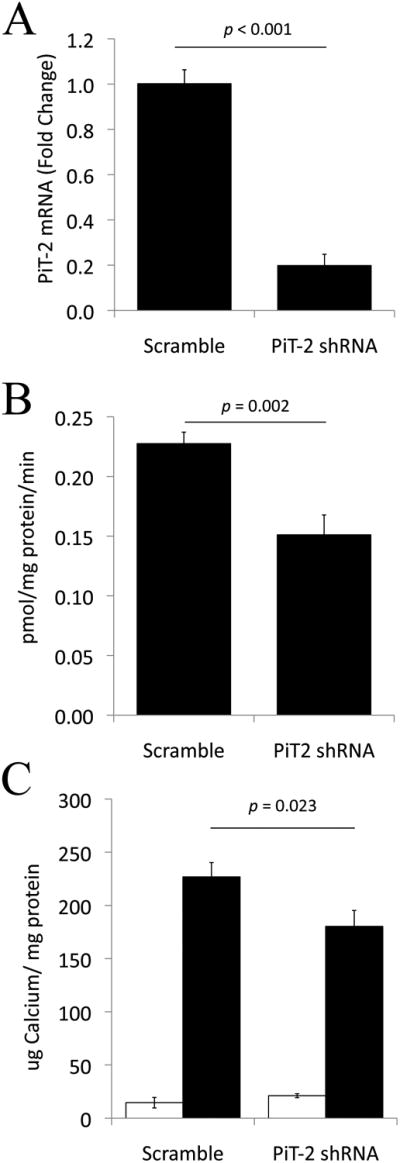
(A) qPCR showing ∼80% knockdown of PiT-2 mRNA in PiT-1 knockdown VSMC (PiT-2 shRNA) compared to scrambled shRNA control VSMC (scrambled). (B) Decreased uptake of radiolabeled phosphate in PiT-2 shRNA expressing human VSMCs compared to scrambled. Cells were incubated in 0.16mmol/L phosphate for 20 minutes. (C) PiT-2 knockdown decreased calcification of PiT-1Δsm VSMCs under high phosphate conditions (PiT-1/PiT-2 double knockdown cells). Open bars = 1mM phosphate; black bars = 2.6mM phosphate. Data are mean ± s.d., n=3 per group.
Discussion
Overall, our studies show that deletion of PiT-1 selectively from VSMCs does not alter the extent of AMC in CKD mice fed a high phosphate diet. Similarly, sodium-dependent phosphate uptake in VSMCs as well as calcification of explanted aortic rings and VSMC primary cultures treated with high phosphate media were equivalent in PiT-1Δsm and PiT-1flox/flox mice. Interestingly, PiT-2 expression was increased two-fold in PiT-1Δsm VSMCs compared to PiT-1flox/flox mice, suggesting that this type III NaPi family member might be compensating for loss of PiT-1. A compensatory role was supported by in vitro studies because overexpression of PiT-2 restored calcification in cultured PiT-1 deficient human VSMCs; additionally, knockdown of PiT-2 in PiT-1Δsm VSMCs diminished calcification. These are the first studies to demonstrate a redundant role for PiT-1 and PiT-2 in vascular calcification.
Previous work in our group demonstrated a role for PiT-1 in phosphate-induced human VSMC calcification and osteochondrogenic transformation in vitro21. Knockdown of PiT-1 by shRNA suppressed phosphate-induced calcification and osteochondrogenic phenotype change, but did not block SMC apoptosis induced by high phosphate under serum free conditions. Others have seen similar results in osteoblast cultures where knockdown of PiT-1 reduced matrix mineralization and reduced expression of osteoblast markers such as OPN35, 36. More recently, Sugita et al37 found that cellular phosphate transport and ATP synthesis mediated by PiT-1 were critical for chondrogenesis in mice. Together, these studies support the hypothesis that PiT-1 is a key player in elevated phosphate-induced calcification. Thus, it was surprising that no differences in SMC phenotype modulation or AMC were observed between PiT-1Δsm and PiT-1flox/flox mice following CKD and high phosphate feeding. Furthermore, no difference was observed in calcification of cultured aortic rings from PiT-1Δsm and PiT-1flox/flox mice, suggesting that compensation by circulating factors was unlikely. Finally, isolated VSMC from PiT-1Δsm and PiT-1flox/flox showed no difference in sodium-dependent phosphate uptake kinetics in vitro. These findings led us to consider a potential cell autonomous compensatory mechanism in smooth muscle cells deficient in PiT-1.
