

Abstract
Introduction
The neurofibromatoses (neurofibromatosis type 1, NF1 and neurofibromatosis type 2, NF2) comprise the most common inherited conditions in which affected children and adults develop tumors of the central and peripheral nervous system. In this review, the authors discuss how the establishment of the Neurofibromatosis Clinical Trials Consortium (NFCTC) has positively impacted on the design and execution of treatment studies for individuals with NF1 and NF2.
Areas covered
Using an extensive PUBMED search in collaboration with select NFCTC members expert in distinct NF topics, the authors discuss the clinical features of NF1 and NF2, the molecular biology of the NF1 and NF2 genes, the development and application of clinically relevant Nf1 and Nf2 genetically engineered mouse models and the formation of the NFCTC to enable efficient clinical trial design and execution.
Expert opinion
The NFCTC has resulted in a more seamless integration of mouse preclinical and human clinical trials efforts. Leveraging emerging enabling resources, current research is focused on identifying subtypes of tumors in NF1 and NF2 to deliver the most active compounds to the patients most likely to respond to the targeted therapy.
Keywords: clinical trials, mouse models, NF1, NF2, preclinical
1. Introduction
Neurofibromatosis type 1 (NF1) and type 2 (NF2) are molecularly distinct conditions clinically characterized by the development of diverse disease manifestations occurring over the lifetime of affected individuals [1]. NF1 is considerably more common, with a birth incidence of approximately 1 in 2600 – 4500 individuals (prevalence, 1:2700), while NF2 has an incidence of 1 in 33,000 individuals and a prevalence of 1:56,000 [2-5]. In this regard, both conditions are considered orphan diseases with great variability in the onset and spectrum of clinical manifestations. Over the past decade, new insights into the molecular pathogenesis and the biological underpinnings of these two disorders have set the stage for the design of rational biologically-informed, molecular-targeted therapies. However, for such therapies to be effective, they have to be evaluated in appropriately-selected patient populations with clinical or molecular features pertinent to the experimental agent. Rigorous outcome measures must be applied and, whenever possible, biomarkers for early readouts of efficacy or futility should be incorporated into treatment trial designs.
2. Clinical targets for therapeutic trials
NF1 and NF2 differ from one another both phenotypically and pathogenetically, and therefore present different clinical targets for therapy and different challenges in design of clinical trials. Clinical and genetic features of the two major neurofibromatoses (NF1 and NF2) are summarized in Table 1 and major complications and treatment targets in Table 2.
Table 1. Major features of the neurofibromatosis and diagnostic criteria.
| NF1 | NF2 | |
|---|---|---|
| Incidence* | 1:2600 – 1:4500 | 1:33,000 |
| Prevalence* | 1:2700 | 1:56,000 |
| Inheritance | Autosomal dominant, complete penetrance | Autosomal dominant, complete penetrance |
| Features | Café-au-lait macules, skinfold freckling, neurofibromas, plexiform neurofibromas, malignant peripheral nerve sheath tumors, optic glioma, skeletal dysplasia, learning disability |
Vestibular schwannomas, other cranial and peripheral nerve schwannomas, meningiomas, ependymomas, cataracts |
| Gene/Protein |
NF1 – chromosome 17/neurofibromin – GTPase activating protein |
NF2 – chromosome 22/merlin – cytoskeletal protein |
| Diagnostic criteria | At least two of: > 6 café-au-lait macules (> 5 mm prepubertal, > 15 postpubertal) |
Definite diagnosis: bilateral VS, or family history of NF2 and unilateral VS or any two of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular cataract |
| 2 or more neurofibromas or 1 plexiform neurofibroma |
||
| iris Lisch nodules | or | |
| skinfold freckling optic glioma characteristic skeletal dysplasia (long bone or sphenoid) |
unilateral VS and any two of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular cataract or |
|
| affected first-degree relative | multiple meningiomas plus unilateral VS or any two of: glioma, neurofibroma, schwannoma, posterior subcapsular cataract |
Table 2. Major treatment targets in the neurofibromatoses.
| Condition | Features | Goals | Age |
|---|---|---|---|
| NF1 | Café-au-lait macules | Improve cosmesis | Any age |
| Dermal neurofibromas | Improve appearance and comfort | Adolescence – adulthood | |
| Spinal neurofibromas | Relieve cord or nerve compression | Any | |
| Plexiform neurofibromas | Relieve disfigurement and/or pain and/or functional impairment |
Most often in childhood; reduce tumor size at any age |
|
| Optic glioma | Avoid loss of vision; treat hypothalamic dysfunction | Childhood | |
| Skeletal dysplasia | Avoid fracture; improve healing | Mostly childhood | |
| Vascular dysplasia | Treat or prevent hypertension; avoid vascular occlusion or dissection |
Any age | |
| Malignancy | Prevent malignant change; diagnose malignancy promptly; improve outcome of treatment |
Any age | |
| NF2 | Vestibular schwannoma | Reduce tumor size; improve hearing; prevent brainstem compression |
Any age, but most often late childhood, adolescence, adulthood |
| Cranial nerve schwannoma | Reduce tumor size and improve cranial nerve function (facial movement, swallowing) |
||
| Spinal schwannoma | Reduce tumor size; relieve pressure on spinal cord | ||
| Ependymoma | Reduce neurological impair/tumor size | ||
| Meningioma | Reduce tumor size, functional impairment, seizures |
NF1: Neurofibromatosis type 1; NF2: Neurofibromatosis type 2.
2.1 Neurofibromatosis type 1
NF1 is the most common form of neurofibromatosis, and the most diverse in terms of clinical features. These include both benign and malignant tumors as well as developmental abnormalities. The hallmark tumor of NF1 is the neurofibroma, a benign tumor comprising Schwann cells, fibroblasts, perineurial cells and mast cells [6]. Neurofibromas occur along peripheral nerves throughout the body and can either be localized lesions ranging in size from millimeters to centimeters or plexiform neurofibromas that may be tens of centimeters in size. The cutaneous form of localized neurofibromas is easily seen (Figure 1A), where they may be flat, pedunculated or create a localized depression in the skin surface. They are usually soft in texture and painless, although they may itch and can cause disfigurement if present in large numbers. Cutaneous and subcutaneous neurofibromas usually start to appear in puberty; however, neurofibromas can also be seen in younger children. They rarely grow to more than a few centimeters and most are much smaller, but some people may be carpeted with innumerable lesions. Localized neurofibromas can also grow internally, where they may compress vital structures, such as the spinal cord or major vessels, and may cause pain. Some nerve root tumors grow in a dumbbell-shaped pattern through the neural foramen (Figure 1B), potentially leading to both radiculopathies and spinal cord compression. Localized neurofibromas are rarely problematic in children, but may accumulate in large numbers beginning in adolescence.
Figure 1. Features associated with NF1.
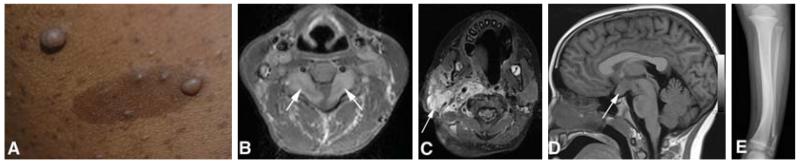
(A) Multiple dermal neurofibromas and café-au-lait macules; (B) bilateral cervical nerve root tumors (arrows) with cord compression; (C) facial plexiform neurofibroma (arrow); (D) glioma involving the optic chiasm (arrow); (E) bowing of the tibia and fracture of the fibula.
Plexiform neurofibromas represent tumors that involve multiple nerve fascicles and can grow along a length of nerve and its branches [7]. They may appear as small plaque-like growths on the skin or can grow to enormous size, leading to disfigurement of the face, trunk or extremities (Figure 1C). Internal plexiform neurofibromas can obstruct major organs or compress the spinal cord. They can also invade the skin and surrounding soft tissues, growing in a diffuse manner. Plexiform neurofibromas usually present in childhood, and may be congenital. They usually exhibit their most rapid growth in childhood [8].
The only established treatment for neurofibromas, whether localized, plexiform, cutaneous or deep, is surgery. Cutaneous tumors can be removed by CO2 laser treatment or electrocauterization, which are essentially surgical treatments. Many adults will choose to have some cutaneous neurofibromas removed; however, the total number of tumors is usually too great for all masses to be removed, and additional tumors accumulate over time. Plexiform neurofibromas can be removed surgically, but their infiltrative nature makes it difficult or impossible to fully resect them, and regrowth is therefore common [9]. In addition, the complete investment of the tumor within the associated nerve puts patients at high risk for iatrogenic nerve injury during the course of surgery.
Central nervous system tumors are also a predominant feature of NF1. Typically, these are pilocytic astrocytomas in children [10,11]; the most common location is the optic pathway, including the optic nerves and/or chiasm (Figure 1D), and sometimes the hypothalamus. Imaging studies reveal optic pathway tumors in 15% of children with NF1 [12], although at least half remain asymptomatic. When symptoms occur, these include loss of vision or hypothalamic disturbances presenting as precocious puberty. Orbital tumors can cause proptosis and pain. Optic pathway tumors usually have their onset in childhood, typically before 7 years of age. Current surveillance includes annual ophthalmological evaluations and neuroimaging when abnormalities are found on examination. Treatment (vincristine/carboplatin chemotherapy) is only indicated for progressive symptomatic tumors [13]. Most patients experience stabilization of their disease, although they may not recover lost vision. A subset will progress despite treatment. Recent studies have shown that young age (< 2 years) and postchiasmal involvement are associated with worse clinical outcome [14]. Pilocytic astrocytomas are also found in the brainstem and also often exhibit an indolent course [15]. Radiation therapy is usually avoided due to an increased risk of secondary malignancy and vascular abnormalities (moyamoya disease) [16].
