

Abstract
Aim:
To develop a novel gastroretentive drug delivery system based on a self-microemulsifying (SME) lipid mixture for improving the oral absorption of the immunosuppressant tacrolimus.
Methods:
Liquid SME mixture, composed of Cremophor RH40 and monocaprylin glycerate, was blended with polyethylene oxide, chitosan, polyvinylpyrrolidone and mannitol, and then transformed into tablets via granulation, with ethanol as the wetting agent. The tablets were characterized in respect of swelling, bioadhesive and SME properties. In vitro dissolution was conducted using an HCl buffer at pH 1.2. Oral bioavailability of the tablets was examined in fasted beagle dogs.
Results:
The tablet could expand to 13.5 mm in diameter and 15 mm in thickness during the initial 20 min of contact with the HCl buffer at pH 1.2. The bioadhesive strength was as high as 0.98±0.06 N/cm2. The SME gastroretentive sustained-release tablets preserved the SME capability of the liquid SME formations under transmission electron microscope. The drug-release curve was fit to the zero-order release model, which was helpful in reducing fluctuations in blood concentration. Compared with the commercially available capsules of tacrolimus, the relative bioavailability of the SME gastroretentive sustained-release tablets was 553.4%±353.8%.
Conclusion:
SME gastroretentive sustained-release tablets can enhance the oral bioavailability of tacrolimus with poor solubility and a narrow absorption window.
Keywords: immunosuppressants, tacrolimus, bioavailability, self-microemulsifying lipid mixture, gastroretentive sustained-release tablet
Introduction
Tacrolimus, a 23-member macrolide lactone, was isolated from Streptomyces tsukubaensis early in 19841, 2. Its use is now well established for primary immunosuppression in liver and kidney transplantation. Meanwhile, experience with its use in other types of solid-organ transplantation, including heart, lung, pancreas and intestinal, as well as its use for the prevention of graft-versus-host disease in allogeneic bone marrow transplantation (BMT), is rapidly accumulating3. However, being a BCS class II drug, the clinical efficacy of tacrolimus is very limited because of its poor water solubility (5–8 μg/mL), which is responsible for its low oral bioavailability2, 4. The dissolution rate of tacrolimus is one of the rate-limiting steps for its in vivo absorption. A self-microemulsifying drug delivery system (SMEDDS) has been used to improve the oral bioavailability of poorly soluble drugs by presenting and maintaining the drug in a dissolved state during its entire transit through the gastrointestinal tract5. Therefore, SMEDDS was designed in this study to improve the dissolution and oral bioavailability of tacrolimus. At present, there are four drug products, Sandimmune®, Sandimmun Neoral® (cyclosporine A), Norvir® (ritonavir), and Fortovase® (saquinavir) on the pharmaceutical market, the active compounds of which have been formulated into the specific SMEDDS. Significant improvement in the oral bioavailability of these drug compounds has been demonstrated for each case, which further justifies this study6.
SMEDDS is usually formulated in a liquid form, which has some disadvantages, especially in the manufacturing process, leading to high production costs. Furthermore, incompatibility problems with the capsule shell are common7. Recently, with the development of new materials and a novel preparation process, there is growing interest in the study of solid self-microemulsifying drug delivery systems (S-SMEDDS)5, 7, 8, 9, 10, 11, 12, 13, 14. Compared with the traditional SMEDDS, S-SMEDDS can increase stability, extend storage time, reduce gastrointestinal irritation, and improve patient compliance. Furthermore, it was reported that the bioavailability of the self-microemulsifying mixture in a solid dosage form was equivalent to that of a liquid form15.
Similar to cyclosporin A, tacrolimus has a very narrow therapeutic window, and it exhibits large intra- and inter-individual variability of bioavailability, ranging from 4% to 89% (mean of around 25%)16, 17. A high blood concentration will not only cause renal toxicity in the patient but also trigger the immune over-infection; however, a low concentration often leads to graft rejection. Post-treatment monitoring of blood levels is an integral part of patient care to maintain drug levels within the therapeutic range to optimize therapy and reduce undesirable toxic effects. This results in a significant inconvenience in the clinical application of tacrolimus. Therefore, designing a sustained-release formulation for tacrolimus that can control the drug blood concentration at a suitable level is quite essential. A tacrolimus sustained-release capsule developed by Astellas was approved both in Europe (Advagraf®) and Japan (Graceptor®). In addition, Life Cycle Pharma A/S has developed a tacrolimus sustained-release tablet, and a clinical phase III trial is in progress18. Some in vitro and in vivo studies have shown that these formulations can reduce blood concentration fluctuations, and the differences in intra- and inter-individual levels have also been improved. However, these sustained-release capsules do not improve the bioavailability of tacrolimus, which is equivalent to the immediate-release capsules. The objective of developing SME sustained-release tablets in our study was not only to reduce the blood concentration fluctuations but also to improve the bioavailability.
