

Abstract
Aim:
To evaluate the pharmacokinetic interactions between theophylline and antofloxacin in vivo and in vitro.
Methods:
A randomized, 5-day treatment and 3-way crossover design was documented in 12 healthy subjects. The subjects were orally administered with antofloxacin (400 mg on d 1 and 200 mg on d 2 to 5), theophylline (100 mg twice a day and morning dose 200 mg on d 1 and 5), or theophylline plus antofloxacin. The plasma and urinary pharmacokinetics of antofloxacin and theophylline were characterized after the first and last dose. The effect of antofloxacin on theophylline metabolism was also investigated in pooled human liver microsomes.
Results:
The 5-day treatment with antofloxacin significantly increased the area of the plasma concentration-time curve and peak plasma concentration of theophylline, accompanied by a decrease in the excretion of theophylline metabolites. On the contrary, theophylline did not affect the pharmacokinetics of antofloxacin. In vitro studies using pooled human hepatic microsomes demonstrated that antofloxacin was a weak reversible and mechanism-based inhibitor of CYP1A2. The clinical interaction between theophylline and antofloxacin was further validated by the in vitro results.
Conclusion:
The results showed that antofloxacin increases the plasma theophylline concentration, partly by acting as a mechanism-based inhibitor of CYP1A2.
Keywords: antofloxacin, theophylline, drug interactions, pharmacokinetics, CYP1A2
Introduction
Antofloxacin (ATFX), developed by Anhui Global Pharmaceutical Limited Co, China, is a newly developed 8-amino derivative of levofloxacin with potent antibacterial activities in vitro and in vivo. It exhibits antibacterial activity that is markedly superior to that of conventional quinolones (ciprofloxacin, ofloxacin, and sparfloxacin) against quinolone-resistant, methicillin-resistant Staphylococcus aureus and Staphylococcus epidermidis1. Clinical pharmacokinetic studies have shown that ATFX is well tolerated and has high oral bioavailability, as well as favorable elimination half-life properties2, 3. It was recently licensed for the treatment of respiratory tract infections and is therefore likely to be administered together with theophylline (TP) in patients requiring bronchodilators.
In humans, TP is mainly metabolized by cytochrome P450s (CYP450s) in the liver to its major metabolites 1,3-dimethyluric acid (1,3-DMU), 3-methylxanthine (3-MX), and 1-methylxanthine (1-MX). 1-MX is further metabolized to 1-methyluric acid (1-MU). The metabolites 1,3-DMU, 3-MX, and 1-MU are mainly excreted in the urine4, 5. Accumulating reports have shown that CYP1A2 is the major enzyme responsible for TP metabolism6, 7, 8, 9, and TP is also a validated CYP1A2 probe that can be used to evaluate the enzymatic kinetics of CYP1A210. TP has a narrow therapeutic plasma concentration range, and it is of the utmost importance to maintain constant concentrations of TP in plasma. Because asthma is often complicated by respiratory infections, the concomitant use of fluoroquinolone antimicrobials and TP may often be necessary. Concomitant use of fluoroquinolones may increase TP concentrations in the plasma, inducing clinically relevant safety issues. In 1984, Wijnands et al first reported severe clinical adverse effects with the concomitant use of TP and enoxacin, and they found that co-administration of enoxacin markedly increased plasma TP concentrations11. Consequently, a series of fluoroquinolones, including ciprofloxacin, tosufloxacin, clinafloxacin, grepafloxacin, and pefloxacin, have been reported to interfere with TP metabolism by inhibiting CYP1A2 activity12, 13, 14, 15. Therefore, it is important to evaluate the interaction between ATFX and TP to understand the pharmacokinetics and safety of these drugs.
A prior study showed that ATFX has little effect on CYP1A2 activity in human liver microsomes16. However, our previous study demonstrated that ATFX was a mechanism-based inhibitor of CYP1A2 in rat liver microsomes through analysis of the formation of acetaminophen from phenacetin17. The aim of this study was to investigate whether a pharmacokinetic interaction between ATFX and TP exists in vivo at clinical dosages and regimens. The interaction between TP and ATFX was further investigated in an in vitro metabolic system with human liver microsomes through analysis of the formation of 1-MU, 1,3-DMU, and 3-MX. The relationship between the in vivo findings and the in vitro results was also evaluated.
Materials and methods
Chemicals
ATFX tablets and an ATFX standard were supplied by Anhui Global Pharmaceutical Co Ltd (Bengbu, China). Sustained-release TP tablets were obtained from Guangzhou Maite Xinhua Pharmaceutical Co Ltd (Guangzhou, China). TP, 1-MX, 1,3-DMU, 1-MU, and 3-MX standards, β-nicotinamide adenine dinucleotide phosphate (NADP), glucose-6-phosphate (G-6-P), and G-6-P dehydrogenase (G-6-PDH, Type V) were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Pooled human liver microsomes were purchased from the Research Institute for Liver Disease Co Ltd (Shanghai, China). Information on the sources of the human liver microsome, including medication history, cause of death and the presence of viral infections was provided by the vendor and deemed suitable for these studies.
Subjects and ethics
The study protocol was approved by the Ethics Committee of the Nanjing First Hospital. The clinical trial was conducted in accordance with the ethical principals in the Declaration of Helsinki and with the requirements in Good Clinical Practice (GCP), including the protocol design and organization, monitoring, auditing, recording, analysis, and reporting. Twelve healthy, non-smoking volunteers (six females and six males) ranging in age from 22 to 29 years within 20% of ideal body weight participated in the study. All were advised of the objectives and possible risks of the study and agreed to participate by signing an informed consent form. All subjects underwent a full medical examination 1 week prior to the study, including a medical history, a physical examination, routine biochemical and hematological tests and a resting 12-lead electrocardiogram. Hematological and biochemical tests were repeated after the study. Pregnancy tests were performed in female subjects before each study period. No drugs (including oral contraceptives) were allowed 1 week prior to and throughout the study.