The major malignant tumor associated with NF1 is the malignant peripheral nerve sheath tumor (MPSNT), which usually arises from a pre-existing plexiform or large nodular neurofibroma. The lifetime risk is 8 – 13%, with presentation most frequently occurring in the second through fifth decades [17]. Symptoms include pain, neurological deficit, sudden tumor growth and change in consistency from soft to hard. Positron emission tomography (PET) imaging with fluorodeoxyglucose may be helpful in distinguishing benign from malignant tumors [18]. Treatment outcome is best for tumors that can be fully resected and is very poor otherwise. Other less common malignant tumors in NF1 patients include juvenile myelomonocytic leukemia, pheochromocytoma and gastrointestinal stromal cell tumors [19]. An increased risk of breast cancer has also been reported [20].
Developmental or dysplastic defects can affect skin, bone, vascular tissue and brain. Skin features include hypermelanotic macules (café-au-lait macules) (Figure 1A) and skinfold freckles, which present in childhood and do not cause harm. Skeletal disorders include bone dysplasia, non-ossifying cysts and generalized osteopenia [21]. The most characteristic site for dysplasia is the tibia (Figure 1E), but other long bones or bones around the orbit may also be involved. Tibial dysplasia occurs congenitally and may lead to fractures that fail to heal spontaneously, creating a ‘false joint’ or pseudarthrosis. Current surgical treatment of these lesions is extremely difficult. Orbital dysplasia is often associated with plexiform neurofibroma arising from the trigeminal nerve and can be disfiguring and disabling as a result of the involvement of local structures. Non-ossifying cysts can lead to fracture, as can osteopenia [22,23]. Vascular dysplasia, such as renal artery stenosis, can cause hypertension in children with NF1; this may be treated medically or by stenting or surgery. Dysplasia of the carotid (moyamoya syndrome) can cause strokes [24], and arterial dysplasias have been reported to lead to dissection and hemorrhage [25].
Neurocognitive disorders occur very commonly, in at least 50% of affected children. A wide range of problems can occur, including attention deficit disorder/hyperactivity, verbal and non-verbal learning disabilities and sometimes intellectual impairment [26]. There may also be hypotonia, speech and articulation problems, and social immaturity. All of these issues take a toll in terms of educational achievement. Attention deficit disorder responds to stimulant medication [27], but the other neurocognitive features are best managed through educational assessment and provision of required support.
2.2 Neurofibromatosis type 2
Most of the features of NF2 are tumor-related; the tumors are benign, but may cause major morbidity. The hallmark tumor is the vestibular schwannoma, which occurs bilaterally (Figure 2A) [28]. Vestibular tumors can present at any time in life, from early childhood through adulthood. They typically present with hearing loss and/or tinnitus, often progressing to deafness. Current treatment is either surgery or stereotactic radiation therapy, although there is concern about malignant transformation due to radiation exposure. Preliminary studies with the angiogenesis inhibitor bevacizumab have shown promising results, but this treatment remains experimental at this time [29-31].
Figure 2. Features associated with NF2.
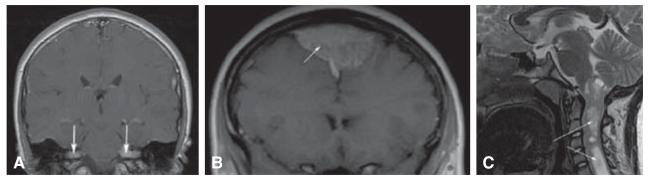
(A) Bilateral vestibular schwannomas (arrows); (B) meningioma (arrow); (C) upper cervical cord ependymoma (arrows).
Schwannomas can arise along other cranial nerves (especially the fifth and the ninth or tenth cranial nerves), spinal nerves (potentially compressing the spinal cord) and skin (presenting as plaque-like lesions). Surgical treatment is the only therapeutic option for symptomatic tumors. Other tumors in NF2 patients include meningiomas and ependymomas. Meningiomas can cause major symptoms due to compression of brain or spinal cord, and surgery is the only effective treatment (Figure 2B). Ependymomas (Figure 2C) often are asymptomatic, but may rarely cause myelopathy, requiring surgical intervention [32].
A major non-tumor manifestation of NF2 is the cataract, usually a posterior subcapsular or cortical wedge opacity [33], which is usually asymptomatic, but can interfere with vision. Rarely, patients with NF2 develop a polyneuropathy associated with Schwann cell proliferation along peripheral nerves [34].
3. Molecular targets for therapeutic trials
With the identification of the NF1 and NF2 genes, we have ushered in an era where individualized, targeted therapies become achievable. The discovery of these causative genes has also led to new insights into the molecular and cellular etiologies of numerous NF1- and NF2-associated clinical problems.
3.1 NF1 gene
The NF1 gene, located on chromosome 17q, was identified in 1990 by positional cloning strategies [35-37]. Encoding the 2818 residue protein, neurofibromin, the NF1 locus includes over 60 coding exons, including three exons exhibiting relative tissue specificity (Figure 3A) [38-40]. Inspection of the predicted coding sequence of neurofibromin revealed a 300 amino acid region containing a catalytic domain similar to proteins that function as negative regulators of the RAS proto-oncogene [41,42]. These RAS GTPase activating protein (GAP) molecules accelerate the conversion of RAS from its active, growth-promoting GTP-bound conformation to its inactive GDP-bound form. In this manner, neurofibromin loss, as found in tumors from individuals with NF1, results in high levels of RAS activity and increased downstream RAS promitogenic signaling (Figure 3B). Active RAS leads to increased RAF/MEK and AKT/mTOR activation, which each can promote cell growth in specific NF1-deficient cell types. In addition to neurofibromin RAS regulation, emerging evidence from both flies and mammals has shown that neurofibromin is also a positive regulator of adenylyl cyclase function and intracellular cyclic adenosine monophosphate (cAMP) generation [43,44]. In cell types with reduced or absent NF1 gene expression, lower levels of cAMP are found [44,45]. Relevant to clinical trial design, each of these neurofibromin signal transduction molecules becomes a viable target for potential therapeutic drug development.
Figure 3. Structure and function of NF1 and NF2 proteins.

(A) Structure of the predicted NF1 protein, neurofibromin. GRD = GAP-related domain. Triangles denote the three known alternatively spliced exons (9a, 23a and 48a). (B) Neurofibromin functions as a negative regulator of the RAS proto-oncogene and inhibits RAS activity and downstream RAS effector (RAF/MEK and AKT/mTOR) signaling. In addition, neurofibromin positive regulates adenylyl cyclase (AC) function and cyclic AMP (cAMP) generation. (C) Structure of the predicted NF2 protein, merlin or schwannomin. FERM = protein 4.1, ezrin, radixin, moesin domain. CTD = carboxyl terminal domain. The triangle denotes the alternatively spliced exon 16. (D) Merlin negatively regulates a number of intracellular signaling molecules, including mTOR, RAC1 and FAK, depending on the cell type examined. The red asterisks denote signaling intermediates that serve as potential targets for therapeutic drug design.
3.2 NF2 gene
The NF2 gene, located on chromosome 22q, was identified in 1993 using a similar cloning strategy [46,47]. Encoding the 595 residue protein, merlin (or schwannomin), the NF2 locus includes 17 coding exons, including one (exon 16) which is alternatively spliced in specific cell types [48]. Inspection of the predicted coding sequence of merlin revealed striking similarity to a family of proteins containing Band 4.1 domains. This large family of molecules includes the cytoskeleton linking proteins, ezrin, radixin and moesin, and the conserved region is thus termed the 4.1-ezrin-radixin-moesin (FERM) domain (Figure 3C). In addition, merlin contains two other domains, a region with a predicted α-helical structure and a carboxyl-terminal domain (CTD). While the function of merlin remains to be fully elucidated, merlin loss (as found in NF2-associated tumors) has been associated with increased mTOR activation, RAC1 activity and FAK signaling [49-55]. In addition, several groups have reported increased activation of receptor tyrosine kinase (RTK) family members in NF2-deficient cells [54,55]. As with neurofibromin, each of these hyperactivated signaling molecules represent possible rational targets for therapeutic drug design (Figure 3D).
3.3 Genetically-engineered mouse models
With these potential targets in hand, the next logical step is the evaluation of drugs that block the hyperactivation of these molecules in preclinical small-animal models of NF-associated clinical abnormalities. Over the past 15 years, a large number of robust genetically engineered mouse (GEM) models have been developed and exploited for preclinical drug studies (Table 3). In this regard, Nf1 GEM strains have been designed to model the cognitive (memory/learning and attention deficits), bone (spine and extremity defects) and tumor phenotypes (optic glioma, malignant peripheral nerve tumor, plexiform neurofibroma, dermal neurofibroma, leukemia, malignant glioma and pheochromocytoma) encountered in individuals with NF1. Similarly, Nf2 GEM strains for schwannoma and meningioma have been developed for small-animal therapeutic studies.