Tacrolimus shows significant site dependence in intestinal permeability, and it is absorbed predominantly in the upper part of the small intestine19, 20. It was reported that differences in P-gp and P450 function in each intestinal site could be a main cause of the site selectivity and large variability in tacrolimus absorption16. The gastroretentive drug delivery system (GRDDS) is a preferable approach to improve the oral bioavailability and variability of a drug with a narrow absorption window in the upper part of the gastrointestinal tract (ie, stomach and small intestine). Prolonged gastric residence is expected to lead to an increased contact interval with the main absorption site of tacrolimus, the mucosa of the upper small intestine. Owing to its improved bioavailability combined with reduced frequency of administration and thus improved patient compliance, gastric retentive devices may also be used as extended-release drug delivery systems21, 22. Several gastroretentive extended-release products are available on the market at present. Glumetza® (metformin hydrochloride) and Proquin® XR (ciprofloxacin hydrochloride) extended-release tables were designed based on the mechanism of expansion and bioadhesion. They could release the drug in the upper gastrointestinal tract and showed higher plasma concentrations than the immediate-release formulation23. In this study, we developed tacrolimus solid SME sustained-release GRDDS combining the advantages of solid SME and sustained-release gastric retention agents to improve the bioavailability and reduce the blood concentration variability of tacrolimus.
Materials and methods
Materials
Tacrolimus (FK506) was purchased from Zhejiang Laiyi Biotechnologies Co, Ltd. Monocaprylin glycerate (GMC) was a gift from Henan Zhengtong Chemical Co, Ltd (Zhengzhou, China). Polyoxyl 40 Hydrogenated Castor Oil (Cremophor RH40) was obtained from the BASF Corporation. Polyethylene oxide (PEO WSR N60K) was gifted from the DOW Chemical Company (Midland, MI, USA). Polyvinylpyrrolidone (PVP) K90 was gifted from the International Specialty Products (ISP) Corporation. Mannitol and chitosan (deacylation rate ≥90.0%, viscosity=150 mPa·s) were purchased from Shandong Jiejing Group Corporation (Rizhao, China) and Sinopharm Chemical Reagent Co, Ltd (Shanghai, China), respectively. The tacrolimus capsules: brand name Prograf; standard 1 mg/tablet; batch number 1D5261A. All other chemicals were of reagent or HPLC grade. Deionized water was obtained from a Millipore® Milli-Q System (Molsheim, France) in the laboratory.
Animals
New Zealand rabbits (body weight 2.5±0.5 kg) were obtained from the Medical Animal Test Center of the Shanghai Institute of Materia Medica (Shanghai, China). Six adult beagle dogs (three male and three female, average weight 8.6±1.5 kg) were provided by the School of the Agriculture of Shanghai Jiao Tong University experiment and teaching practice field. License No: SCXK (Shanghai) 27–0004. All experiments were performed according to the Shanghai Institute of Materia Medica guidelines for experimental animal care.
Preparation of the solid-state self-microemulsifying dosage form
The solid self-microemulsifying drug delivery system (S-SMEDDS) of tacrolimus was prepared as follows: Cremophor RH40 and GMC at a ratio of 4:6 were accurately weighed into a glass vial, melted in a water bath at 40 °C and mixed by a vortex to form a homogenous mixture. Tacrolimus was then dispersed into this mixture of oil and surfactant by vortex mixing and shaking at 37 °C until a transparent solution of SMEDDS was obtained. Microemulsion adsorbed granular material was obtained from a mixture of SMEDDS solution, PEO N60K, PVP K90, mannitol and chitosan by constant stirring with ethanol as a wetting agent. The dried granules were lubricated with magnesium stearate and compressed into tablets on a single station tablet press (TDP-I, Shanghai Huamao Industrial & Commercial Co, Shanghai, China).
The amounts of PEO, chitosan, mannitol and PVP added to the formulation to make one tablet containing 102 mg of SMEDDS are listed in Table 1.
Table 1. Composition of the investigated tablets.
| Ingredients | Composition (mg/tablet) |
|---|---|
| Tacrolimus | 2 |
| GMC | 60 |
| Cremophor RH40 | 40 |
| Polyethylene oxide | 300 |
| Chitosan | 300 |
| Mannitol | 250 |
| Polyvinylpyrrolidone | 50 |
Expanding study
The swelling of the tablets can be measured by their ability to absorb water and the degree to which they swell. The swelling property of the formulation was determined by various techniques. Swelling index, water absorption rate and exposed size parameter are essential parameters to predict the gastroretentive performance of the expandable systems24. In this study, the exposed size parameter was chosen to measure the tablets' swelling. The water uptake study of the tablets was performed according to the Chinese Pharmacopoeia (CP) XC basket method. The medium used was 500 mL of an HCl buffer at pH 1.2 rotated at 50 rotations per minute (r/min). The medium was maintained at 37±0.5 °C throughout the study. At a selected time interval, the tablets were withdrawn to measure their diameter and thickness.