We discovered that PiT-2 mRNA expression was upregulated in aortas in response to developmental loss of PiT-1 specifically in mouse SMCs. This finding was distinct from our previous findings in human VSMC, where levels of PiT-2 were low and unchanged following shRNA knockdown of PiT-1, pointing to a potential developmental compensatory mechanism (discussed in more detail below). The increase in PiT-2 expression in PiT-1Δsm aortas was selective, since no change in other known sodium-dependent phosphate cotransporters was detected. The increase in PiT-2 compensated for PiT-1 in phosphate transport and vascular calcification in vitro. Overexpression of PiT-2 in PiT-1 deficient human SMC restored phosphate uptake as well as calcification following elevated phosphate treatment. Furthermore, siRNA knockdown of PiT-2 in PiT-1Δsm VSMCs diminished both phosphate uptake and calcification. Together, these data support the hypothesis that PiT-2 could compensate for PiT-1 in both sodium-dependent phosphate uptake and phosphate-induced calcification.
While we were able to knockdown PiT-2 mRNA levels by about 80% in PiT-1Δsm VSMC, we observed only a 35% decrease in sodium dependent phosphate uptake and 20% decrease in phosphate-induced calcification. There are several possible explanations for this observation. First, a low density of PiT-2 transporter may be sufficient, within the duress of a high phosphate milieu, to initiate the signaling cascade that results in VSMC calcification. Second, since decline in phosphate uptake and phosphate-induced calcification were comparable, our data might suggest that the phosphate uptake function of PiT-2 is most critical for VSMC calcification. Finally, a combination of these mechanisms may be acting to explain the results.
A review of the literature shows that regulation of PiT-1 and PiT-2 expression is specific to the cell type, physiological state and treatment agent. Byskov32 (in MC3T3-E1 and NIH3T3 cell lines) and our lab (unpublished data in mouse VSMCs) have shown that expression of PiT-1 and PiT-2 is variable based on cell density in vitro, whereby increased cell density results in lower PiT-1 and PiT-2 mRNA expression. BMP-2 has been shown to increase PiT-1 but not PiT-2 mRNA expression in MC3T3-E1 osteoblast cell and human VSMC in vitro35, 38. Likewise, PDGF-BB increased PiT-1 mRNA and protein in rat VSMCs with no change in PiT-239. Our data showing PiT-2 upregulation in VSMCs in response to PiT-1 deletion is in agreement with other reports. Beck et al24 demonstrated that in mice with a global deletion of PiT-1, PiT-2 was upregulated in the embryonic liver and Byskov et al32 showed PiT-2 mRNA upregulation in MC3T3-E1 following PiT-1 shRNA knockdown. However, we are the first to demonstrate that PiT-2 can functionally compensate for PiT-1 in VSMC calcification.
As mentioned above, PiT-2 mRNA expression was upregulated in VSMCs in response to developmental loss of PiT-1 specifically in mouse SMCs and this finding was in contrast to results obtained when shRNA was used to more acutely knockdown PiT-1 in cultured human SMCs. Though the underlying mechanisms of developmental compensation are often enigmatic, the increased level of PiT-2 expression observed in the PiT-1 knockout mice, together with published literature, suggests at least two potential compensatory mechanisms. In the first hypothetical mechanism a decrease in PiT-1 triggers a regulatory switch, which in turn results in increased transcriptional activation of PiT-2, either through increased expression or posttranslational stabilization of a transcriptional activator, or increased accessibility of the PiT-2 promoter. Molecular cloning and sequence analysis have revealed several potential cis-acting elements in the PiT-2 promoter, including a CACCC (KLF) binding site, thyroid hormone response elements, serum response elements, retinoic acid response elements, and vitamin D3-response elements 40. Examination of the endogenous activity of these candidate upstream regulators and their interaction with the PiT-2 promoter in vivo may reveal important information about the mechanism of compensation. Importantly, a KLF family member was recently shown to regulate expression of PiT-1 during erythroid maturation 41. Alternately, the compensatory mechanism may be independent of transcriptional regulation. A decrease in phosphate levels is known to result in enhanced mRNA stability of PiT-2 42, 43. It is possible that the decrease in phosphate intake caused by the absence of PiT-1 during development results in a prolonged half-life of PiT-2 and in turn the higher levels of PiT-2 mRNA that we observed by qPCR (Figure 4A). Future analysis of PiT-1/PiT-2 double knock out models along with evaluation of candidate PiT-2 transcriptional regulators and examination of PiT-2 mRNA stability in vivo will be required to test these two hypotheses. Finally, potential species differences in PiT compensation cannot be ruled out.