Table 3. GEM models of NF-associated clinical problems.
| NF1 | ||
|---|---|---|
| Memory and learning deficits | Nf1 +/− mice | [93] |
| Nf1fiox/mut; GFAP-Cre mice | [94] | |
| Attention deficit | Nf1 +/− mice | [93] |
| Nf1fiox/mut; GFAP-Cre mice | [95] | |
| Nf1fiox/wt; Syn1-Cre mice | [96] | |
| Nf1fiox/wt; Dix6-Cre mice | [96] | |
| Optic glioma | Nf1flox/mut; GFAP-Cre | [63,64] |
| Nf1 +/−; LSL-KRas; GFAP-Cre mice | [97] | |
| Malignant glioma | Nf1flox/flox. p53flox/flox; ptenflox/flox mice | [98] |
| Nf1flox/flox; p53flox/flox mice | [99] | |
| Dermal neurofibroma | Nf1flox/mut; PLP-CreER mice | [100] |
| Plexiform neurofibroma | Nf1flox/flox; Dhh-Cre mice | [101] |
| Nf1flox/flox; plp-Cre mice | [100] | |
| Nf1flox/mut; Krox20-Cre mice | [61] | |
| Malignant peripheral nerve sheath tumor | Nf1 +/−; p53+/− mice | [102,103] |
| Long bone abnormalities | Nf1flox/flox; Prx1-Cre mice | [104] |
| Nf1flox/mut mice + Cre | [105] | |
| Nf1flox/flox; Col2α1-Cre mice | [106] | |
| Skeletal abnormalities | Nf1flox/mut; Col2.3-Cre mice | [107] |
| Leukemia | Nf1flox/flox; Msx1-Cre mice | [108] |
| Pheochromocytoma | Nf1 +/− mice | [109] |
|
| ||
| NF2 | ||
|
| ||
| Schwannoma | Nf2flox/flox mice +Ad-Cre | [110] |
| Nf2Δ39-121; P0-Cre mice | [111] | |
| Meningioma | Nf2flox/flox; PGDS-Cre mice | [112] |
| Nf2flox/flox mice +Ad-Cre | [113] | |
| Ependymoma | None to date | |
| Mesothelioma | Nf2flox/flox; p16flox/flox mice +Ad-Cre | [114] |
| Nf2flox/flox; p53flox/flox mice +Ad-Cre | [114] | |
| Cataracts | Nf2flox/flox mice +Ad-Cre | [110] |
| Neuropathy | Nf2exon16 knockout mice | [115] |
GEM: Genetically engineered mouse; NF: Neurofibromatosis.
While each model has inherent limitations, they have been enormously instructive for human clinical trial design. First, the use of these GEM strains has revealed that the precise mechanism by which neurofibromin (or merlin) regulates cell growth or function varies from tissue to tissue. For example, neurofibromin growth regulation in NF1-deficient leukemic cells is primarily related to increased RAS/MEK signaling [56,57], whereas in Nf1-deficient astrocytes, neurofibromin growth control reflects RAS/mTOR hyperactivation [58,59]. Additionally, in some NF1-associated tumors (plexiform neurofibromas), both Ras/MEK and Ras/mTOR hyperactivation underlie increased proliferation [56,57,60]. Similarly, Nf2-deficient glial cells depend on merlin inhibition of ErbB2 activation [54], while other Nf2-deficient cell types are dependent on merlin suppression of a related RTK, the EGFR [55]. The diversity of signal transduction pathways regulated by merlin and neurofibromin necessitates a more comprehensive analysis of the molecular mechanism(s) that underlie the function of these tumor suppressor proteins in the specific cell types to be targeted. As such, we have to appreciate the molecular ‘currency’ important for cell growth control in any given tissue in order to identify compounds with the highest likelihood of success.
Second, Nf1 GEM strains have revealed a requirement for non-neoplastic cells in the tumor microenvironment in the process of tumorigenesis and continued growth. Specifically, Nf1 loss in Schwann cell or astroglial cell precursors alone is insufficient for neurofibroma or optic glioma formation in mice, respectively [61,62]. However, Nf1+/− mice (harboring one non-functional and one functional Nf1 allele) in which both copies of the Nf1 gene are inactivated in Schwann cell or astroglial cell precursors develop neurofibromas or optic gliomas [61,63,64]. In the case of neurofibromas, mast cells represent one of the primary stromal cell types critical for tumor formation and growth (Figure 4A) [65,66], while microglia in optic gliomas are important for the development and continued expansion of these low-grade brain tumors (Figure 4B) [67]. The need for non-neoplastic stromal Nf1+/− cells creates a new opportunity for therapeutic drug development, in which targeting these microenvironmental elements (mast cells, microglia) may deprive the NF1-deficient tumor cells of important mitogens that drive their expansion during tumorigenesis and govern their continued growth. In addition to the Nf1+/− cellular microenvironment, there are also genomic influences that predispose susceptible GEM strains to glioma formation, such that the same genetic changes in mice from a different strain dramatically modify the risk of glioma or MPNST formation [68]. Recent studies have begun to reveal some of these genomic modifiers [69,70]. As is true for molecular currency, it is equally important to appreciate the cellular and genomic context in which NF1 loss in progenitors leads to tumor development and progression.
Figure 4. NF1-associated low-grade tumors are composed of a heterogeneous collection of cell types.

(A) NF1-associated neurofibromas contain neoplastic NF1-deficient Schwann cells as well as NF1+/− mast cells, endothelial cells and fibroblasts that each contribute to neurofibroma formation and growth. (B) Similarly, NF1-associated optic gliomas contain NF1-deficient neoplastic glial cell types (astrocytes, glioma stem cells) coupled with NF1+/− astrocytes, neurons and microglia, which also participate in gliomagenesis and continued growth. Both the neoplastic and non-neoplastic cell types represent targets for therapeutic drug targeting.
Third, GEM strains provide opportunities to develop a battery of preclinical models that represent the spectrum of clinical symptomatology in NF1 and NF2. In this respect, no single Nf1 optic glioma model is representative of the various types of optic pathway tumors observed in children with NF1. For example, some children with NF1 who harbor optic pathway gliomas located in the postchiasmatic radiations are more likely to lose vision, whereas others with large tumors in their prechiasmatic optic nerves may never experience visual decline. Future use of Nf1 optic glioma GEM models might evaluate proposed targeted therapies on a collection of strains that model the various presentations of optic pathway glioma encountered in children with NF1. This approach would enable the identification of individualized treatments that selectively target the subgroup of patients most likely to respond.
Fourth, GEM strains can be employed as proof-of-concept model systems to evaluate a class of drug for its efficacy against a specific tumor type or clinical abnormality. In this manner, the results from such preclinical trials can be used to decide whether a given signaling molecule or cell type is a viable target for further exploration. In addition, Nf1 and Nf2 mouse models can be highly instructive in determining whether a specific drug reaches the intended tissue (e.g., brain) and exhibits good target engagement (e.g., MEK inhibition) as well as to perform pharmacodynamic and pharmacokinetic studies. Similarly, these studies can define the dose relevant to future human clinical studies in terms of systemic exposure, toxicity and efficacy.
Although these are powerful models for clinical translation, it is incumbent on the preclinical and clinical research communities to establish standards for the informed use of GEM strains in translational investigation studies. One reason for the perceived failure of mouse models to yield potent drugs that succeed in human clinical trials may be the low bar that is often set for success in preclinical experimentation. Attention should be paid to the number of mice exhibiting significant tumor shrinkage, the actual percent of tumor volume reduction and the durability of the observed effect. Studies using drugs in which 25% of the mice exhibit a 20% reduction in tumor size may yield statistically significant results, but are unlikely to succeed in human clinical trials. Scientists and clinicians need to work closely to develop outcome expectations that more effectively translate to human tumor trial design.
4. Clinical trials for NF1 and NF2
The goal of all therapeutics research, preclinical to clinical, is to identify therapies that safely and reliably improve the disease state for patients. Improving the ‘disease state’ has many broad definitions in any medical setting, but the potential ambiguity of this goal is exemplified in the setting of NF1 and NF2, given that patients with these syndromes can have multiple different pathologic manifestations, at different times, each with variable clinical expression. Recognizing this, it becomes critical to focus the efforts of any NF clinical trial on a single, clinically meaningful and measurable outcome. This chosen outcome is the primary end point, which dictates all other aspects of the clinical study design.
A meaningful study end point is determined by several factors including: i) the purpose of the study (i.e., dose finding, biologic activity, signal of clinical efficacy or proof of clinical benefit), ii) the clinical expression of the disease being targeted, iii) the mechanism of action of the therapy to be tested and iv) the feasibility and reproducibility of the planned assessment measures. The success or failure of early and late efficacy studies depends not only on the treatment being tested, but on the end points chosen to demonstrate efficacy and the power of the study design to detect a meaningful difference in outcome based on treatment. In a well constructed study, the results may be ‘negative’; however, such ‘negative’ outcomes may be almost as informative as ‘positive’ results to the ultimate goal of developing effective therapies.
In order to obtain meaningful end points in NF1 and NF2, several response criteria for the multiple manifestations of these syndromes must be developed. These criteria are both specific to the manifestation of the disease (i.e., cognitive function, pain, neurologic disability), the phase of clinical investigation (i.e., exploratory efficacy vs confirming efficacy) and the mechanism of action of the investigational therapy. In this regard, the newly formed Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) International Consortium (led by Dr. Scott Plotkin, Massachusetts General Hospital) is generating response criteria across radiologic and functional domains that impact patients with NF1 and NF2. To accomplish this, several working groups have been formed within REiNS to specifically address neurologic functional outcomes, visual outcomes, neurocognitive outcomes, patient reported outcomes, tumor volume and whole body magnetic resonance imaging (MRI). Each of these groups is charged with evaluating, and when needed, developing, the tools needed for measuring clinical end points in patients with NF1 and NF2. For example, the neurological functional outcomes group recently recommended that speech discrimination (word recognition score) should be the primary hearing endpoint in therapeutic trials designed to assess the ability to restore hearing or prevent hearing loss in patients with NF2-associated vestibular schwannomas [71,72]. This endpoint was recommended, since it is both clinically meaningful and represents a reliable measure for assessment [73]. Similar recommendations are currently in development for visual outcomes in patients with NF1 who have optic pathway gliomas [14]. Such functional outcomes require careful consideration and validation of the measures to be used and consensus building across multiple fields, but, once established, are likely to become the most potent tools for demonstrating clinical benefit. By contrast, imaging endpoints, such as tumor volume and whole body tumor burden, are well developed and widely agreed upon as reproducible and reliable measures, and hence are valuable marked of biologic effect, but may not necessarily correlate with clinical benefit [74,75].
Volumetric MRI assessment of well-demarcated tumors, such as plexiform neurofibromas, MPNST, peripheral schwannomas and vestibular schwannomas, has been accepted as the standard imaging assessment in clinical investigations in NF1 and NF2. This conclusion is based on results from independent analyses of linear versus volumetric measurements showing that, for both plexiform neurofibromas and vestibular schwannomas, volumetric assessments are far more sensitive to change (growth or regression) than traditional one- or two-dimensional measures [76,77]. Recently, whole body MRI (WBMRI) has been applied to patients with NF1 and NF2 to assess total tumor burden. Although WBMRI is far less technologically developed or widely available as regional MRI, it is very effective at demonstrating large tumors that cross traditional body regions for longitudinal assessment [78-80].