Bioadhesive strength study
The adhesive strength of the tablets to rabbit stomach mucosa was evaluated using a self-made system with pallet scales. As shown in Figure 1, a hanging platform consisting of a cord and a glass slide attached with stomach mucosa was fixed to the pallet scales by a thick iron wire with a hook. A flat surfaced steel block was used as a lower static platform. The mucosa was mounted onto the lower platform using a medical rubber adhesive. The tablets were attached to the bottom of the hanging platform. The hanging platform with the tablets was brought down and placed over the surface of the mucosa with an applied force of 100 G for 15 min, and the force required to detach the tablets from the mucosal surface was determined by the weight of the added water. The test was performed at room temperature (23–25 °C), and the mean of the three measurements was used as the mucoadhesive strength of the tablets.
Figure 1.

Schematic diagram of the bioadhesion testing device.
Self-microemulsifying study
To assess the self-microemulsifying properties, the tablet was introduced into 250 mL of 37 ºC HCl buffer at pH 1.2 under a gentle agitation of 50 r/min in a rotating basket dissolution apparatus. When the tablet was completely eroded, a sample was withdrawn and investigated using the transmission electron microscope (JEM-1230, JEOL, Japan). The sample was stained with phosphotungstic acid for visualization and placed on copper grids for viewing at 25±2 ºC. The droplet size distribution of the resultant emulsion was also determined by photon correlation spectroscopy (PCS) using a PSS Nicomp 380 ZLS (PSS Nicomp, Santa Barbara, CA, USA). Particle size distribution was expressed in a nicomp distribution. The sample was filtered through a 0.45-μm micropore filter.
Drug-release study
Drug redispersibility and the release profiles of tacrolimus from the self-microemulsifying tablets and reference commercial immediate capsules were determined using the CP XC rotating basket method (RCZ-8B dissolution tester, RZQ-8A automatic sampler, RDB-8A peristaltic pump, Tian Da Tian Fa, Tianjin, China) at 37 °C. The rotating speed was 50 r/min, and the dissolution medium was 900 mL of HCl buffer at pH 1.2 containing 0.005% (w/v) of hydroxypropylmethyl cellulose (HPC-M). Dissolution studies were conducted over 12 h to evaluate the sustained-release properties of the preparations.
Samples (8 mL) were withdrawn at predetermined time intervals and were assayed for tacrolimus by HPLC at 210 nm. The HPLC system was composed of an autosampler (G1313A ALS), a pump (G1311 Quatpump), a column oven (G1316A Column), a UV detector (G1314A VWD) and data processing software (HP Chemstation Rev.A.10.01). Briefly, tacrolimus was analyzed using Agilent Zorbax XDB C8 (150×4.6 mm, 5 μm) a reverse phase chromatography column. The mobile phase consisted of acetonitrile-0.25% phosphoric acid (65:35, v/v) and was pumped at a flow rate of 1.0 mL/min.
In vitro release data of the sustained-release tablets were analyzed using the zero-order release model (Q=kt), Higuchi model (Q=kts1/2) and the first-order release model [ln(100-Q)=ln100–kt], where Q is the percentage of drug released at t time and k is the release rate constant.
In vivo pharmacokinetics study in beagle dogs
Six healthy beagle dogs that had fasted but had free access to water for 12 h prior to the experiment were used in this study. They were allocated at random to two treatment groups and were orally administered gastroretentive tablets and two reference commercial capsules of tacrolimus once a day in a crossover design with a 1-week washout period between dosing. The dose of tacrolimus administered was 2 mg.
At a predetermined time interval, a blood sample (3.0 mL) was withdrawn at 0, 1, 2, 4, 6, 8, 10, 12, 24, and 36 h for the gastroretentive tablet group and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, and 24 h for the reference capsule group. Blood samples collected from the thigh vein were syringed into centrifuge tubes containing heparin and then kept frozen at -20 °C until analysis. The tacrolimus concentrations in the whole blood samples were determined by high performance liquid chromatography mass spectrometry (HPLC-MS) with a low quantitation limit of 0.2 ng/mL.
A selective, rapid and sensitive HPLC-MS method was developed for the quantification of tacrolimus in dog whole blood. A two-step liquid-liquid extraction was included in the sample pretreatment. Then, 1 mL of a 70% acetonitrile solution was added to 1 mL of the whole blood. After vortex mixing for 30 s, 4 mL of dichloromethane solution containing 1% isoamyl alcohol was added and vortexed for 15 min. After centrifugation at 4000 r/min for 10 min, the lower organic phase was collected, and the residue was extracted with 4 mL of dichloromethane solution containing 1% isoamyl alcohol. The lower organic phase was combined and dried under nitrogen gas at 70 °C. The residue was reconstituted with 200 μL of the mobile phase. Separation was carried out on an Alltech-AlltimaTM-C18 column (150 mm×2.1 mm, 3 μm) with methanol as the mobile phase at a flow rate of 0.2 mL/min.