Study design
This was a randomized three-period crossover trial using a Latin-square design. Each treatment period was 5 d. The investigational medication, all given orally, was as follows: treatment A, ATFX alone; treatment B, TP alone; and treatment C, TP plus ATFX. For the ATFX treatment, subjects received 200 mg of ATFX once daily on d 2 to 5 of the 5-day study period and 400 mg of ATFX on d 1. For the TP treatment, subjects orally received 200 mg of TP twice daily on d 2 to 4 of the 5-day period and received 200 mg of TP as a single morning doses on d 1 and 5. The study periods were separated by a 1-week washout interval. No alcohol- or methylxanthine-containing beverages or food were permitted for 48 h prior to the start of each study period and for the duration of the study period.
The plasma and urinary pharmacokinetics of ATFX and TP were characterized after the first and last doses. The morning dose was administered after an overnight fast (at approximately 8:00) and the evening dose was given 12 h later (at approximately 20:00). Venous blood samples for the determination of ATFX and TP were drawn at the following time points: pre-dose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 96.5, 97, 97.5, 98, 99, 100, 102, 104, 108, 120, 132, and 144 h after the first dose. At each time point, 3 mL of venous blood was collected in heparinized tube and centrifuged. The plasma samples were transferred to polypropylene tubes and stored at -20 °C until further analysis.
Urine was collected in the following fractions: pre-dose assay blank and then at 0 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 10, 10 to 12, 12 to 24, 96 to 98, 98 to 100, 100 to 102, 102 to 104, 104 to 108, 108 to 120, 120 to 132, and 132 to 144 h time intervals after dose. The volume of each fraction was recorded, and a part of the aliquot was stored at -20 °C until assays were performed.
Registration of adverse events
Subjects were asked to document their adverse events in a diary. The diary entries were tabulated and compared among treatments.
Effect of ATFX on CYP1A2 activity in human liver microsomes
The effect of ATFX (0, 0.24, 0.48, 1.96, 1.92, and 2.40 mmol/L) on the metabolism of TP (1, 2, 4, 5, and 8 mmol/L) in pooled human liver microsomes was investigated. Enzyme activities were evaluated by analysis of the formation of 1-MX, 3-MX and 1,3-DMU. The incubation mixture consisted of 1 mg/mL human liver microsomes, an NADPH-regenerating system (0.5 mmol/L NADP, 10 mmol/L G-6-P, 1 U/mL G-6-PDH and 10 mmol/L MgCl2) and designated concentrations of ATFX and TP in 100 mmol/L potassium phosphate buffer at pH 7.4. The final volume was 200 μL. After incubation at 37 °C for 30 min, the reaction was terminated by adding 100 μL of 10% perchloric acid.
The time- and concentration-dependent inhibition of TP by ATFX was also observed. An NADPH-generating system and different concentrations of ATFX (0, 0.24, 0.48, 0.96, and 1.92 mmol/L) were added to human liver microsomes (1 mg/mL) in a total volume of 180 μL and pre-incubated at 37 °C for 0, 20, 30, 40, and 60 min. The enzyme reaction was initiated by adding 20 μL of TP at the designated time. The final concentration of TP was set at 5 mmol/L. After incubation at 37 °C for 30 min, the reaction was terminated by adding 100 μL of 10% perchloric acid.
Drug analysis
TP and its metabolites were measured using an HPLC-based method, as previously described18. The methods were validated according to the currently accepted US Food and Drug Administration (FDA) bioanalytical method validation guidelines on specificity, linearity, sensitivity, accuracy, and precision.
To analyze TP in plasma, briefly, 10 μL of solution containing 5 μg/mL propylthiouracil (internal standard) was added to the plasma (100 μL), and the mixture was deproteinized by adding 100 μL of 10% perchloric acid. After vigorous mixing and centrifugation, 20 μL of the supernatant was injected into an HPLC system with a mobile phase of water (0.1% acetic acid) and methanol (76:24 v/v). The calibration graphs were linear from 0.15 to 15 μg/mL.
To analyze TP and its metabolites in urine, a 5 mL of a mixture of dichloromethane and isopropanol (8:2) was added to 1.0 mL of urine. The mixture was vortexed for 2 min and centrifuged for 10 min at 4000×g. The organic layer (3.5 mL) was obtained and evaporated to dryness under a stream of N2 at 45 °C. The residues were reconstituted in 200 μL of mobile phase containing 25 μg/mL of epigoitrin (internal standard). An aliquot of 20 μL was injected into an HPLC system with a mobile phase of water (0.1% acetic acid) and acetonitrile (95:5 v/v). The calibration graphs were linear from 0.31 to 40 μg/mL for TP, from 0.15 to 10 μg/mL for 1-MU, from 0.08 to 10 μg/mL for 1,3-DMU and from 0.16 to 20 μg/mL for 3-MX.
1-MX, 3-MX, and 1,3-DMU were assessed in the reaction mixtures by centrifugation at 15 000×g for 10 min, and 20 μL of supernatant was injected into an HPLC system. The mobile phase consisted of water (0.1% formic acid) and acetonitrile (95:5, v/v). The calibration graphs were linear from 15.6 to 125 ng/mL for 1-MX, from 7.8 to 125 ng/mL for 3-MX and from 31.25 to 500 ng/mL for 1,3-DMU.
ATFX concentrations in plasma and urine were measured by HPLC with UV detection, as previously described19. The calibration graphs were linear from 0.1 to 12.5 μg/mL for plasma and from 0.40 to 25.0 μg/mL for urine.