There is an extensive history of clinical trials for patients with NF1 and more recently, patients with NF2. In NF1, clinical trials have largely focused on plexiform neurofibromas, MPNSTs, optic pathway gliomas and cognitive function, with a handful of trials targeting cutaneous neurofibromas (Table 4). By far, the greatest number of clinical trials has been for plexiform neurofibromas (16 trials to date), since these tumors represents one of most common complications in NF1, frequently requiring intervention in the absence of a known effective standard intervention. By contrast, although optic pathway gliomas are common in children with NF1, only one-third of patients require intervention and most will respond to first-line conventional therapies.
Table 4. Clinical trials for NF1-associated medical problems.
| Drug | Phase | N | Target | Age (year) | End point | Results |
|---|---|---|---|---|---|---|
| Plexiform neurofibroma | ||||||
| Thalidomide | I | 20 | Angiogenesis | ≥ 5 | 2D | Few patients with minor decrease in tumor, 12 patients with symptom improvement [116] |
| CRA or IFN-α-2a | II | 57 | Differentiation, angiogenesis | Children & adults |
2D | Few patients with minor decrease in tumor or symptom improvement [117] |
| Tipifarnib (I, II) | I, II | 17, 60 | Farnesyl transferase inhibitor | 3 – 25 | 2D; TTP; 3D | No responses; inactive [81,118] |
| Pirfenidone | II | 24 | Tumor-associated fibroblasts | 18 – 70 | 3D | Some patients with minor decrease in tumor or symptom improvement [119] |
| Pirfenidone | I, II | 16, 36 | 3 – 21 | TTP; 3D | No response; inactive [120] | |
| Photodynamic therapy | I | Tumor tissue disruption and pro-inflammatory |
3 – 21 | Tolerability | Advanced to efficacy study [121] | |
| Sirolimus | II | 12 | mTOR | ≥ 3 | TTP; 3D | Ongoing; no responses; TTP pending [122] |
| PEG-IFN-α-2b (ClinicalTrials. gov: NCT00396019) |
I, II | 30, 69 | Immune modifier, angiogenesis |
1.5 – 21 | TTP; 3D; symptom |
Some patients with minor decrease in tumor or symptom improvement; doubles TTP [123] |
| Sorafenib | I | 9 | Raf, PDGFRfS, c-Kit, VEGFR2 | 3 – 18 | Tox, PK, 3D | No maximum tolerated; dose limiting toxicity is pain in tumor [124] |
| Imatinib | II | 36 | c-Kit, PDGFRβ | 3 – 65 | 3D | Some patients with minor decrease in tumor; symptom improvement [65] |
| Niiotinib (ClinicalTrials.gov: NCT01275586) |
II | 20 | c-Kit, BCR-ABL, MAPK11, PDGFRβ |
≥ 18 | 3D | Ongoing |
| Methotrexate & vinblastine (ClinicalTrials.gov: NCT00030264) |
II | 35 | Cytotoxic | < 25 | TTP | Ongoing |
| Sunitinib (ClinicalTrials.gov: NCT01402817) |
II | 42 | PDGFR, VEGFR, c-Kit | 7 – 65 | 3D | Ongoing |
| AZD6244 (ClinicalTrials.gov: NCT01362803) |
I | MEK | 3 – 18 | Toxicity, PK, dose finding |
Ongoing | |
| Malignant peripheral nerve sheath tumor | ||||||
| Erlotinib | II | 24 | EGFR | ≥ 18 | Response rate | Completed, no objective response, progression-free survival was 2 months [125] |
| Sorafenib | II | 15 | VEGF, PDGFR, C-Raf | ≥ 18 | Response rate | Completed, no responders [126] |
| Imatinib | II | 11 | c-Kit | ≥ 18 | Response rate | Completed, no responders [127] |
| Dasatinib | II | 502 (all sarcomas) |
Dual Src/Abl inhibitor and multiple other kinases |
≥ 13 | Response rate | Completed, no responders [128] |
| Sorafenib and dacarbazine (ClinicalTrials.gov: NCT00837148) |
II | 37 | VEGF, PDGFR, C-Raf and cytotoxic therapy |
≥ 18 | Response rate | Enrollment completed |
| Doxorubicin hydrochloride, etoposide, ifosfamide preceding radiation or surgery (ClinicalTrials. gov: NCT00304083) |
II | 74 | Cancer cell cytotoxic therapy | ≥ 18 | Response rate | Completed, results pending |
| BEV and RAD001 (SARC016) (ClinicalTrials.gov: NCT01661283) |
II | 25 | VEGFand mTOR | ≥ 18 | Response rate | Ongoing |
| PLX3397 (ClinicalTrials.gov: NCT01004861) |
I | FMS, c-kit and Fit3-ITD | ≥ 18 | Toxicity, PK, dose finding |
Ongoing | |
| Cognitive deficits | ||||||
| Lova statin | I | 24 | Modification of Ras pathway | 10 – 17 | Safety and tolerability |
No toxicity [84] |
| Lovastatin (ClinicalTrials.gov: NCT00352599) |
I/II | 50 | Modification of Ras pathway | 10-50 | Non-verbal learning |
Completed; results pending |
| Lovastatin (ClinicalTrials.gov: NCT00853580) |
II/III | 142 | Modification of Ras pathway | 10 – 15 | Visual spatial learning and memory scores |
Active enrollment |
| Cutaneous neurofibroma | ||||||
| Ketotifen fumarate | II | 20 | Anti-histamine | Children & adults |
Symptoms (itchiness) |
No clear change in tumor, some symptom improvement [129] |
| Topical rapamycin | I | Topical mTOR inhibitor | ≥ 13 | PK, tolerability, safety |
Well tolerated with minimal systemic penetration, NF1 data pending, improvement in facial angiofibromas [130] |
|
| Photodynamic therapy with 5-aminolevulinic acid (ClinicalTrials.gov: NCT01682811) |
I | ≥ 18 | Uptake and maximum tolerated dose |
Ongoing | ||
| Topical imiquimod 5% cream (ClinicalTrials.gov: NCT00865644) |
II | 11 | Immune modifier | ≥ 18 | Tumor volume | Ongoing |
| Er-YAG laser vaporization (ClinicalTrials.gov: NCT00921037) |
II/III | 12 | Ablative therapy | All | QOL | Ongoing |
| Ranibizumab (ClinicalTrials. gov: NCT00657202) |
II | 11 | VEGF | ≥ 18 | Tumor volume | Ongoing |
| Optic pathway gliomas | ||||||
| Cyclophosphamide | II | 15 | Cytotoxic | ≤ 10 | Radiographic response |
5/15 with progressive disease, study closed due to lack of efficacy [131] |
| Everolimus (RAD001) (ClinicalTrials.gov: NCT01158651) |
II | 23 | mTOR | ≤ 21 | Radiographic response |
Ongoing |
| Tarceva & rapamycin (ClinicalTrials.gov: NCT00901849) | I | 72 | EGFR and mTOR | ≥ 21 | Safety; tumor and clinical measurements |
Ongoing |
| Vinblastine & carbopiatin (ClinicalTrials.gov: NCT00352495) |
I | 18 | Cytotoxic | ≥ 21 | Maximum tolerated dose; safety; tumor and clinical measurements |
Ongoing |
2D: 2-dimensional tumor measurement; 3D: 3-dimensional tumor measurement; BCR-ABL: Breakpoint cluster region-Abelson fusion gene; CRA: 13-cis-retinoic acid; Er:YAG: Erbium:yttrium-aluminum-garnet; I: Phase 1 study; II: Phase II study; MAPK11: Mitogen-activated protein kinase 11; MEK: MAP2K or mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; PD: Pharmacodynamics; PDGFRβ: Platelet-derived growth factor receptor beta; PK: Pharmacokinetics; QOL: Quality of life; Raf: Rapidly accelerated fibrosarcoma kinase; Tox: Toxicity; TTP: Time to progression; c-Kit: Tyrosine-protein kinase Kit.
2D: 2-dimensional tumor measurement; 3D: 3-dimensional tumor measurement; BCR-ABL: Breakpoint cluster region-Abelson fusion gene; CRA: 13-cis-retinoic acid; Er:YAG: Erbium:yttrium-aluminum-garnet; I: Phase 1 study; II: Phase II study; MAPK11: Mitogen-activated protein kinase 11; MEK: MAP2K or mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; PD: Pharmacodynamics; PDGFRβ: Platelet-derived growth factor receptor beta; PK: Pharmacokinetics; QOL: Quality of life; Raf: Rapidly accelerated fibrosarcoma kinase; Tox: Toxicity; TTP: Time to progression; c-Kit: Tyrosine-protein kinase Kit.
2D: 2-dimensional tumor measurement; 3D: 3-dimensional tumor measurement; BCR-ABL: Breakpoint cluster region-Abelson fusion gene; CRA: 13-cis-retinoic acid; Er:YAG: Erbium:yttrium-aluminum-garnet; I: Phase 1 study; II: Phase II study; MAPK11: Mitogen-activated protein kinase 11; MEK: MAP2K or mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; PD: Pharmacodynamics; PDGFRβ: Platelet-derived growth factor receptor beta; PK: Pharmacokinetics; QOL: Quality of life; Raf: Rapidly accelerated fibrosarcoma kinase; Tox: Toxicity; TTP: Time to progression; c-Kit: Tyrosine-protein kinase Kit.