Detection was performed by a Thermo Finnigan LTQ HPLC-MS (Thermo Finnigan, USA). The mass spectrometer was operated with an electrospray ionization (ESI) interface in positive ionization mode and with multiple-reaction monitoring mode. The selected reaction monitoring (SRM) of tacrolimus was m/z 826.48. The concentration of tacrolimus was determined by a standard linear calibration curve in the concentration range of 0.2–20 ng/mL.
The maximum whole blood concentration (Cmax), time to Cmax (tmax), and the area under the whole blood concentration versus time curve (AUC) were calculated from observed data points with the Drug and Statistics (DAS 2.1.1) pharmacokinetic software. Statistical analysis (Cmax, tmax, AUC) was also conducted with the DAS 2.1.1 software. The relative bioavailability (F) of the gastroretentive tablet to the commercial capsule (reference) was calculated using the following equation:
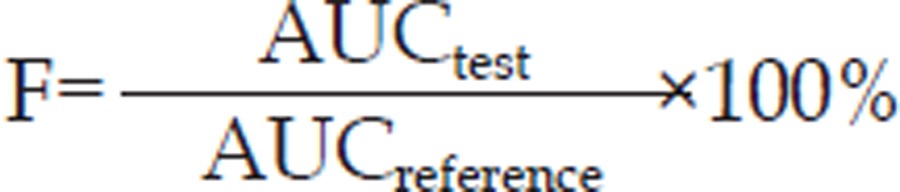 |
Statistical analysis
Results are expressed as the mean±standard deviation. Statistical comparisons were performed using a bilateral t-test (Cmax, AUC) and a non-parametric test (tmax) with the DAS 2.1.1 software. A P value <0.05 was considered statistically significant.
Results
Development of the tablet
Self-microemulsifying systems form fine oil-water emulsions with only gentle agitation upon their introduction into aqueous media. Therefore, the selection of the oil, surfactant, and mixing ratio play an important role in the formulation of the microemulsion. In the present study, Cremophor RH40 and GMC were tested for phase behavior. The influence of the oil and surfactant ratio on the particle size distribution was evaluated. As seen in Figure 2, Cremophor RH40 gave a wide microemulsion region. Increasing the oil ratio resulted in a larger particle size. Cremophor RH40 and monocaprylin glycerate at a ratio of 4:6 have the optimal particle size distribution, so it was selected for the formulation study.
Figure 2.

Phase diagram and particle size distribution of SMEDDS containing Cremophor RH40 and monocaprylin glycerate.
On the basis of previous studies on the swelling properties of matrix tablets, we concluded that the polyethylene oxide (PEO) polymer is an excellent vehicle for swelling tablets25, 26, 27. Earlier studies have demonstrated that PEO also has good adhesion. Therefore, PEO was chosen as the skeleton material of the tablet, which provided swelling and adhesion capability for the tablet. PVP is a nonirritant material that is extensively used as a tablet binder27. It is well known that the expanded form must maintain its integrity and have adequate strength to withstand the force in the stomach28. Moreover, the high molecular weight grades of PVP offer higher binding capacity. Therefore, PVP K90 was introduced as a tablet binder and was simultaneously used to increase the ability of the resistance to the gastric motility of the swelling tablet.
The goals of gastric retention and controlling release are not always compatible. PEO is a matrix material that possesses the characteristics of both swelling and controlling release; however, PEO takes a very long time to completely erode when it is used at the amount needed for sufficient swelling to achieve gastric retention. Some other matrix materials can also swell but offer the benefit of faster and more even erosion in the gastric environment, which means that the dosage form made with these materials can pass through the gastrointestinal tract more predictably after a few hours of drug release. One such material is chitosan, which swells, but not to the same degree as PEO. Chitosan can generate synergies with PEO in the expansion of matrix tablets, and the releases that are controlled by the erosion of the polymeric matrix are also easier. Moreover, chitosan also has very strong adhesion, which has a prominent role in enhancing the adhesion force of the tablet. Hence, we developed a PEO and chitosan combination gastric retention tablet, which not only maintained good swelling of the tablets but also insured that the tablets completely eroded at the scheduled time, taking double advantage of the chitosan. In this study, mannitol was used to further regulate the drug-release rate.