Data analysis
The plasma and urinary pharmacokinetics of ATFX and TP on d 1 (single dose) and d 5 (steady-state) were analyzed by non-compartmental methods using the Bioavailability Program Package (BAPP, version 3.1, China Pharmaceutical University, Nanjing, China). The maximum plasma concentration (Cmax) and time to Cmax (Tmax) were taken directly from the plasma-time profiles. The terminal elimination constant (λZ) was estimated by the least squares linear regression analysis of the concentration-time data obtained over the terminal in-linear phase after the last dose. Terminal half-lives (t1/2) were calculated as 0.693/λZ. The area under the plasma concentration-time curve (AUC0-t) was calculated using the linear trapezoidal rule. Total clearance (Cl/F) was calculated as Cl/F=Dose/AUCss0–t. The accumulated amounts (Ae) in designed intervals were also calculated. Renal clearance (Clr) was calculated as Aet/AUCtn, where Aet and AUCtn were the accumulated amounts excreted into urine and the AUC values during the interval, respectively. The non-renal clearance (Clnr) was designed to be Cl/F-Clr. Analysis of the differences between the pharmacokinetic parameters of drug alone and the combination of drugs was handled as an equivalence problem20. Comparison of pharmacokinetic parameters was carried out with analysis of variance (ANOVA)-based 90% confidence intervals (CI) after log-transformation. Those were calculated for the test (drug combination) and reference (drug alone) for all parameters, except Tmax and t1/2, for which the differences were used. A lack of interaction was assumed if the values of CI for the ratios μtest/μreference were completely within the 0.80–1.25 equivalence range.
Reversible inhibition enzymatic constants (Km, Vmax, and KI) were estimated from the observed metabolite formation rate (v), the TP concentration [S] and inhibitor concentration [I] by nonlinear regression analysis using the extended Michaelis-Menten equation:
 |
The parameters of the corresponding enzyme inactivation (kobs,i, kinact,i and Kapp,i) by ATFX were estimated as follows. The logarithm of the enzymatic activity (formation rate of metabolite) was plotted against the pre-incubation time, and the observed inactivation rate constant (kobs,i) was calculated from the slope of the initial linear phase. The reciprocal of the kobs,i obtained was plotted against the reciprocal of the ATFX concentration to determine kinact,i and Kapp,i, according to equation 221.
 |
where kinact,i is maximum inactivation rate constant for the ith metabolite, [I] is the initial concentration of inhibitor and Kapp,i is the inhibitor concentration when kobs,i=kinact,i/2.
Simulation
The in vitro data describing the mechanism-based inhibition of CYP1A2 by ATFX were further used to rationalize the observed clinical drug interactions. The following assumptions were made: TP is well absorbed; the substrate exhibits linear pharmacokinetics; the drug is eliminated via both metabolism in the liver and excretion in the kidney; and the inhibitor has no effect on the absorption or kidney clearance of the substrate. The hepatic clearance of the substrate is adequately described by the well-stirred model, and degradation of the enzyme is a first order process. Furthermore, the inhibitor has no effect on the rate of synthesis of the enzyme.
An attempt was made to evaluate the effects on the oral pharmacokinetics of TP involved in multi-elimination pathways, according to equation 322.
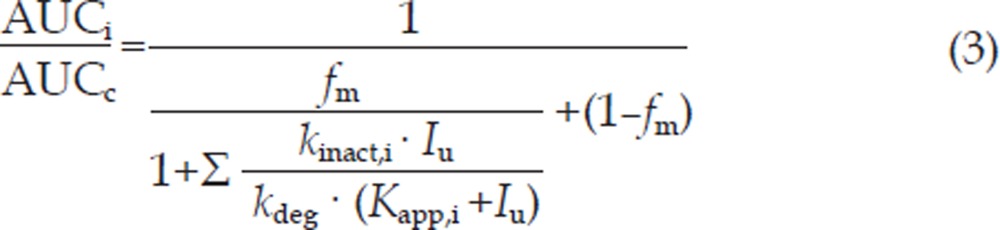 |
where AUCi and AUCc are the AUC values of TP in the presence and absence of inhibitors, respectively. fm represents the fraction of total hepatic elimination via CYPs. Kapp,i and kinact,i are the parameters describing inactivation of the enzyme that forms the ith metabolite. kdeg represents the rate constant of degradation of the enzyme. Estimates of the concentration of the inhibitor at the enzyme (Iu) were based on the free plasma concentrations. To determine the effect of ATFX on CYP1A2, the in vitro Kapp,i and kinact,i data obtained in the study were used in the simulations. Values of kdeg were set to be 0.000769 min−1 or 0.00128 min−1, as cited by Wang et al22. The steady-state concentrations of ATFX in plasma (Cmax and Cav) obtained in the study were used for simulation after being corrected for fu=0.825. The simulated AUC change was compared with that observed in the clinic.
Results
Clinical observations
All 12 enrolled volunteers completed the study in accordance with the protocol. Adverse events were reported by 7 subjects. The numbers of subjects reporting adverse events were 4 for ATFX, 3 for TP and 7 for TP plus ATFX. The adverse events included neurological symptoms (mainly drowsiness, headache and sleeplessness) and gastrointestinal complaints (diarrhea, nausea, lack of appetite and stomach disorders). Neurological symptoms were reported by 3, 3, and 6 subjects; gastrointestinal events were reported by 0, 4, and 6 subjects in periods with TP alone, ATFX alone and TP plus ATFX, respectively. The results showed that the incidence of adverse events was higher in the period with TP plus ATFX. Neither serious nor severe adverse experiences were observed, and most of the adverse experiences were mild and transient. All symptoms had resolved by the time of the follow-up examination. No clinically relevant changes were observed in the laboratory safety tests either during the study or at the follow-up examination.