Several important lessons have emerged from early clinical trials for NF1-associated plexiform neurofibromas. First, initial clinical studies did not have the benefit of detailed preclinical biological data specifically relevant to NF1, and hence, employed compounds with biological rationale for tumors in general. Second, in the absence of a clear natural history of tumor behavior, it was difficult to interpret the true impact of any given treatment on tumor behavior. Recognizing the lack of a reliable natural history that would support the use of single arm studies moving forward, Widemann and colleagues executed a placebo-controlled study of tipifarnib for clinically-progressive plexiform neurofibromas in children using a crossover design [81]. In this study, patients were randomized in a double-blind fashion to receive tipifarnib or placebo (Phase A) and were followed for disease progression. At time of disease progression, patients crossed over to the opposite treatment (Phase B), and again followed until disease progression. The primary end point was time to progression (TTP), as measured by volumetric MRI, with a doubling in the TTP set as the goal. Tipifarnib was well tolerated and prolonged the TTP in Phase A (19.2 months for tipifarnib vs 10.6 months for placebo); however it did not reach the stated goal of doubling the TTP. In Phase B, there was no significant difference between tipifarnib and placebo (14.5 vs 13.3 months). Despite yielding an overall negative result, this study was highly successful on two counts: i) it definitively demonstrated that tipifarnib did not have the minimum desired clinical effect and ii) it provided important natural history information regarding the TTP for plexiform neurofibromas. Ongoing and planned future studies are incorporating the lessons learned from these completed trials in order to best evaluate new biologically based therapies that derive from basic science laboratory investigation. The coupling of improved clinical trial designs specific to NF1-associated features with exciting preclinical data is highly likely further to increase the odds of identifying successful compounds for the treatment of plexiform neurofibromas.
For patients with NF2, the therapeutic trials to date have focused exclusively on progressive vestibular schwannomas, since these are the tumors frequently associated with significant clinical morbidity and mortality in this population. However, translational studies targeting peripheral schwannoma and meningioma are in progress (Table 5). Somewhat divergent from the experience in NF1 and NF2 clinical experiences are heavily influencing the direction of basic laboratory exploration. For example, the anti-angiogenesis inhibitor bevacizumab was found to exhibit remarkable clinical efficacy in some patients with progressive NF2-associated vestibular schwannomas, but the mechanism for this response is unknown [30,31]. This clinical observation has led to new fundamental science research aimed at understanding how merlin controls angiogenesis relevant to Schwann cell tumorigenesis (schwannomas) [82,83]. As with NF1, future therapies for tumors arising in NF2 will require ongoing dialogs between basic researchers and clinical trialists in order to match the best therapies with the patients most likely to benefit.
Table 5. Clinical trials for NF2-associated tumors.
| Drug | Phase | Target | Age (year) |
End point | Status | Results |
|---|---|---|---|---|---|---|
| PTC299 | II | VEGF | ≥ 18 | VS: tumor volume and/or word recognition |
Stage 1 completed; study suspended |
Some responses; rare but serious toxicity |
| Lapatinib | 0, translational |
EGFR/ErBb2 | ≥ 18 | VS: tumor PK, molecular & gene mutation studies |
Enrolling | Pending: preliminary evidence of good drug concentration |
| Lapatinib | II | EGFR/ErBb2 | 4 - 80 | VS: 15% volume reduction |
Completed | 4/17 patients with > 15% volume reduction [132] |
| RAD001 | II | mTOR | ≥ 3 | VS and MEN: 15% volume reduction |
Stage 1 enrollment (9 patients) completed |
Pending |
| RAD001 | II | mTOR | ≥ 15 | VS: TTP (MRI) | Open in France, and Los Angeles |
Pending |
| RAD001 | 0, translational |
mTOR | ≥ 18 | VS and MEN: tumor PK, molecular & gene mutation analysis |
In development | N/A |
| BEV | II | VEGF | ≥ 12 | VS: word recognition score |
Completed enrollment |
Pending |
| Nilotinib | II | PDGF, c-Kit | ≥ 18 | VS: 20% volume reduction or TTP |
Enrolling | Pending |
| Sorafenib & nilotinib |
0, translational |
PDGF, VEGR, c-Kit |
≥ 18 | Cutaneous SWN: PK, molecular studies |
Enrolling | Pending |
BEV, Bevacizumab; c-Kit: Tyrosine-protein kinase Kit; MEN: Multiple endocrine neoplasia; MRI: Magnetic resonance imaging; mTOR: Mammalian target of rapamycin; NF2: Neurofibromatosis type 2; PDGF: Platelet-derived growth factor; PK: Pharmacokinetics; SWN: Schwannomas; TTP: Time to progression; VS: Vestibular schwannomas.
5. Development of the NF Clinical Trials Consortium
Given the inherent variability of these conditions and the relative rarity of these syndromes and their associated tumors, it is unlikely that any one institution or even, for that matter, a small group of institutions will be able to efficiently perform therapeutic clinical trials. Moreover, until recently, prior trials were limited by relatively small sample sizes, retrospective trial designs and non-uniform eligibility and outcome measures. The ability to more effectively evaluate new therapies for individuals affected with NF1 and NF2 requires i) scientifically rational selections of the most promising agents for investigation, ii) prospective innovative trial designs, iii) specific measures to assess endpoints and outcomes and iv) appropriately powered studies that match the patients most likely to benefit to specific therapeutic interventions. This approach requires broad and deep expertise in neuroscience, cell biology, genetics, oncology, biostatistics and clinical investigation. In order to create such a pool of expertise and address the complex requirements of effective clinical trial development for patients with NF1 and NF2, in 2005 the United States Department of Defense gathered experts in NF research and clinical trial development to make recommendations aimed at expediting clinical investigations. This culminated in the formation of the Department of Defense-sponsored Neurofibromatosis Clinical Trials Consortium (NFCTC). The NFCTC was created to accelerate the development and completion of biologically informed, statistically sound translational clinical trials for children and adults with NF1 and NF2. This cooperative group was patterned after cooperative clinical trials consortia that have successfully treated children with cancer over the past 40 years, such as the Children’s Oncology Group.
The NFCTC was charged with the responsibility of leveraging its capabilities in rapidly performing NF clinical trials to attract academic and industry partners. A ‘request for applications’ was issued for potential sites, and through peer review, the initial NFCTC sites were chosen (Table 6). The criteria for inclusion in the NFCTC included having a large well-characterized NF population, an infrastructure which could support clinical trial execution, expertise in clinical trial performance and a multidisciplinary program experienced in NF research. The sites were initially chosen predominantly based on the size of their pediatric NF1 population, as it was believed that the majority of patients initially entered on clinical trials would be children with NF1. After this initial selection of sites, the Department of Defense expanded the purview of the NFCTC to include adult patients and the development of studies for patients with NF2. In a separate selection process, the University of Alabama at Birmingham was chosen as the NFCTC operations office and statistical center.
Table 6. Neurofibromatosis consortium sites.
| 2007 – 2012 | 2012 – 2017 |
|---|---|
| University of Alabama (Birmingham, AL) | University of Alabama (Birmingham, AL) |
| Children’s National Medical Center (Washington, DC) | Children’s National Medical Center (Washington, DC)/Johns Hopkins University (Baltimore, MD) |
| Cincinnati Children’s Hospital Medical Center | Cincinnati Children’s Hospital Medical Center (Cincinnati, OH)/Ohio |
| (Cincinnati, OH) | State University (Columbus, OH)* |
| Children’s Hospital of Philadelphia (Philadelphia, PA) | Children’s Hospital of Philadelphia (Philadelphia, PA) |
| University of Chicago (Chicago, IL) | University of Chicago (Chicago, IL)/Lurie Children’s Hospital of Chicago* |
| Washington University (St. Louis, MO) | Washington University (St. Louis, MO) |
| Primary Children’s Medical Center (Salt Lake City, UT) | Primary Children’s Medical Center (Salt Lake City, UT) |
| National Cancer Institute | National Cancer Institute |
| Boston Children’s Hospital (Boston, MA)/Massachusetts | Boston Children’s Hospital (Boston, MA)/Massachusetts General |
| General Hospital (Boston, MA)* | Hospital (Boston, MA)* |
| New York University (New York, NY)‡ | |
| Indiana University (Bloomington, IN)‡ | |
| Children’s Hospital Los Angeles (Los Angeles, CA)‡ |
New affiliates.
New sites.
The NFCTC opened in 2007, and early in its process, it identified the need to create committees focused on disease manifestations of NF1 as well as other committees to provide needed expertise for the Consortium, such as the Biology Committee and Imaging Committee (Figure 5). Based on their relative high incidence and associated morbidity, the NF1-associated clinical manifestations chosen for initial clinical studies included plexiform neurofibromas, learning and behavior deficits and gliomas. Although the NFCTC was restricted financially to a limited number of member sites, study proposals were welcomed from any interested investigator. If a study was chosen from a non-consortium investigator, the site of the investigator was then invited to join the study. Similarly, mechanisms were developed to allow other sites to join, if required for timely completion of a specific study. The initial study opened by the NFCTC utilized a biologic agent, rapamycin (mTOR inhibitor), for children and adults with progressive and symptomatic plexiform neurofibromas. This study stratified patients into those having radiographic-progressive symptomatic lesions and a second group with symptomatic tumors without documented radiographic progression. The primary outcome measure for those patients with radiographic progression was TTP, which was compared with a cohort of patients evaluated on two recently completed negative studies, one of which included a placebo control group. For those patients without documentation of pre-study entry radiographic progression, the primary outcome was radiographic response. The NFCTC utilized centralized volumetric neuroradiologic review to assess response and progression, which provides more sensitive and more reliable estimates of tumor size than standard one- or two-dimensional tumor measures [77].
Figure 5. Organization of the NF Clinical Trials Consortium.

The second NFCTC study assessed the ability of lovastatin, which, among its many actions, inhibits RAS signaling, to improve neurocognitive function in children with NF1 [84]. This was a prospective randomized clinical trial utilizing detailed pre-treatment screening and rigorous computer-based cognitive outcome measures. The third trial, which opened late in the 5-year initial grant cycle, was an evaluation of RAD001, another mTOR inhibitor, in children and adults with progressive low-grade gliomas, based on promising preclinical data in one Nf1 GEM optic glioma model [59].