The idea that both PiT-1 and PiT-2 may be important regulators of vascular calcification is further strengthened by recent findings in rare human genetic syndromes. Wider et al44 found that mutations in PiT-2 associated with impaired phosphate transport caused familial idiopathic basal ganglia calcification, a condition characterized by mineralization of capillaries as well as small arteries and veins of the basal ganglion region44. While the exact mechanism for this effect is unknown, the authors speculated that loss of PiT-2 function and regional phosphate homeostasis might lead to increased PiT-1 function in VSMC, thereby promoting osteochondrogenic differentiation and calcification. Furthermore, overexpression of PiT-1 has been reported in fibroblasts derived from patients with Werner syndrome, an autosomal recessive disorder caused by mutations in RecQ DNA helicase that is characterized by premature aging and soft tissue calcification45.
In summary, while we have shown that the extent of AMC in high phosphate fed, uremic mice was not different between the PiT-1Δsm and PiT-1flox/flox, nor in vitro using VSMC primary culture from these mice, we discovered increased PiT-2 expression in the PiT-1Δsm VSMCs. Using in vitro methods, we demonstrated that PiT-2 overexpression could compensate for phosphate uptake and phosphate-induced calcification in PiT-1 deficient VSMCs, while knockdown of PiT-2 reduced phosphate uptake and calcification. Taken together our data suggests redundant roles for PiT-1 and PiT-2 in phosphate-driven AMC.
Supplementary Material
Significance.
Vascular calcification (VC) is a major risk factor for cardiovascular morbidity and mortality, and elevated phosphate has been identified as a key inducer of VC via procalcific effects on vascular smooth muscle cells (SMCs). We identified a novel function for the sodium dependent phosphate transporter, PiT-2, as a mediator of elevated-phosphate induced VC in vitro and in vivo. Moreover, we provide mechanistic insight into compensatory mechanisms that operate in SMCs to protect against phosphate transporter deficiency. Importantly, as our studies were in progress, PiT-2 was identified as the causative gene for idiopathic basal ganglion calcification in people, and thus our studies noting compensatory mechanisms for phosphate transporters may help to explain how mutation of PiT-2 might lead to compensatory changes that actually facilitate VC. Clinically, our data provide a cautionary note regarding compensatory pathways that should be considered when attempting to translate inhibition of phosphate transport to clinical therapies.
Acknowledgments
Sources of funding: This study was funded by NIH grants to CM Giachelli (R01 HL62329 and R01 HL081785). WL Lau and MH Crouthamel received funding from T32 HL007828, and WL Lau was also funded by T32 DK007467.
Nonstandard Abbreviations
- AMC
arterial medial calcification
- CKD
chronic kidney disease
- Pi
inorganic phosphate
- PiT-1
type III sodium-dependent phosphate cotransporter 1 (SLC20A1)
- PiT-2
type III sodium-dependent phosphate cotransporter 2 (SLC20A2)
- VSMC
vascular smooth muscle cell
Footnotes
Disclosures: Nothing to disclose.
References
- 1.U.S. Renal data system (usrds) 2011 annual data report. Atlas of End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: http://www.usrds.org/adr.htm. [Google Scholar]
- 2.Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938–942. doi: 10.1161/hy1001.096358. [DOI] [PubMed] [Google Scholar]
- 3.Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–1483. doi: 10.1056/NEJM200005183422003. [DOI] [PubMed] [Google Scholar]
- 4.Schlieper G, Aretz A, Verberckmoes S, et al. Ultrastructural analysis of vascular calcifications in uremia. J Am Soc Nephrol. 2010;21:689–696. doi: 10.1681/ASN.2009080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, Hiorns M, Donald AE, Deanfield J, Rees L, Shanahan CM. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 6.Ibels LS, Alfrey AC, Huffer WE, Craswell PW, Anderson JT, Weil R., 3rd Arterial calcification and pathology in uremic patients undergoing dialysis. Am J Med. 1979;66:790–796. doi: 10.1016/0002-9343(79)91118-5. [DOI] [PubMed] [Google Scholar]
- 7.Dhingra R, Sullivan L, Fox C, Wang T, D'Agostino RS, Gaziano J, Vasan R. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167:879–885. doi: 10.1001/archinte.167.9.879. [DOI] [PubMed] [Google Scholar]