Expanding study
It is reported that the dosage form size has a great influence on its residual effects in the stomach. Due to the retropulsion reflex, gastroretentivity may simply be achieved by large dimensions that are physically unable to pass through the pyloric sphincter29. The pyloric sphincter has a diameter of 12.8±7 mm in humans29. To achieve a size adequate for preventing passage through the pylorus yet small enough to be swallowed requires very significant expansion in at least two dimensions28. The tablet we developed expanded to 13.5 mm in diameter and 15 mm in thickness after 20 min, and then to 16 mm in diameter and 20 mm in thickness after 3 h of contact with an HCl buffer at pH 1.2. The two dimensions of the size of the tablet at different times are shown in Figure 3. The results revealed that the tablet swelled rapidly when immersed in the HCl buffer at pH 1.2, and the tablet maintained this large size for at least 8 h. These results indicated that the tablet had a very good capacity to improve the effectiveness of gastric retention.
Figure 3.

Gel layer diameter and thickness of the self-microemulsifying gastroretentive sustained-release tablets during contact with the HCl buffer at pH 1.2. n=6. Mean±SD.
Bioadhesive strength study
Gastroretention can be achieved by the swelling property of the tablet, and bioadhesion may be an important property for further strengthening of the gastroretentive feature of tablets. PEO polymers are reported to have potential bioadhesive properties. To reinforce the mucoadhesion feature, chitosan, which is widely used as a bioadhesive polymer, was added to the formulation. Its mucoadhesive properties are mediated by ionic interactions of the positively charged amino groups and negatively charged substructures of the gastrointestinal mucus, mainly sialic acid30. The dissolution time and bioadhesion force with and without chitosan are shown in Table 2. As depicted in Table 2, the result of the bioadhesion study indicated that chitosan had a more significant effect than PEO on bioadhesion. This may be due to different adhesion mechanisms in PEO and chitosan, which resulted in different contributions to the adhesion of the tablet. Moreover, tablets with chitosan took less time to completely erode. The bioadhesive strength of PEO N60K tablet with chitosan was 0.98±0.06 N/cm2, which ensured that the tablet could adhere to the gastric mucosa.
Table 2. Bioadhesion and time for 95% drug release of PEO tablets. n=6. Mean±SD.
| Code# | Composition | Bioadhesion (N/cm2) | Time for 95% drug release (h) |
|---|---|---|---|
| 1 | PEO 600 mg | 0.81±0.09 | 21.8±1.2 |
| 2 | PEO 300 mg+Chitosan 300 mg | 0.98±0.06 | 12.1±0.8 |
PEO, polyethylene oxide. Formation #1 and #2 also include 2 mg tacrolimus, 60 mg monocaprylin glycerate, 40 mg Cremophor RH40, 250 mg mannitol, 50 mg polyvinylpyrrolidone.
Self-microemulsifying study
The incorporation of the self-microemulsifying mixture into a solid dosage form is desirable, but challenging, because self-microemulsifying properties are harder to achieve with solid materials7. Therefore, it is necessary to determine whether or not the tablet that is composed of a self-microemulsifying mixture and solid materials can self-microemulsify when it contacts water or SGF. The droplet size nicomp distribution of the liquid SMEDDS and solid SMEDDS are shown in Figure 4. The average dispersing droplet size of the liquid SMEDDS and solid SMEDDS were 28.9 nm and 26.2 nm, respectively. These results indicated that the solid self-microemulsifying formulation (SMEF) preserved the self-microemulsification performance of the liquid SMEF. TEM (Figure 5) showed that the reconstituted microemulsions were released from the solid SMEF when exposed to SGF. The size range was narrow, and the droplet size was very small (approximately 20–30 nm). These results were consistent with the results of the PCS analysis. TEM showed the quality of the microemulsion that was formed. Emulsion droplet size is a decisive factor in self-microemulsifying formulation performance because it determines the rate and the extent of drug release. Smaller droplet size improves the drug release and provides a larger interfacial area across which the drug can diffuse into the gastrointestinal fluids, and thus, it increases drug absorption31, 32. TEM and nicomp distribution results confirmed that the tablet could self-microemulsify in the gastrointestinal tract, and the droplet size of the emulsion drops was small, which could achieve the purpose of increasing drug solubility and improving drug absorption.
Figure 4.

Microemulsion droplet size distribution: solid SMEDDS (blue line); liquid SMEDDS (red line).
Figure 5.

Transmission electron micrograph of the reconstituted microemulsion released from the gastroretentive sustained-release tablet.
Drug-release study
For drug candidates with lower aqueous solubility, the drug-release rate is dependent mainly on the erosion of the polymeric matrix33, 34. As demonstrated in Figure 6, about 100% of the tacrolimus is released within 2 h from the commercial capsules. In contrast, for the tacrolimus self-microemulsifying gastroretentive sustained-release tablets, the concentration of drug released was very low within two hours, and almost no drug was released in the first hour. This was because the matrix swelling was much faster compared with the erosion process during the initial contact with the dissolution medium; therefore, matrix erosion did not occur during this time.