Pharmacokinetics of TP
The mean plasma concentration-time courses of TP during the administration of multiple doses (complete profiles after morning doses on d 1 and 5, morning trough concentrations on d 2 to 5) for TP treatment alone and for TP treatment with ATFX are shown in Figure 1. The individual accumulated amounts (0 to 24 h) of TP and its three metabolites excreted into urine were calculated. The corresponding pharmacokinetic parameters for the first dose and last dose are summarized in Table 1. Comparison of treatments was carried out with analysis of variance-based 90% confidence intervals (CI) as an equivalence problem. After the first dose (on d 1), estimated plasma pharmacokinetic parameters for the two treatments were equivalent in terms of AUC0–24 (106.6%, CI: 94.4% to 120.4%) and Cmax(106.4%, CI: 88.7% to 124.6%). The accumulated amounts of TP and its three metabolites in the urine in the two treatments were similar. However, after multiple doses (on d 5), the mean CSSmax of 9.32 μg/mL, mean CSSmin of 6.02 μg/mL and AUC0–12SS of 92.4 μg·h/mL in the period of TP alone significantly increased to a CSSmax of 12.02 μg/mL (132.1%, CI: 114.0% to 153.1%), CSSmin of 8.04 μg/mL (139.2%, CI: 122.4% to 156.0%) and AUC0–12SS of 117.7 μg·h/mL (129.4%, CI: 113.7% to 147.3%) in the period of co-administration with ATFX, respectively, accompanied by a decrease in clearance (2.36±0.81 L/h in periods of TP alone vs 1.78±0.39 L/h in the period of combination). For comparison of the pharmacokinetic parameters of TP between the two treatments at steady state, the parametric point estimates of the μtest/μreference ratios and corresponding CI for AUC, Cl, and Cmax were partially or completely outside the predefined interval of no interaction (0.80 to 1.25).
Figure 1.

Comparison of TP concentration in plasma after an oral dose of TP alone (open circle) and co-administration of ATFX (filled circle). For TP, on d 1 and 5, 200 mg of TP was given to subjects as a single morning dose, and on d 2 to 4, 400 mg of TP was given to subjects twice daily (morning dose and evening dose). For ATFX, on d 1, 400 mg was given to subjects as a single morning dose, and on d 2 to 5, 200 mg of ATFX was given to subjects once daily (morning dose). The results are expressed as the mean±SD (n=12).
Table 1. Pharmacokinetic parameters of TP alone and the co-administration of ATFX.
| Parameters | TP alone | TP plus ATFX | Point estimate (90% CI) | |
|---|---|---|---|---|
| 1st day | AUC0–24 (μg·h/mL) | 50.00±14.49 | 51.77±10.27 | 106.6% (94.4%–120.4%) |
| Cmax (μg /mL) | 3.00±0.91 | 3.23±1.30 | 106.4% (88.7%–124.6%) | |
| Tmax (h) | 4.8±2.8 | 6.2±1.8 | / | |
| Cl (L/h) | 4.45±1.74 | 4.01±0.81 | 95.7% (86.3%–105.2%) | |
| Ae(0–24) | / | |||
| 1-MX (mg) | 3.82±3.22 | 5.19±2.67 | / | |
| 3-MX (mg) | 6.13±2.12 | 5.36±1.39 | / | |
| 1,3-DMU (mg) | 5.93±3.15 | 6.60±5.21 | / | |
| TP (mg) | 21.46±5.83 | 20.21±4.33 | / | |
| 5th day | AUC0–12 (μg·h/mL) | 92.64±27.08 | 117.72±27.77b | 129.4% (113.7%–147.3%) |
| AUC0–24 (μg·h/mL) | 152.95±43.06 | 200.04±525b | 132.7% (118.7%–148.5%) | |
| Cmax (μg/mL) | 9.32±2.70 | 12.02±2.41b | 132.1% (114.0%–153.1%) | |
| Tmax (h) | 3.0±1.2 | 3.3±0.9 | / | |
| t1/2 (h) | 17.93±5.08 | 18.66±5.04 | / | |
| Cl (L/h) | 2.36±0.81 | 1.78±0.39b | 77.2% (67.8%–87.9%) | |
| Clr (L/h) | 0.34±0.07 | 0.32±0.08 | 93.6% (78.9%–111.3%) | |
| Clnr (L/h) | 2.03±0.78 | 1.46±0.37c | 74.4% (65.7%–84.3%) | |
| Ae(0–24) | / | |||
| 1-MX (mg) | 25.49±21.17 | 9.76±5.07b | / | |
| 3-MX (mg) | 28.75±13.38 | 19.44±5.06b | / | |
| 1,3-DMU (mg) | 39.24±29.70 | 20.77±15.60b | / | |
| TP (mg) | 53.13±17.23 | 66.75±15.69b | / |
For TP, on d 1 and 5, 200 mg of TP was given to subjects as a single morning dose. On d 2 to 4, 400 mg of TP was given to subjects twice daily (morning dose and evening dose). For ATFX, on d 1, 400 mg was given to subjects as a single morning dose, and on d 2 to 5, 200 mg of ATFX was given to subjects once daily (morning dose). The results are expressed as the mean±SD (n=12).
bP<0.05,
cP<0.01 vs TP alone.
Urinary excretion of TP and its metabolites were measured both on d 1 and 5 of TP administration alone and co-administration with ATFX. It was found that a 1-day period of co-administration with 400 mg of ATFX did not alter the excretion levels of TP, 3-MX, 1-MU, or 1,3-DMU. However, a 5-day period of the co-administration with 200 mg of ATFX resulted in decreases of all urinary TP metabolites and increases in urinary TP (Table 1). The renal clearance (Clr) and non-renal clearance (Clnr) of TP were also calculated. It was observed that the non-renal clearances of TP in the period of combination significantly decreased, but no alteration of TP renal clearance was noted. The decrease of Clnr was associated with decreased amounts of the three TP metabolites in urine.