As expected, the NFCTC experienced considerable growing pains. The first study took over 1 year to open, and study site approvals were slower than expected, as investigators had to become comfortable with cooperative consortium operations and the requirements for clinical trials performance. Although all sites did have large pediatric NF1 patient populations and experienced NF researchers, clinical and therapeutic studies were not easily executed at some centers, and the mindset of investigators had to be refocused to meet the rigor of these types of investigations and the need to recruit patients for studies. In addition, it was discovered that certain endpoints currently in wide clinical use (such as certain cognitive measures) could not be reliably performed in the clinical trial setting, thus requiring the re-design of some trials.
Other challenges were soon recognized by NFCTC leadership. Although all the investigators were committed to performing the studies expeditiously, identification of an adequate number of patients with any specific manifestation of NF1 within a specific age range and exhibiting active disease required for enrollment was considerably more challenged than initially appreciated. Furthermore, while the NFCTC included excellent centers, the number of centers was probably too few to expeditiously complete needed accruals. Centers chosen were also predominantly located in the eastern part of the USA and, to a lesser extent, in the Midwest; patients in the western part of the USA found it difficult to access studies, further limiting enrollment. Another major issue was the ability of the NFCTC to organize trials for patients with NF2. As the NFCTC was initially chosen on the basis of its ability to perform trials in children with NF1, some of the sites lacked active NF2 patient populations, and, overall, the consortium was inappropriately sized to execute NF2 clinical studies.
In agreement with the Department of Defense, the NFCTC was expanded in 2011 – 2012 (the last year of original granting period) to allow the addition of sites with NF2 expertise, the use of regional networks to support some of the sites and the addition of new institutions and centers. These changes resulted in better geographic balance and expansion of clinically appropriate populations (Table 6; Figure 6). The new and expanded capabilities of the consortium led to a reassessment of what trials required development, resulting in the new focus on NF2 and vestibular schwannomas.
Figure 6. Distribution of NF Clinical Trials Consortium sites in the USA.

Stars denote participating university sites
In 2012, the expanded NFCTC received 5 years of additional funding through the Department of Defense, with the re-stated goal of performing biologically informative trials for children and adults with NF1 and NF2. To ensure that all studies chosen were biologically sound and met the objectives of the consortium to perform statistically sound, clinically important studies, the ‘renewed’ consortium added an External Review Committee to review all studies before submission to the DOD External Advisory Board for final approval. The expanded consortium opened, as its first study, a trial of bevacizumab in children and adults with NF2 and progressive/symptomatic vestibular schwannomas using an innovative hearing outcome measure. A second study, planned during the initial funding period, opened in collaboration with the SARC consortium (a multi-institution sarcoma-focused alliance) to evaluate the efficacy of combined bevacizumab and RAD001 therapy for children and adults with recurrent MPNSTs. For children with plexiform neurofibromas, two trials are presently near initiation and will utilize volumetric responses as primary outcome measures. This change in primary outcome measure will greatly facilitate completion of the studies, so that multiple studies can be performed. The initial study for children with NF1 and plexiform neurofibromas used progression-free survival as its outcome measure and took nearly 4 years to complete. In addition to these planned studies, the NFCTC is rapidly moving forward to open a novel prospective study evaluating the impact of new agents on the re-fracture rate in patients with NF1-associated tibial dysplasia. The NFCTC is also developing a successor study for the lovastatin neurocognitive study.
As the NFCTC matures, it has become better sized to perform adequately powered, yet relatively rapid, clinical studies. It has created important relationships with industry to facilitate study development, and also supports the financial realities of performing these types of molecularly targeted trials. Without industry support, these trials are financially impossible. Finally, leveraging the biology committee and preclinical consortium studies from individual investigators as well as those funded by the Children’s Tumor Foundation, unique biologically informed trials are being planned.
6. Expert opinion
As we move forward with the translation of promising drugs from mouse model testing to clinical trials, we are creating more seamless integration of mouse preclinical and human clinical trials efforts. The NFCTC has established regular communication with the Children’s Tumor Foundation NF Preclinical Consortium (NFPC) including the ability to critically evaluate preclinical mouse data prior to publication. This is made possible by incorporating individuals from the NFPC into the NFCTC Biology Committee responsible for the review of proposed human clinical trials, including researchers on the various clinical protocol design committees, and involving NFCTC leadership representation in NFPC meetings. This bi-directional feedback facilitates the development of translatable outcomes as well as provides a forum to cross-inform each group, similar to the co-clinical trial infrastructures operational for other cancer types [85,86].
It is important to continue to refine the thinking about clinical trial design to incorporate lessons learned about the disease heterogeneity from preclinical mouse modeling (Figure 7). GEM strains have been highly instructive in demonstrating the critical role of non-neoplastic cell types in NF1-associated tumorigenesis [66,67], the impact of cell of origin and timing of tumor suppressor gene inactivation [87-89], the contribution of genomic background to tumorigenesis [68-70] and the diversity of tumor suppressor protein signaling pathway regulation [90]. In this respect, not all schwannomas (NF2) or plexiform neurofibromas (NF1) are identical tumor types. Similar to other tumor types [91,92], there are likely distinct subgroups of schwannomas or plexiform neurofibromas that are defined by unique molecular signatures, cellular composition, genomic landscape or patient age. The ability to develop effective therapies is contingent on the ability to appreciate these subgroups and design trials that allow for testing of the right compound in the right group at the right time. For this reason, enabling resources and technologies are required to discern these subtypes of tumors, including clinically-annotated tissue repositories and more detailed genetic, molecular and cellular analyses. In addition, given that subgroups will by definition include smaller and smaller study populations, novel clinical trial design will be required to most efficiently develop compounds for the right patients.
Figure 7. NF disease heterogeneity.

Future treatments for NF1 and NF2 patients should incorporate emerging basic science and clinical observations demonstrating that multiple factors influence clinical feature development, progression and response to therapy. For example, optic pathway gliomas arising in children with NF1 likely comprise a heterogeneous population of tumors defined by age, brain location, glioma composition, patient genomic background and other factors. In this regard, optic gliomas may represent several distinct diseases, each with a more predictable outcome and response to targeted therapy.
Article highlights.
Neurofibromatoses (NF1 and NF2) are clinically-distinct inherited cancer predisposition syndromes in which patients manifest a spectrum of heterogeneous clinical features, each with different outcome measures and biological therapeutic targets.
Current treatments for NF1 and NF2 largely have adapted therapies used for individuals with sporadic tumors and do not target the specific growth control pathways de-regulated in NF1 and NF2.
A major obstacle to successful clinical trials execution for NF was the absence of a coordinated clinical trials consortium.
New biologically targeted therapies for NF1 and NF2 have emerged from basic science discoveries aimed at understanding how the NF1 and NF2 proteins control cell growth.
Prioritization of clinical trials has leveraged robust preclinical models of NF1- and NF2-associated clinical features.
Future clinical trials need to incorporate rational biologically targeted therapies supported by positive preclinical data designed to address the inherent clinical heterogeneity in NF1 and NF2.
Acknowledgments
The NFCTC is funded by grants from the Department of Defense.
Footnotes
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
Papers of special note have been highlighted as either of interest
(•) or of considerable interest
(••) to readers.
- 1.Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–7. [PubMed] [Google Scholar]
- 2.Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327–32. doi: 10.1002/ajmg.a.33139. [DOI] [PubMed] [Google Scholar]
- 3.Huson SM, Compston DAS, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in southeast Wales. 1. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989:26704–11. doi: 10.1136/jmg.26.11.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71–4. doi: 10.1001/archderm.141.1.71. [DOI] [PubMed] [Google Scholar]
- 5.Evans DG, Huson SM, Donnai D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29(12):841–6. doi: 10.1136/jmg.29.12.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999;89:31–7. doi: 10.1002/(sici)1096-8628(19990326)89:1<31::aid-ajmg7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 8.Tucker T, Friedman JM, Friedrich RE, et al. Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009;46:81–5. doi: 10.1136/jmg.2008.061051. [DOI] [PubMed] [Google Scholar]
- 9.Needle MN, Cnaan A, Dattilo J, et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children’s Hospital of Philadelphia experience, 1974 –1994. J Pediatr. 1997;131:678–82. doi: 10.1016/s0022-3476(97)70092-1. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–9. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis RA, Gerson LP, Axelson KA, et al. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91:929–35. doi: 10.1016/s0161-6420(84)34217-8. [DOI] [PubMed] [Google Scholar]
- 12••.Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis 1: controversies and recommendations. Ann Neurol. 2007;61:189–98. doi: 10.1002/ana.21107. This review outlines the current recommendations for monitoring children with NF1-associated optic glioma.
- 13.Packer RJ, Ater J, Allen J, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747–54. doi: 10.3171/jns.1997.86.5.0747. [DOI] [PubMed] [Google Scholar]
- 14••.Fisher MJ, Loguidice M, Gutmann DH, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol. 2012;14:790–7. doi: 10.1093/neuonc/nos076. Important study assessing visual outcomes in children with NF1 following treatment.