- 8.Kestenbaum BR, Adeney KL, de Boer IH, Ix JH, Shlipak MG, Siscovick DS. Incidence and progression of coronary calcification in chronic kidney disease: The multi-ethnic study of atherosclerosis. Kidney Int. 2009;76:991–998. doi: 10.1038/ki.2009.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonelli M, Curhan G, Pfeffer M, Sacks F, Thadhani R, Melamed ML, Wiebe N, Muntner P. Relation between alkaline phosphatase, serum phosphate, and all-cause or cardiovascular mortality. Circulation. 2009;120:1784–1792. doi: 10.1161/CIRCULATIONAHA.109.851873. [DOI] [PubMed] [Google Scholar]
- 10.Jono S, McKee M, Murry C, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli C. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 11.Giachelli CM, Jono S, Shioi A, Nishizawa Y, Mori K, Morii H. Vascular calcification and inorganic phosphate. Am J Kidney Dis. 2001;38:S34–37. doi: 10.1053/ajkd.2001.27394. [DOI] [PubMed] [Google Scholar]
- 12.Lomashvili K, Cobbs S, Hennigar R, Hardcastle K, O'Neill W. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J Am Soc Nephrol. 2004;15:1392–1401. doi: 10.1097/01.asn.0000128955.83129.9c. [DOI] [PubMed] [Google Scholar]
- 13.Lau WL, Pai A, Moe SM, Giachelli CM. Direct effects of phosphate on vascular cell function. Adv Chronic Kidney Dis. 2011;18:105–112. doi: 10.1053/j.ackd.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ Res. 2011;109:697–711. doi: 10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steitz S, Speer M, Curinga G, Yang H, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli C. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 16.Reimer RJ, Edwards RH. Organic anion transport is the primary function of the slc17/type i phosphate transporter family. Pflugers Arch. 2004;447:629–635. doi: 10.1007/s00424-003-1087-y. [DOI] [PubMed] [Google Scholar]
- 17.Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2010;299:F285–296. doi: 10.1152/ajprenal.00508.2009. [DOI] [PubMed] [Google Scholar]
- 18.Werner A, Dehmelt L, Nalbant P. Na+-dependent phosphate cotransporters: The napi protein families. J Exp Biol. 1998;201:3135–3142. doi: 10.1242/jeb.201.23.3135. [DOI] [PubMed] [Google Scholar]
- 19.Kavanaugh M, Miller D, Zhang W, Law W, Kozak S, Kabat D, Miller A. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci U S A. 1994;91:7071–7075. doi: 10.1073/pnas.91.15.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villa-Bellosta R, Bogaert Y, Levi M, Sorribas V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler Thromb Vasc Biol. 2007;27:1030–1036. doi: 10.1161/ATVBAHA.106.132266. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Yang H, Giachelli C. Role of the sodium-dependent phosphate cotransporter, pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–912. doi: 10.1161/01.RES.0000216409.20863.e7. [DOI] [PubMed] [Google Scholar]
- 22.Olah Z, Lehel C, Anderson W, Eiden M, Wilson C. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J Biol Chem. 1994;269:25426–25431. [PubMed] [Google Scholar]
- 23.Bøttger P, Hede SE, Grunnet M, Høyer B, Klaerke DA, Pedersen L. Characterization of transport mechanisms and determinants critical for na+-dependent pi symport of the pit family paralogs human pit1 and pit2. Am J Physiol Cell Physiol. 2006;291:C1377–1387. doi: 10.1152/ajpcell.00015.2006. [DOI] [PubMed] [Google Scholar]
- 24.Beck L, Leroy C, Beck-Cormier S, Forand A, Salaün C, Paris N, Bernier A, Ureña-Torres P, Prié D, Ollero M, Coulombel L, Friedlander G. The phosphate transporter pit1 (slc20a1) revealed as a new essential gene for mouse liver development. PLoS One. 2010;5:e9148. doi: 10.1371/journal.pone.0009148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Abbadi MM, Pai AS, Leaf EM, Yang HY, Bartley BA, Quan KK, Ingalls CM, Liao HW, Giachelli CM. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009;75:1297–1307. doi: 10.1038/ki.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol. 2011;178:764–773. doi: 10.1016/j.ajpath.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, Giachelli CM. Vitamin d receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–1270. doi: 10.1038/ki.2012.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivandic BT, Utz HF, Kaczmarek PM, Aherrahrou Z, Axtner SB, Klepsch C, Lusis AJ, Katus HA. New dyscalc loci for myocardial cell necrosis and calcification (dystrophic cardiac calcinosis) in mice. Physiol Genomics. 2001;6:137–144. doi: 10.1152/physiolgenomics.2001.6.3.137. [DOI] [PubMed] [Google Scholar]