Pharmacokinetics of ATFX
The mean plasma concentration-time courses of ATFX during the administration of multiple doses (complete profiles after morning dose on d 1 and 5, morning trough concentrations on d 2 to 5) of ATFX treatment alone and ATFX treatment with TP are shown in Figure 2. The accumulated amounts (0 to 24 h) of ATFX excreted into the urine were also calculated.
Figure 2.

Comparison of ATFX concentrations in plasma after an oral dose of ATFX alone (open circle) and co-administration of TP (filled circle). For TP, on d 1 and 5, 200 mg of TP was given to subjects as a single morning dose, and on d 2 to 4, 400 mg of TP was given to subjects twice daily (morning dose and evening dose). For ATFX, on d 1, 400 mg was given to subjects as a single morning dose, and on d 2 to 5, 200 mg of ATFX was given to subjects once daily (morning dose). The results are expressed as the mean±SD (n=12).
The respective pharmacokinetic parameters for the first and last doses of the treatment are summarized in Table 2. After the first dose, the plasma pharmacokinetics of ATFX for the two treatments were equivalent in terms of AUC0–24 (98.9%, CI: 86.5% to 113.1%) and Cmax (98.6%, CI: 83.2% to 116.8%). The mean Tmax was also similar for the two treatments. The amount of ATFX excreted into the urine showed a trend of increasing in the period of combination, even though no significance was found. After multiple doses, similar plasma pharmacokinetic parameters were observed in the two treatments. Parametric point estimates of the μtest/μreference ratios and the corresponding CI for Cmax, AUC, and Cl following the first and last doses were within the equivalence interval from 0.80 to 1.25. The results indicated that TP does not affect the pharmacokinetics of ATFX in humans.
Table 2. Pharmacokinetic parameters of ATFX alone and the co-administration of TP.
| Parameters | ATFX | ATFX plus TP | Point estimate (90% CI) | |
|---|---|---|---|---|
| 1st day | AUC0–24 (μg·h/mL) | 39.93±8.86 | 38.46±8.36 | 98.9% (86.5%–113.1%) |
| Cmax (μg/mL) | 2.94±0.78 | 2.90±0.85 | 98.6% (83.2%–116.8%) | |
| Tmax (h) | 1.3±1.0 | 2.1±1.1 | / | |
| Cl (L/h) | 10.76±2.41 | 10.83±2.17 | 104.1% (91.8%–116.5%) | |
| Ae(0–24) (mg) | 226.40±56.26 | 266.42±46.82 | / | |
| 5th day | AUC0–24SS (μg·h/mL) | 24.32±6.03 | 27.57±6.98 | 113.5% (104.9%–122.8%) |
| CSSmax (μg /mL) | 2.10±0.39 | 2.36±0.48 | 112.5% (104.6%–121.0%) | |
| Cav (μg /mL) | 1.03±0.25 | 1.15±0.29 | 113.5% (104.9%–122.8%) | |
| Tmax (h) | 1.5±0.7 | 1.8±1.2 | / | |
| t1/2 (h) | 13.56±3.75 | 15.01±4.11 | / | |
| Cl (L/h) | 8.62±1.83 | 7.57±1.44 | 88.1% (81.4%–95.4%) | |
| Ae(0–24) (mg) | 193.36±59.48 | 209.35±26.61 | / | |
| Clr (L/h) | 8.55±2.42 | 7.60±1.91 | 90.6% (80.3%–110.6%) |
For ATFX, on d 1, 400 mg was given to subjects as a single morning dose, and on d 2 to 5, 200 mg of ATFX given to subjects once daily (morning dose). For TP, on d 1 and 5, 200 mg of TP was given to subjects as a single morning dose. On d 2 to 4, 400 mg of TP was given to subjects twice daily (morning dose and evening dose). The results are expressed as the mean±SD (n=12).
In vitro inhibition of TP metabolism by ATFX in human liver microsomes
The effects of ATFX on the formation of 1-MX, 3-MX, and 1,3-DMU from TP were investigated with human liver microsomes in the presence of an NADPH-generating system. The reversible inhibition parameters were estimated (Table 3). To measure accurate kinetic parameters, the concentration of the given substrate (TP in this study) should approach Km23. We established TP concentrations for in vitro study according to several previous studies24, 25. Additionally, ATFX concentrations for the in vitro study were selected according to our previous study17.
Table 3. Inhibitory effect of ATFX on TP metabolites in human liver microsomes in vitro. Data represent mean±SD of three experiments.
| KI (μmol/L) | Kapp,i (μmol/L) | kinact,i (min−1) | kinact,i/Kapp,i (×10−5) (min−1·M−1) | |
|---|---|---|---|---|
| 3-MX | 1209.2±66.3 | 265.88±18.81 | 0.0097±0.0016 | 3.68±0.84 |
| 1-MX | 1033.2±78.9 | 353.29±16.20 | 0.0073±0.0012 | 2.06±0.37 |
| 1,3-DMU | 1519.4±56.6 | 430.21±14.19 | 0.0047±0.0080 | 1.08±0.15 |
It was noted that ATFX may inhibit TP metabolism in human liver microsomes. The inhibition of TP metabolism by ATFX is pathway-dependent. The inhibition is more selective for 3-MX than for 1-MX and 1,3-DMU. For example, co-incubation of 1 mg/mL ATFX with 2 mmol/L TP induced decreases in 3-MX, 1-MX, and 1,3-DMU formation from the controls by 63%, 54%, and 38%, respectively. The estimated reversible inhibition index (KI) values for the formation of the three metabolites were over 1000 μmol/L (corresponding 412 μg/mL) (Table 3); however, these concentrations do not occur in the human body. These results indicated that ATFX was a weak, reversible inhibitor of CYP1A2, and this mechanism-based inhibition was further investigated. When pre-incubated with NADPH-fortified human microsomes in the absence of the substrate, ATFX was shown to have inhibitory effects on the metabolism of TP to 1-MX, 3-MX, and 1,3-DMU. The effect was NADPH-, time-, and concentration-dependent (Figure 3). It is well known that time-dependent inhibition of enzymes is one of the characteristics of mechanism-based inhibition. Therefore, our data suggested that ATFX behaves as a mechanism-based inhibitor of CYP1A2. Upon further study, the maximal inactivation rate constant (kinact,i), apparent rate constant Kapp,i and ratios of kinact,i/Kapp,i were estimated (Table 3). The mechanism-based inhibition was also pathway-dependent, and the kinact,i/Kapp,i ratios were estimated to be 0.0000368 min−1·μM−1 for 3-MX, 0.0000206 min−1·μM−1 for 1-MX and 0.0000108 min−1·μM−1 for 1,3-MDU, which indicated that the mechanism-based inhibition was favorable for 3-MX.