Figure 6.

Dissolution profiles of tacrolimus from the sustained-release self-microemulsifying gastroretentive tablets and the commercial immediate-release capsules. (2 mg tacrolimus; n=6, arithmetic mean±SD).
The fitting equations of the drug-release curve are shown in Table 3. According to the correlation coefficient of the fitting curve, the release curve was fit to the zero-order release model, which was helpful in reducing fluctuations in blood concentrations. The release mechanism of tacrolimus from the PEO, chitosan and mannitol coupling gastric retention matrix tablets was mainly based on erosion.
Table 3. The release pattern of tacrolimus self-microemulsifying sustained release tablets.
| Release pattern | Fitting equation | R2 | Adj R2 |
|---|---|---|---|
| Zero order | Q=8.70426t–9.30741 | 0.9938 | 0.9925 |
| First order | Ln (100–Q)=−0.23325t+5.13975 | 0.8768 | 0.8522 |
| Higuchi | Q=38.55712t1/2–45.72334 | 0.9586 | 0.9503 |
In vivo pharmacokinetics study in beagle dogs
Mean whole blood levels of tacrolimus at each time point are summarized in Figure 7. From Table 4, it can be seen that the AUC(0–∞) was approximately five times greater when tacrolimus was administered as gastroretentive tablets as compared to the commercial capsules. The mean value of Cmax for the gastroretentive tablets (8.86 ng/mL) was 2.3 times greater than the Cmax obtained with the same dose of tacrolimus administered as the commercial capsules (3.78 ng/mL). Gastroretentive tablets resulted in an average tmax of 6.67 h, which was obviously longer than the tmax of the commercial capsules (1 h). The gastroretentive tablets resulted in a significant absorption of tacrolimus compared to the commercial capsules (P<0.05).
Figure 7.

Whole blood concentration versus time curve (arithmetic mean±SD, n=6) of tacrolimus (2 mg per dog) in beagle dogs after oral administration of the self-microemulsifying gastroretentive sustained-release tablets (Δ) or commercial capsules (□).
Table 4. Pharmacokinetic parameters of tacrolimus from self-microemulsifying gastricretentive sustained release tablets and commerical capsules.
| No | Order of administration | AUC(0–t) (ng·h·mL−1) |
AUC(0–∞) (ng·h·mL−1) |
tmax(h) |
Cmax (ng·mL−1) |
F% | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| SEGTa | CCb | SEGT | CC | SEGT | CC | SEGT | CC | |||
| 1 | SMEGSRT/CC | 88.20 | 28.54 | 126.22 | 40.56 | 4 | 1 | 6.61 | 4.82 | 309.0 |
| 2 | SMEGSRT/CC | 206.91 | 18.67 | 252.52 | 25.68 | 8 | 1 | 11.54 | 2.50 | 1107.8 |
| 3 | SMEGSRT/CC | 74.48 | 43.99 | 85.39 | 68.74 | 6 | 1 | 6.02 | 5.01 | 169.3 |
| 4 | CC/SMEGSRT | 209.91 | 32.11 | 320.47 | 48.21 | 10 | 1 | 11.23 | 3.98 | 653.7 |
| 5 | CC/SMEGSRT | 82.73 | 26.21 | 103.03 | 35.65 | 4 | 1 | 7.80 | 3.47 | 315.6 |
| 6 | CC/SMEGSRT | 194.35 | 25.41 | 265.86 | 33.14 | 8 | 1 | 9.96 | 2.88 | 764.9 |
| Mean | 142.76 | 29.16 | 192.25 | 42.00 | 6.67 | 1 | 8.86 | 3.78 | 553.4 | |
| SD | 67.12 | 8.51 | 99.23 | 15.10 | 2.42 | 0 | 2.38 | 1.02 | 353.8 | |
aSMEGSRT self-microemulsifying gastricretentive sustained release tablets.
bCC commercial capsules.
Discussion
There is a clear pharmaceutical need for advanced delivery systems that continuously supply drugs with narrow absorption window to their absorption site for an extended time period. This approach would provide effective and sustained drug concentrations in the blood for prolonged periods of time. It would also eliminate the need for frequent drug administration and the use of inconvenient modes of administration. This is particularly true for tacrolimus to prolong the release of drug in the duodenum by a gastroretentive dosage form (GRDF).