- 15.Pollack IF, Shultz B, Mulvihill JJ. The management of brainstem gliomas in patients with neurofibromatosis 1. Neurology. 1996;46:1652–60. doi: 10.1212/wnl.46.6.1652. [DOI] [PubMed] [Google Scholar]
- 16.Ullrich NJ, Robertson R, Kinnamon DD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. 2007;68(12):932–8. doi: 10.1212/01.wnl.0000257095.33125.48. [DOI] [PubMed] [Google Scholar]
- 17.Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–14. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warbey V, Ferner R, Dunn J, et al. [(18)F]FDG PET/CT in the diagnosis of malignant peripheral nerve sheath tumours in neurofibromatosis type-1. Eur J Nucl Med Mol Imaging. 2009;13:36751–7. [Google Scholar]
- 19.Korf BR. Malignancy in neurofibromatosis type 1. Oncologist. 2000;5:477–85. doi: 10.1634/theoncologist.5-6-477. [DOI] [PubMed] [Google Scholar]
- 20.Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007;44:481–4. doi: 10.1136/jmg.2007.049346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elefteriou F, Kolanczyk M, Schindeler A, et al. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A. 2009;149A:2327–38. doi: 10.1002/ajmg.a.33045. [DOI] [PubMed] [Google Scholar]
- 22.Stevenson DA, Moyer-Mileur LJ, Murray M, et al. Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr. 2007;150:83–8. doi: 10.1016/j.jpeds.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunetti-Pierri N, Doty S, Hicks J, et al. Generalized metabolic bone disease in neurofibromatosis type I. Mol Genet Metab. 2008;94:105–11. doi: 10.1016/j.ymgme.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cairns A, North K. Cerebrovascular Dysplasia in neurofibromatosis type 1. J Neurol Neurosurg Psychiatry. 2008;79:1165–70. doi: 10.1136/jnnp.2007.136457. [DOI] [PubMed] [Google Scholar]
- 25.Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4:105–11. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Hyman SL, Arthur Shores E, North KN. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48:973–7. doi: 10.1017/S0012162206002131. [DOI] [PubMed] [Google Scholar]
- 27.Mautner VF, Kluwe L, Thakker SD, Leark RA. Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol. 2002;44:164–70. doi: 10.1017/s0012162201001876. [DOI] [PubMed] [Google Scholar]
- 28.Evans GR, Lloyd SK, Ramsden RT. Neurofibromatosis type 2. Adv Otorhinolaryngol. 2011;70:91–8. doi: 10.1159/000322482. [DOI] [PubMed] [Google Scholar]
- 29.Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12:14–18. doi: 10.1093/neuonc/nop010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plotkin SR, Stemmer-Rachamimov AO, Barker FG, II, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358–67. doi: 10.1056/NEJMoa0902579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plotkin SR, Merker VL, Halpin C, et al. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: a retrospective review of 31 patients. Otol Neurotol. 2012;33:1046–52. doi: 10.1097/MAO.0b013e31825e73f5. [DOI] [PubMed] [Google Scholar]
- 32.Plotkin SR, O’Donnell CC, Curry WT, et al. Spinal ependymomas in neurofibromatosis type 2: a retrospective analysis of 55 patients. J Neurosurg Spine. 2011;14:543–7. doi: 10.3171/2010.11.SPINE10350. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser-Kupfer MI, Freidlin V, Datiles MB, et al. The association of posterior capsular lens opacities with bilateral acoustic neuromas in patients with neurofibromatosis type 2. Arch Ophthalmol. 1989;107:541–4. doi: 10.1001/archopht.1989.01070010555030. [DOI] [PubMed] [Google Scholar]
- 34.Hagel C, Lindenau M, Lamszus K, et al. Polyneuropathy in neurofibromatosis 2: clinical findings, molecular genetics and neuropathological alterations in sural nerve biopsy specimens. Acta Neuropathol. 2002;104:179–87. doi: 10.1007/s00401-002-0535-7. [DOI] [PubMed] [Google Scholar]
- 35.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 36.Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–92. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 37.Marchuk DA, Saulino AM, Tavakkol R, et al. cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product. Genomics. 1991;11:931–40. doi: 10.1016/0888-7543(91)90017-9. [DOI] [PubMed] [Google Scholar]
- 38.Gutmann DH, Zhang Y, Hirbe A. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann Neurol. 1999;46:777–82. doi: 10.1002/1531-8249(199911)46:5<777::aid-ana15>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 39.Gutmann DH, Andersen LB, Cole JL, et al. An alternatively-spliced mRNA in the carboxy terminus of the neurofibromatosis type 1 (NF1) gene is expressed in muscle. Hum Mol Genet. 1993;2:989–92. doi: 10.1093/hmg/2.7.989. [DOI] [PubMed] [Google Scholar]
- 40.Gutmann DH, Geist RT, Rose K, et al. Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. Dev Dyn. 1995;202:302–11. doi: 10.1002/aja.1002020309. [DOI] [PubMed] [Google Scholar]
- 41.Ballester R, Marchuk D, Boguski M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–9. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 42.Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in hematopoietic cells. Nat Genet. 1996;12:144–8. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 43.Tong J, Hannan F, Zhu Y, et al. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–6. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- 44.Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–54. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci. 2010;30:5579–89. doi: 10.1523/JNEUROSCI.3994-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- 47.Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causing neuro-fibromatosis type 2. Nature. 1993;363:515–21. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 48.Gutmann DH, Sherman L, Seftor L, et al. Increased expression of the NF2 tumor suppressor gene product, merlin, impairs cell motility, adhesion, and spreading. Hum Mol Genet. 1999;8:267–75. doi: 10.1093/hmg/8.2.267. [DOI] [PubMed] [Google Scholar]
- 49.Jukam D, Desplan C. Binary regulation of Hippo pathway by merlin/NF2, Kibra, Lgl, and melted specifies and maintains post-mitotic neuronal fate. Dev Cell. 2011;21:874–87. doi: 10.1016/j.devcel.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper J, Li W, You L, et al. Merlin/NF2 functions upstream of the nuclear E3 ubiquitin ligase CRL4DCAF1 to suppress oncogenic gene expression. Sci Signal. 2011;4(pt6) doi: 10.1126/scisignal.2002314. [DOI] [PubMed] [Google Scholar]
- 51.Yi C, Kissil JL. Merlin in organ size control and tumorigenesis: hippo versus EGFR? Genes Dev. 2010;24:1673–9. doi: 10.1101/gad.1964810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi C, Wilker EW, Yaffe MB, et al. Validation of the p21-activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68:7932–7. doi: 10.1158/0008-5472.CAN-08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.James MF, Han S, Polizzano C, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29:4250–61. doi: 10.1128/MCB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Houshmandi SS, Emnett RJ, Giovanini M, et al. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an Erb2- and Src-dependent manner. Mol Cell Biol. 2009;29:1472–86. doi: 10.1128/MCB.01392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Curto M, Cole BK, Lallemand D, et al. Contact-dependent inhibition of EGFR signaling by Nf2/merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lauchle JO, Kim D, Le DT, et al. Response and resistance to MEK inhibition in leukemias initiated by hyperactive Ras. Nature. 2009;461:411–14. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–9. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dasgupta B, Yi Y, Chen DY, et al. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–60. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 59•.Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68:1520–8. doi: 10.1158/0008-5472.CAN-07-5916. First preclinical report demonstrating that rapamycin inhibits the growth of murine Nf1 optic glioma.
- 60.Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123(1):340–7. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Ghosh P, Charnay P, et al. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bajenaru ML, Zhu Y, Hedrick NM, et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–13. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Y, Harada T, Liu L, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–88. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–7. [PubMed] [Google Scholar]
- 65.Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol. 2012;13:1218–24. doi: 10.1016/S1470-2045(12)70414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66••.Yang FC, Ingram DA, Chen S, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/− and c-kit-dependent bone marrow. Cell. 2008;135:437–48. doi: 10.1016/j.cell.2008.08.041. Landmark study demonstrating that targeting the tumor microenvironment provides an efficient therapy for NF1-associated plexiform neurofibroma.
- 67.Daginakatte G, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 68•.Reilly KM, Loisel DA, Bronson RT, et al. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. Important study highlighting the existence of genomic modifiers in Nf1 mouse high-grade astrocytoma.
- 69.Amlin-Van Schaick J, Kim S, Broman KW, Reilly KM. Scram1 is a modifier of spinal cord resistance for astrocytoma on mouse Chr 5. Mamm Genome. 2012;23:277–85. doi: 10.1007/s00335-011-9380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reilly KM, Tuskan RG, Christy E, et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci USA. 2004;101:13008–13. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Blakeley JO, Evans DG, Adler J, et al. Consensus recommendations for current treatments and accelerating clinical trials for patients with neurofibromatosis type 2. Am J Med Genet A. 2011 doi: 10.1002/ajmg.a.34359. Epub 2011 Dec 2. Important review of the current recommendations for treating individuals with NF2.
- 72.Plotkin SR, Halpin C, Blakeley JO, et al. Suggested response criteria for phase II antitumor drug studies for neurofibromatosis type 2 related vestibular schwannoma. J Neurooncol. 2009;93:61–77. doi: 10.1007/s11060-009-9867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Halpin C, Rauch SD. Using audiometric thresholds and word recognition in a treatment study. Otol Neurotol. 2006;27:110–16. doi: 10.1097/00129492-200601000-00020. [DOI] [PubMed] [Google Scholar]
- 74.O’Shaughnessy JA, Wittes RE, Burke G, et al. Commentary Concerning Demonstration of Safety and Efficacy of Investigational Anticancer Agents in Clinical Trials. J Clin Oncol. 1991;9:2225–32. doi: 10.1200/JCO.1991.9.12.2225. [DOI] [PubMed] [Google Scholar]
- 75.U.S. Department of Health and Human Services Food. Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Guidance for Industry Clinical Trail Endpoints for the Approval of Cancer Drugs and Biologics. 2007 Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071590.pdf.
- 76.Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007;68:643–7. doi: 10.1212/01.wnl.0000250332.89420.e6. [DOI] [PubMed] [Google Scholar]
- 77•.Harris GJ, Plotkin SR, Maccollin M, et al. Three-dimensional volumetrics for tracking vestibular schwannoma growth in neurofibromatosis type II. Neurosurgery. 2008;62:1314–19. doi: 10.1227/01.neu.0000333303.79931.83. Excellent analysis of the value of volumetric MRI in assessing vestibular schwannoma growth.