Figure 3.

Kinetics of inactivation of microsomal TP metabolism by ATFX (n=5). Human liver microsomes were pre-incubated for the indicated times in an NADPH-generating system, and ATFX, followed by CYP1A2 activity, was measured. Symbols: open circle, 0 μg/mL ATFX; filled circle, 100 μg/mL ATFX; open square, 200 μg/mL ATFX; filled square, 400 μg/mL ATFX, and open triangle, 800 μg/mL ATFX. The insets correspond to the double reciprocal of the inactivation rates as a function of ATFX concentration.
Retrospective in vitro-in vivo correlations
After it was determined that ATFX behaves as a mechanism-based inhibitor of CYP1A2 in human liver microsomes, an attempt was made to relate the in vitro data (Kapp,i and kinact,i) to the pharmacokinetics of ATFX. In turn, the effect of ATFX on the pharmacokinetics (AUC) of TP (a CYP1A2 substrate) was simulated, assuming that the degradation of CYP1A2 was a first order process and that kdeg was set to be 0.000769 min−1 or 0.00128 min−1, as cited by Wang et al22. fm (fraction of total hepatic elimination via CYPs) values were estimated to be 0.86 using the ratio of Clnr to total clearance of TP in the absence of ATFX in the study. After correction for binding to human plasma proteins, fu=0.825 (unpublished data), the Cmax (5.73 μmol/L≈2.36 μg/mL) at steady state and Cav (2.79 μmol/L≈1.15 μg/mL) of ATFX obtained in the study were used for simulating AUC changes. Upon implementation, good simulation was found using Cmax (5.73 μmol/L) and kdeg (0.000769 min−1). The estimated change of AUC was 1.32-fold, which was close to the 1.29-fold change observed in the study. However, Cav (2.79 μmol/L) or kdeg (0.00128 min−1) gave an underestimation of AUC changes, as the estimated changes were 1.16- and 1.20-fold, respectively.
Discussion
This study is different from a traditional three-period crossover trial. A randomized, 5-day treatment and 3-way crossover design was documented in 12 healthy subjects. The oral medications prescribed were ATFX, TP, or TP plus ATFX. For the ATFX treatment, subjects received 200 mg of ATFX once daily on d 2 to 5 of the 5-day study period and 400 mg of ATFX on d 1. For the TP treatment, subjects orally received 200 mg of TP twice daily on d 2 to 4 of the 5-day period and 200 mg of TP as a single morning dose on d 1 and 5. The purpose of the study was to investigate the pharmacokinetic interaction between TP and ATFX in healthy Chinese volunteers. After the three-period trial, we compared either ATFX alone with ATFX plus TP, or TP alone with ATFX plus TP. However, we could not compare ATFX alone with TP alone. The significant difference between the three periods was not our emphasis for the purpose of the study.
The present study clearly demonstrated that ATFX increased the Cmax and AUC of TP at steady state by 32% and 29%, respectively. According to the rule of Niki's classification26, ATFX belongs to the class II quinolones because the increase in Cmax and AUC of TP is between 15% and 39%. A significant decrease of formation of TP metabolites was also found, as evidenced by decreased urinary excretion. It is well known that TP is mainly metabolized via CYP1A2. These results indicated that ATFX may elicit an increase in the concentration of TP in plasma by inhibiting hepatic microsomal CYP1A2. However, the effect of ATFX on the TP concentration was time-dependent. This was in contrast to the findings at steady state (d 5) that the co-administration of ATFX on d 1 did not alter the plasma concentrations of TP or the urinary excretion of TP metabolites (Figure 1 and Table 1), although the ATFX concentrations in plasma on d 1 were higher than those on d 5 (Figure 2 and Table 2). Similar phenomena were found for the effects of tosufloxacin on TP plasma concentrations and the effects of enoxacin on caffeine concentrations20, 27. The in vitro results using human liver microsomes showed that ATFX is a weak reversible inhibitor of CYP1A2, but ATFX appears to be a clinically relevant inhibitor. The clinical observations cannot be reconciled with the in vitro results, as the in vivo findings were simply not explained using a reversible inhibition of CYP1A2. Instead, ATFX may be a mechanism-based inhibitor of CYP1A2 activity. Further experiments were designed to verify the hypothesis using human liver microsomes to analyze 1-MX, 3-MX, and 1,3-DMU formation, and the results were positive. Inhibition of the formation of the three metabolites of TP by ATFX was time-, concentration- and NADPH-dependent, which is characteristic of mechanism-based inhibition. Similar findings were observed using rat liver microsomes to assess the formation of acetaminophen from phenacetin17.