The current investigation presents a novel and promising sustained-release GRDF based on the combination of large dimensions, bioadhesive strength, sustained release of the loaded drug and self-microemulsification. In this study, the sustained-release GRDF of tacrolimus was prepared using a combination of PEO and chitosan as a solid carrier. The SME gastroretentive sustained-release tablets preserved the self-microemulsification performance of the liquid SMEDDS. The optimized formulation followed zero-order release kinetics. Swelling studies indicated significant water uptake and swelling, which may be significant in gastroretention. The bioadhesive properties were also studied, and the developed formulation showed significant bioadhesion. Thus, by combining these approaches of gastroretention together, the in vivo gastroretention could be predicted more reliably. Based on these promising in vitro results, in vivo studies in beagle dogs were carried out to determine various pharmacokinetic parameters. The relative oral bioavailability of the model drug, tacrolimus, was 5.5-fold compared to the commercial capsules. Gastroretentive tablets resulted in an average tmax of 6.67 h, which was obviously longer than the tmax obtained from the commercial capsules (1 h).
The higher bioavailability of hydrophobic drugs incorporated in a SMEDDS has been reported elsewhere35, 36, 37. The contribution of the GRDF approach extended the length of the absorption phase in comparison with the non-gastroretentive dosage form. This allows the desired therapeutic concentration to be achieved for prolonged periods of time. Thus, a combination of SMEDDS and GRDF can usually further improve the bioavailability of drugs. However, in terms of some rapid metabolism of drugs in the gastrointestinal tract, GRDF, which inputs the drug into the intestinal epithelial cells with the sustained mode, may enhance the efficacy of first pass metabolism in the intestinal wall. This phenomenon may reduce the bioavailability of drugs in some cases. Therefore, whether the sustained-release gastroretentive tablets can enhance the bioavailability of self-microemulsifying agents needs further study according to the specific absorption and metabolism characteristics of drugs.
Several factors have been suggested as possible determinants of the low oral bioavailability of tacrolimus. These include a low solubility in the intestine, extensive metabolism by CYP3A4 in the gut, and P-gp-mediated drug efflux. Regional differences in the functional expression of P-gp were investigated in the human intestine, indicating that there is higher activity of P-gp in the ileum and the colon than in the jejunum16. Therefore, the sustained-release gastroretentive tablets we prepared in this study can avoid extensive CYP3A4 metabolism and P-gp-mediated efflux by releasing tacrolimus in the upper part of the small intestine, promote the drug absorption, and further enhance the bioavailability of SME. Both SME, which increased drug solubility, and gastroretention technology, which prolonged the time that the drug remains in the upper part of the gastrointestinal tract, are responsible for the enhanced bioavailability of tacrolimus.
In conclusion, the current study demonstrates that a self-microemulsifying, swellable and bioadhesive gastroretentive delivery system, which has great potential to reduce the blood concentration fluctuations and increase the overall bioavailability of the model drug tacrolimus, has been successfully developed. The presence of highly swellable and strongly bioadhesive polymers within the developed systems provided rapid swelling and significant bioadhesive force. Soluble mannitol made the matrix more liable to erosion, which resulted in drug release in a predetermined manner by varying the mannitol concentration. The drug release was characterized by a substantial zero-order profile, which was helpful in reducing blood concentration fluctuations. Complete dissolution or erosion of the formulation matrix in a timely manner is important for gastroretentive dosage forms because very slow erosion or dissolution may lead to expulsion and other gastrointestinal safety issues. The tablet we developed could completely erode within approximately 12 h, so it would avoid the safety problems that were previously mentioned. We can come to the conclusion that PEO in combination with chitosan is a promising carrier for gastroretentive drug delivery systems and solid self-microemulsifying drug delivery systems. SME gastroretentive sustained-release tablets are potential vehicles for drugs with poor solubility and a narrow absorption window. Moreover, it possesses broad prospects for industrial application.
Author contribution
This research was designed by Yong GAN and Xin-xin ZHANG. The experiments were performed by Yan-ping WANG. The manuscript was written by Yan-ping WANG and Xin-xin ZHANG.
Acknowledgments
We are grateful to DOW Chemical for the generous gift of PEO WSR N60K and to the ISP Corporation for kindly providing PVP K90.