- 29.Festing M, Speer M, Yang H, Giachelli C. Generation of mouse conditional and null alleles of the type iii sodium-dependent phosphate cotransporter pit-1. Genesis. 2009;47:858–863. doi: 10.1002/dvg.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Miano J, Cserjesi P, Olson E. Sm22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78:188–195. doi: 10.1161/01.res.78.2.188. [DOI] [PubMed] [Google Scholar]
- 31.Beck L, Leroy C, Salaün C, Margall-Ducos G, Desdouets C, Friedlander G. Identification of a novel function of pit1 critical for cell proliferation and independent of its phosphate transport activity. J Biol Chem. 2009;284:31363–31374. doi: 10.1074/jbc.M109.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byskov K, Jensen N, Kongsfelt IB, Wielsøe M, Pedersen LE, Haldrup C, Pedersen L. Regulation of cell proliferation and cell density by the inorganic phosphate transporter pit1. Cell Div. 2012;7:7. doi: 10.1186/1747-1028-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravera S, Virkki L, Murer H, Forster I. Deciphering pit transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol. 2007;293:C606–620. doi: 10.1152/ajpcell.00064.2007. [DOI] [PubMed] [Google Scholar]
- 34.Virkki L, Biber J, Murer H, Forster I. Phosphate transporters: A tale of two solute carrier families. Am J Physiol Renal Physiol. 2007;293:F643–654. doi: 10.1152/ajprenal.00228.2007. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki A, Ghayor C, Guicheux J, Magne D, Quillard S, Kakita A, Ono Y, Miura Y, Oiso Y, Itoh M, Caverzasio J. Enhanced expression of the inorganic phosphate transporter pit-1 is involved in bmp-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21:674–683. doi: 10.1359/jbmr.020603. [DOI] [PubMed] [Google Scholar]
- 36.Yoshiko Y, Candeliere GA, Maeda N, Aubin JE. Osteoblast autonomous pi regulation via pit1 plays a role in bone mineralization. Mol Cell Biol. 2007;27:4465–4474. doi: 10.1128/MCB.00104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugita A, Kawai S, Hayashibara T, Amano A, Ooshima T, Michigami T, Yoshikawa H, Yoneda T. Cellular atp synthesis mediated by type iii sodium-dependent phosphate transporter pit-1 is critical to chondrogenesis. J Biol Chem. 2011;286:3094–3103. doi: 10.1074/jbc.M110.148403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Yang H, Giachelli C. Bmp-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis. 2008;199:271–277. doi: 10.1016/j.atherosclerosis.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villa-Bellosta R, Levi M, Sorribas V. Vascular smooth muscle cell calcification and slc20 inorganic phosphate transporters: Effects of pdgf, tnf-alpha, and pi. Pflugers Arch. 2009;458:1151–1161. doi: 10.1007/s00424-009-0688-5. [DOI] [PubMed] [Google Scholar]
- 40.Bai L, Collins JF, Xu H, Xu L, Ghishan FK. Molecular cloning of a murine type iii sodium-dependent phosphate cotransporter (pit-2) gene promoter. Biochim Biophys Acta. 2001;1522:42–45. doi: 10.1016/s0167-4781(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 41.Forand A, Beck L, Leroy C, Rousseau A, Boitez V, Cohen I, Courtois G, Hermine O, Friedlander G. Eklf-driven pit1 expression is critical for mouse erythroid maturation in vivo and in vitro. Blood. 2013;121:666–678. doi: 10.1182/blood-2012-05-427302. [DOI] [PubMed] [Google Scholar]
- 42.Richardson C, Bank A. Developmental-stage-specific expression and regulation of an amphotropic retroviral receptor in hematopoietic cells. Mol Cell Biol. 1996;16:4240–4247. doi: 10.1128/mcb.16.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chien ML, O'Neill E, Garcia JV. Phosphate depletion enhances the stability of the amphotropic murine leukemia virus receptor mrna. Virology. 1998;240:109–117. doi: 10.1006/viro.1997.8933. [DOI] [PubMed] [Google Scholar]
- 44.Wider C, Dickson DW, Schweitzer KJ, Broderick DF, Wszolek ZK. Familial idiopathic basal ganglia calcification: A challenging clinical-pathological correlation. J Neurol. 2009;256:839–842. doi: 10.1007/s00415-009-5025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honjo S, Yokote K, Fujimoto M, Takemoto M, Kobayashi K, Maezawa Y, Shimoyama T, Satoh S, Koshizaka M, Takada A, Irisuna H, Saito Y. Clinical outcome and mechanism of soft tissue calcification in werner syndrome. Rejuvenation Res. 2008;11:809–819. doi: 10.1089/rej.2007.0649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.