The inhibitory effect of ATFX on metabolite formation in human liver microsomes was amenable to kinetic analysis, yielding estimates of kinact,i, Kapp,i, and kinact,i/Kapp,i ratios (Table 3). The kinact,i/Kapp,i ratios of ATFX were considerably lower than those reported for furafylline (0.229 min−1·μM−1)28 and zileuton (0.0003 min−1·μM−1)29. We sought to relate the in vitro inhibition parameters of ATFX to rationalize the observed interaction between ATFX and TP in the study. Efforts have been made to evaluate the mechanism-based inhibition of CYPs using physiologically based models17, 21, 30, 31. Overall, it is accepted that the net effect of inhibitors is dependent on the kinact,i/Kapp,i ratio, kdeg and concentration of inhibitor at the enzyme active site. For the sake of simplicity, retrospective analysis of the ATFX data were based on a kinetic model (Equation 3) using the values of kdeg cited by Wang et al22 (0.00128 min−1 or 0.000769 min−1). The parameters of inactivation of the enzyme with Cmax at steady state or Cav (corrected for fu) are listed in Table 3. It was noted that the simulated AUC changes were in agreement with the observed values using Cmax at steady state and a kdeg of 0.000769 min−1, which indicated that it is possible to reconcile the clinical drug interactions between TP and ATFX with the in vitro results according to a mechanism-based inhibition. The mechanism-based inhibition may explain why ATFX causes an increase in TP levels in plasma and why the inhibitory effect of ATFX is time-dependent.
A recent report has demonstrated that treatment with ciprofloxacin is associated with a significant increase in the risk of TP toxicity in elderly patients32. Our study confirmed the idea that the co-administration of ATFX increases the plasma levels of TP in young populations. These results indicate that ATFX probably decreases TP clearance and increases the risk of TP toxicity in elderly patients. Further studies are required to investigate the pharmacokinetic interactions between ATFX and TP in elderly populations.
Based on the results of our study, it was concluded that ATFX appears to be a clinical mechanism-based inhibitor of CYP1A2. Co-administration of ATFX may increase TP concentrations in plasma, which would raise the risk of TP toxicity in patients. We suggest that when patients receiving TP require treatment with antibiotics, avoidance of ATFX may be clinically appropriate. Alternatively, close monitoring for TP concentrations and toxicity is warranted in cases where the use of ATFX is required.
Author contribution
Li LIU and Xian PAN performed the research; Hai-yan LIU and Hui-wen YANG measured the plasma concentrations of theophylline and antofloxacin; Li LIU wrote the manuscript; Xiao-dong LIU and Lin XIE reviewed and revised the manuscript; and Jun-lin CHENG, Hong-wei FAN, and Da-wei XIAO participated in the clinical study and reviewed the manuscript.
Acknowledgments
This work was supported by the “Eleventh five-year Plan”–Technical Platform for Drug Development from the Ministry of Science and Technology of the People's Republic of China (No 2009ZX09304-001).
References
- Ye H, Wu JM, Yang YS, Chen KX, Ji RY. Antibacterial activities of the derivatives-YH54 and YH57 of levofloxacin in vitro. Chin Pharmacol Bull. 2002;18:112–3. [Google Scholar]
- Xiao Y, Lu Y, Kang Z, Hou F, Wang S, Li T, et al. Tolerability and pharmacokinetics of antofloxacin hydrochloride after multiple oral dose administration in healthy Chinese male volunteers. Int J Clin Pharmacol Ther. 2008;46:172–9. doi: 10.5414/cpp46172. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Lu Y, Kang Z, Zhang M, Liu Y, Zhang M, et al. Pharmacokinetics of antofloxacin hydrochloride, a new fluoroquinolone antibiotic, after single oral dose administration in Chinese healthy male volunteers. Biopharm Drug Dispos. 2008;29:167–72. doi: 10.1002/bdd.600. [DOI] [PubMed] [Google Scholar]
- Gundert-Remy U, Hildebrandt R, Hengen N, Weber E. Non-linear elimination processes of theophylline. Eur J Clin Pharmacol. 1983;24:71–8. doi: 10.1007/BF00613930. [DOI] [PubMed] [Google Scholar]
- Tang-Liu DD, Williams RL, Riegelman S. Nonlinear theophylline elimination. Clin Pharmacol Ther. 1982;31:358–69. doi: 10.1038/clpt.1982.46. [DOI] [PubMed] [Google Scholar]
- Campbell ME, Grant DM, Inaba T, Kalow W. Biotransformation of caffeine, paraxanthine, theophylline and theobromine by polycyclic aromatic hydrocarbon-inducible cytochrome P450 in human liver microsomes. Drug Metab Dispos. 1987;15:237–49. [PubMed] [Google Scholar]
- Ha HR, Chen J, Freiburghaus AU, Follath F. Metabolism of theophylline by cDNA expressed human cytochromes P-450. Br J Clin Pharmacol. 1995;39:321–6. doi: 10.1111/j.1365-2125.1995.tb04455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar MA, Hunt C, Guzelian PS, Karnes HT. Characterization of human liver cytochrome P-450 involved in theophylline metabolism. Drug Metab Dispos. 1992;20:31–7. [PubMed] [Google Scholar]
- Tjia JF, Colbert J, Back DJ. Theophylline metabolism in human liver microsomes: inhibition studies. J Pharmacol Exp Ther. 1996;276:912–7. [PubMed] [Google Scholar]