- 78.Plotkin SR, Bredella MA, Cai W, et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012;7:e35711. doi: 10.1371/journal.pone.0035711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kluwe L, Nguyen R, Vogt J, et al. Internal tumor burden in neurofibromatosis type I patients with large NF1 deletions. Genes Chromosomes Cancer. 2012;51:447–51. doi: 10.1002/gcc.21931. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen R, Kluwe L, Fuensterer C, et al. Plexiform neurofibromas in children with neurofibromatosis type 1: frequency and associated clinical deficits. J Pediatr. 2011;159:652–5. doi: 10.1016/j.jpeds.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 81.Widemann BC, Salzer WL, Arceci RJ, et al. Phase 1 trial and pharmacokinetic study of the farnesyl transferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type 1 and plexiform neurofibromas. J Clin Oncol. 2009;24:507–16. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- 82.Wong HK, Lahdenranta J, Kamoun WS, et al. Anti-vascular endothelial growth factor therapies as a novel therapeutic approach to treating neurofibromatosis-related tumors. Cancer Res. 2010;70:3483–93. doi: 10.1158/0008-5472.CAN-09-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yener U, Avsar T, Akgün E, et al. Assessment of antiangiogenic effect of imatinib mesylate on vestibular schwannoma tumors using in vivo corneal angiogenesis assay. J Neurosurg. 2012;117:697–704. doi: 10.3171/2012.6.JNS112263. [DOI] [PubMed] [Google Scholar]
- 84.Acosta MT, Kardel PG, Walsh KS, et al. Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. Ped Neurol. 2011;45:241–5. doi: 10.1016/j.pediatrneurol.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 85.Nardella C, Lunardi A, Patnaik A, et al. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011;1:108–16. doi: 10.1158/2159-8290.CD-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Z, Cheng K, Walton Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–17. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Le LQ, Liu C, Shipman T, et al. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer Res. 2011;71:4686–95. doi: 10.1158/0008-5472.CAN-10-4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–63. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–8. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weber JD, Gutmann DH. Deconvoluting mTOR biology. Cell Cycle. 2012;11:236–48. doi: 10.4161/cc.11.2.19022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Northcutt PA, Jones DT, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818–34. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Atkinson JM, Shelat AA, Carcaboso AM, et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell. 2011;20:384–99. doi: 10.1016/j.ccr.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Silva AJ, Frankland PW, Marowitz Z, et al. A mouse model for the learning and memory deficits associated with neurofibromatosis type 1. Nat Genet. 1997;15:281–4. doi: 10.1038/ng0397-281. [DOI] [PubMed] [Google Scholar]
- 94.Diggs-Andrews KA, Tokuda K, Izumi Y, et al. Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann Neurol. doi: 10.1002/ana.23793. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95••.Brown JA, Emnett RJ, White CR, et al. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum Mol Genet. 2010;19:4515–28. doi: 10.1093/hmg/ddq382. First study to demonstrate the neurochemical basis for attention system deficits in Nf1 GEM.
- 96.Cui Y, Costa RM, Murphy GG, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–60. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. 2005;65:236–45. [PubMed] [Google Scholar]
- 98.Kwon CH, Zhao D, Chen J, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–94. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mayes DA, Rivzi TA, Cancelas JA, et al. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res. 2011;71:4675–85. doi: 10.1158/0008-5472.CAN-10-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu J, Williams JP, Rizvi TA, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–16. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–6. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 103.Vogel KS, Klesse LJ, Velasco-Miguel S, et al. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–9. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.El-Khassawna T, Toben D, Kolanczyk M, et al. Deterioration of fracture healing in the mouse model of NF1 long bone dysplasia. Bone. 2012;51:651–60. doi: 10.1016/j.bone.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 105.El-Hoss J, Sullivan K, Cheng T, et al. A murine model of neurofibromatosis type 1 tibial pseudarthrosis featuring proliferative fibrous tissue and osteoclast-like cells. J Bone Miner Res. 2011 Sep 29; doi: 10.1002/jbmr.528. Epub. [DOI] [PubMed] [Google Scholar]
- 106.Wang W, Nyman JS, Ono K, et al. Mice lacking Nf1 in osteochondroprogenitor cells display skeletal dysplasia similar to patients with neurofibromatosis type 1. Hum Mol Genet. 2011;20:3910–24. doi: 10.1093/hmg/ddr310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang W, Rhodes SD, Zhao L, et al. Primary osteopathy of vertebrae in a neurofibromatosis type 1 murine model. Bone. 2011;48:1378–87. doi: 10.1016/j.bone.2011.03.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myelodysplastic disorder. Blood. 2004;103:4243–50. doi: 10.1182/blood-2003-08-2650. [DOI] [PubMed] [Google Scholar]
- 109.Tischler AS, Shih TS, Williams BO, Jacks T. Characterization of pheochromocytomas in a mouse strain with a targeted disruptive mutation of the neurofibromatosis gene Nf1. Endocr Pathol. 1995;6:323–35. doi: 10.1007/BF02738732. [DOI] [PubMed] [Google Scholar]
- 110•.Giovannini M, Robanus-Maandag E, van der Valk M, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–30. Development of the first mouse model of NF2-associated schwannoma.
- 111.Giovannini M, Robanus-Maandag E, Niwa-Kawakita M, et al. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13:978–86. doi: 10.1101/gad.13.8.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kalamarides M, Stemmer-Rachamimov AO, Niwa-Kawakita M, et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene. 2011;30:2333–44. doi: 10.1038/onc.2010.609. [DOI] [PubMed] [Google Scholar]
- 113.Kalamarides M, Niwa-Kawakita M, Leblois H, et al. Nf2 inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16:1060–5. doi: 10.1101/gad.226302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jongsma J, van Montfort E, Vooiks M, et al. A conditional mouse model for malignant mesothelioma. Cancer Cell. 2008;13:261–71. doi: 10.1016/j.ccr.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 115.Schulz A, Baader SL, Niwa-Kawakita M, et al. The role of merlin isoform 2 in neurofibromatosis type 2-associated polyneuropathy. Nat Med. doi: 10.1038/nn.3348. in press. [DOI] [PubMed] [Google Scholar]
- 116.Gupta A, Cohen B, Ruggierri P, et al. A phase I study of thalidomide for the treatment of plexiform neurofibroma in patients with neurofibromatosis 1 (NF1) Neurology. 2000;54:12–13. doi: 10.1212/01.wnl.0000042321.94839.78. [DOI] [PubMed] [Google Scholar]
- 117.Packer R, Gutmann D, Rubenstein A, et al. Plexiform neurofibromas in NF1: toward biologic-based therapy. Neurology. 2002;58:1461–70. doi: 10.1212/wnl.58.10.1461. [DOI] [PubMed] [Google Scholar]
- 118.Widemann B, Dombi E, Gillespie A, et al. Phase II randomized, flexible cross-over, double-blinded, placebo-controlled trial of the farnesyl transferase inhibitor (FTI) tipifarnib (R115777) in pediatric patients (pts) with neurofibromatosis type 1 (NF1) and progressive plexiform neurofibromas (PN); Proceedings of Children’s Tumor Foundation Meeting; Portland, OR. 2009. [Google Scholar]
- 119.Babovic-Vuksanovic D, Ballman K, Michels V, et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology. 2006;67:1860–2. doi: 10.1212/01.wnl.0000243231.12248.67. [DOI] [PubMed] [Google Scholar]
- 120.Babovic-Vuksanovic D, Widemann BC, Dombi E, et al. Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol. 2007;36:293–300. doi: 10.1016/j.pediatrneurol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 121.Fisher MJ. Photodynamic therapy in children with NF1 and plexiform neurofibromas; Proceedings of Children’s Tumor Foundation Meeting; Portland OR. 2009. [Google Scholar]
- 122.Weiss B, Fisher M, Dombi E, et al. Phase II study of the mTOR inhibitor sirolimus for non-progressive NF1-associated plexiform neurofibromas: a Neurofibromatosis Consortium Study; Proceedings of the ISPNO Meeting; Vienna Austria. 2010. [Google Scholar]
- 123.Jakacki RI, Bouffet E, Adamson PC, et al. A phase 1 study of vinblastine in combination with carboplatin for children with low-grade gliomas: a Children’s Oncology Group phase 1 consortium study. Neuro Oncol. 2011;13:910–15. doi: 10.1093/neuonc/nor090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim A, Dombi E, Tepas K, et al. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2012 doi: 10.1002/pbc.24281. Epub Sep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Albritton KH, Rankin C, Coffin CM, et al. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST) J Clin Oncol. 2006;24:524s. [Google Scholar]
- 126.Maki RG, D’Adamo DR, Koehan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:133–40. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a Bayesian hierarchical statistical model. J Clin Oncol. 2009;27:3148–53. doi: 10.1200/JCO.2008.20.5054. [DOI] [PubMed] [Google Scholar]
- 128.Schuetze S, Wathen JK, Choy E, et al. Results of a sarcoma alliance for research through collaboration (SARC) phase II trial of dasatinib in previously treated, high-grade, advanced sarcoma. J Clin Oncol. 2009;28:15s. [Google Scholar]
- 129.Riccardi VM. A controlled multiphase trial of ketotifen to minimize neurofibroma-associated pain and itching. Arch Dermatol. 1993;129:577–81. [PubMed] [Google Scholar]
- 130.Koenig MK, Hebert AA, Roberson J, et al. Topical rapamycin therapy to alleviate the cutaneous manifestations of tuberous sclerosis complex: a double-blind, randomized, controlled trial to evaluate the safety and efficacy of topically applied rapamycin. Drugs R D. 2012;12:121–6. doi: 10.2165/11634580-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kadota RP, Kun LE, Langston JW, et al. Cyclophosphamide for the treatment of progressive low-grade astrocytoma: a Pediatric Oncology Group phase II study. J Pediatr Hematol Oncol. 1999;21:198–202. doi: 10.1097/00043426-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 132.Karajannis MA, Legault G, Hagiwara M, et al. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro Oncol. 2012;14:1163–70. doi: 10.1093/neuonc/nos146. [DOI] [PMC free article] [PubMed] [Google Scholar]