- Bachmann K, White D, Jauregui L, Schwartz JI, Agrawal NG, Mazenko R, et al. An evaluation of the dose-dependent inhibition of CYP1A2 by rofecoxib using theophylline as a CYP1A2 probe. J Clin Pharmacol. 2003;43:1082–90. doi: 10.1177/0091270003257454. [DOI] [PubMed] [Google Scholar]
- Wijnands WJA, Van Herwaarden CLA, Vreeet TB. Enoxacin raises plasma theophylline concentrations. Lancet. 1984;2:108–9. doi: 10.1016/s0140-6736(84)90283-6. [DOI] [PubMed] [Google Scholar]
- Edwards DJ, Bowles SK, Svensson CK, Rybak MJ. Inhibition of drug metabolism by quinolone antibiotics. Clin Pharmacokinet. 1988;15:194–204. doi: 10.2165/00003088-198815030-00004. [DOI] [PubMed] [Google Scholar]
- Fuhr U, Anders EM, Mahr G, Sörgel F, Staib AH. Inhibitory potency of quinolone antibacterial agents against cytochrome P450IA2 activity in vivo and in vitro. Antimicrob Agents Chemother. 1992;36:942–8. doi: 10.1128/aac.36.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi CM, Parker BM, Cusack BJ, Vestal R. Individual and combined effects of cimetidine and ciprofloxacin on theophylline metabolism in male nonsmokers. Br J Clin Pharmacol. 1993;36:195–200. doi: 10.1111/j.1365-2125.1993.tb04216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuschka PR, Vissing RS. Clinafloxacin-theophylline drug interaction. Ann Pharmacother. 1995;29:378–80. doi: 10.1177/106002809502900407. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wei MJ, Zhao CY, Qi HM. Determination of the inhibitory potential of 6 fluoroquinolones on CYP1A2 and CYP2C9 in liver microsomes. Acta Pharmacol Sin. 2008;29:1507–14. doi: 10.1111/j.1745-7254.2008.00908.x. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Liao J, Xie L, Wang GJ, Liu XD. Mechanism-based inhibition of CYP1A2 by antofloxacin, an 8-NH2 derivative of levofloxacin in rats. Xenobiotica. 2009;39:293–301. doi: 10.1080/00498250802709428. [DOI] [PubMed] [Google Scholar]
- Nie SL, Liu HY, Xie L. Reversed-phase high performance liquid chromatographic method for simultaneous determination of theophylline and its two metabolites in human urine. Chin J Clin Pharmacol Ther. 2009;14:1386–91. [Google Scholar]
- Pang XY, Xie L, Xu X, Liu HY, Liu L, Liu XD. Pharmacokinetics of ATFX in rats coadministered with non-steroidal anti-inflammatory drugs. Chin J New Drugs. 2007;16:600–3. [Google Scholar]
- Kinzig-Schippers M, Fuhr U, Zaigler M, Dammeyer J, Rüsing G, Labedzki A, et al. Interaction of pefloxacin and enoxacin with the human cytochrome P450 enzyme CYP1A2. Clin Pharmacol Ther. 1999;65:262–74. doi: 10.1016/S0009-9236(99)70105-0. [DOI] [PubMed] [Google Scholar]
- Mayhew BS, Jones DR, Hall SD. An in vitro model for predicting in vivo inhibition of cytochrome P450 3A4 by metabolic intermediate complex formation. Drug Metab Dispos. 2000;28:1031–7. [PubMed] [Google Scholar]
- Wang YH, Jones DR, Hall SD. Prediction of cytochrome P450 3A inhibition by verapamil enantiomers and their metabolites. Drug Metab Dispos. 2004;32:259–66. doi: 10.1124/dmd.32.2.259. [DOI] [PubMed] [Google Scholar]
- Fowler S, Zhang H. In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: current status on methodologies and their utility for predicting drug-drug interactions. AAPS J. 2008;10:410–24. doi: 10.1208/s12248-008-9042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar M, Polk RE, Guzelian PS, Hunt C, Karnes HT. In vitro effect of fluoroquinolones on theophylline metabolism in human liver microsomes. Antimicrob Agents Chemother. 1990;34:594–9. doi: 10.1128/aac.34.4.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Suh OK, Lee MG. Pharmacokinetics of intravenous theophylline in mutant Nagase analbuminemic rats. Life Sci. 2003;72:1231–45. doi: 10.1016/s0024-3205(02)02373-1. [DOI] [PubMed] [Google Scholar]
- Niki Y, Watanabe S, Yshida K, Miyashita N, Nakajima M, Matsushima T. Effect of pazufloxacin mesilate on the serum concentration of theophylline. J Infect Chemother. 2002;8:33–6. doi: 10.1007/s101560200003. [DOI] [PubMed] [Google Scholar]
- Niki Y. Pharmacokinetics and safety assessment of tosufloxacin tosilate. J Infect Chemother. 2002;8:1–18. doi: 10.1007/s101560200000. [DOI] [PubMed] [Google Scholar]
- Fairman DA, Collins C, Chapple S. Progress curves analysis of CYP1A2 inhibition: a more information approach to assessment of mechanism-based inactivation. Drug Metab Dispos. 2007;35:2159–65. doi: 10.1124/dmd.107.017236. [DOI] [PubMed] [Google Scholar]
- Lu P, Schrag ML, Slaughter DE, Raab CE, Shou M, Rodrigues AD. Mechanism-based inhibition of human liver microsomal cytochromes P450 1A2 by zileuton, a 5-lipoxygenase inhibitor. Drug Metab Dispos. 2003;31:1352–60. doi: 10.1124/dmd.31.11.1352. [DOI] [PubMed] [Google Scholar]
- Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50:387–411. [PubMed] [Google Scholar]
- Ito K, Iwatsubo T, Kanamitsu S, Itoh T. Prediction of the in vivo interaction between midazolam and macrolides based on in vitro studies using human liver microsomes. Drug Metab Dispos. 2003;31:945–54. doi: 10.1124/dmd.31.7.945. [DOI] [PubMed] [Google Scholar]
- Antoniou T, Gomes T, Mamdani MM, Juurlink DN. Ciprofloxacin-induced theophylline toxicity: a population-based study. Eur J Clin Pharmacol. 2011;67:521–6. doi: 10.1007/s00228-010-0985-0. [DOI] [PubMed] [Google Scholar]