References
- Yamashita K, Nakate T, Okimoto K, Ohike A, Tokunaga Y, Ibuki R, et al. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int J Pharm. 2003;267:79–91. doi: 10.1016/j.ijpharm.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, et al. FK 506, a novel immunosuppressant isolated from a Streptomyces I. Fermentation, isolation and physico-chemical and biological characteristics. J Antibiot. 1987;42:1249–55. doi: 10.7164/antibiotics.40.1249. [DOI] [PubMed] [Google Scholar]
- Plosker GL, Foster RH. Tacrolimus-a further update of its pharmacology and therapeutic use in the management of organ transplantation. Drugs. 2000;59:323–89. doi: 10.2165/00003495-200059020-00021. [DOI] [PubMed] [Google Scholar]
- Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS PharmSciTech. 2008;9:13–21. doi: 10.1208/s12249-007-9014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdalla A, Klein S, Mader K. A new self-emulsifying drug delivery system (SEDDS) for poorly soluble drugs: characterization, dissolution, in vitro digestion and incorporation into solid pellets. Eur J Pharm Sci. 2008;35:457–64. doi: 10.1016/j.ejps.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Abdalla A, Mader K. Preparation and characterization of a self-emulsifying pellet formulation. Eur J Pharm Biopharm. 2007;66:220–6. doi: 10.1016/j.ejpb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Nazzal S, Khan MA. Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm. 2006;315:110–21. doi: 10.1016/j.ijpharm.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Patil P, Joshi P, Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004;5:e42. doi: 10.1208/pt050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S.inventors; AluhaRx Inc., assigneeSolid self-emulsifying dosage form for improved delivery of poorly soluble hydrophobic compounds and the process for preparation there ofUS Patent 10252158. 2002 Sep 23
- Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discovery Today. 2008;13:606–12. doi: 10.1016/j.drudis.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Serratoni M, Newton M, Booth S, Clarke A. Controlled drug release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur J Pharm Biopharm. 2007;65:94–8. doi: 10.1016/j.ejpb.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Balakrishnan P, Lee BJ, Oh DH, Kim JO, Hong MJ, Jee JP, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS) Eur J Pharm Biopharm. 2009;72:539–45. doi: 10.1016/j.ejpb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Nazzal S, Nutan M, Palamakula A, Shah R, Zaghloul AA, Khan MA. Optimization of a self-nanoemulsified tablet dosage form of ubiquinone using response surface methodology: effect of formulation ingredients. Int J Pharm. 2002;240:103–14. doi: 10.1016/s0378-5173(02)00130-8. [DOI] [PubMed] [Google Scholar]
- Tuleu C, Newton M, Rose J, Euler D, Saklatvala R, Clarke A, et al. Comparative bioavailability study in dogs of a self-emulsifying formulation of progesterone presented in a pellet and liquid form compared with an aqueous suspension of progesterone. J Pharm Sci. 2004;93:1495–502. doi: 10.1002/jps.20068. [DOI] [PubMed] [Google Scholar]
- Tamura S, Tokunaga Y, Ibuki R, Amidon GL, Sezaki H, Yamashita S. The site-specific transport and metabolism of tacrolimus in rat small intestine. J Pharmacol Exp Ther. 2003;306:310–6. doi: 10.1124/jpet.103.050716. [DOI] [PubMed] [Google Scholar]
- Venkataramanan R, Swaminathan A, Prasad T, Jain A, Zuckerman S, Warty V, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29:404–30. doi: 10.2165/00003088-199529060-00003. [DOI] [PubMed] [Google Scholar]
- Holm P, Norling T, Lademann AM.inventors; LIFECYCLE PHARMA A/S, assigneeOnce daily oral dosage form comprising tacrolimusPatent WO2008/145143. 2008 May 30
- Kagayama A, Tanimoto S, Fujisaki J, Kaibara A, Ohara K, Iwasaki K, et al. Oral absorption of FK506 in rats. Pharm Res. 1993;10:1446–50. doi: 10.1023/a:1018967107612. [DOI] [PubMed] [Google Scholar]
- Shigeki T, Atsuo O, Rinta I, Gordon L, Shinji Y. Tacrolimus is a class II low-solubility high-permeability drug: the effect of P-glycoprotein efflux on regional permeability of tacrolimus in rats. J Pharm Sci. 2002;91:719–29. doi: 10.1002/jps.10041. [DOI] [PubMed] [Google Scholar]
- Streubel A, Siepmann J, Bodmeier R. Gastroretentive drug delivery systems. Expert Opin Drug Deliv. 2006;3:217–33. doi: 10.1517/17425247.3.2.217. [DOI] [PubMed] [Google Scholar]
- Strubing S, Abboud T, Contri RV, Metz H, Mader K. New insights on poly(vinyl acetate)-based coated floating tablets: Characterisation of hydration and CO2 generation by benchtop MRI and its relation to drug release and floating strength. Eur J Pharm Biopharm. 2008;69:708–17. doi: 10.1016/j.ejpb.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Ramji AKA, Chandra SRG, Prabhakar RV. Formulation and evaluation of swellable and floating gastroretentive ciprofloxacin hydrochloride tablets. AAPS PharmSciTech. 2009;10:220–6. doi: 10.1208/s12249-009-9200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh DC, Amin AF. In vitro and in vivo techniques to assess the performance of gastro-retentive drug delivery systems: a review. Expert Opin Drug Deliv. 2008;5:951–65. doi: 10.1517/17425247.5.9.951. [DOI] [PubMed] [Google Scholar]
- Gusler G, Berner B, Chau M, Berner B.inventors; Depomed, Inc, assigneeOptimal polymer mixtures for gastricPatent AU2002337974. 2002 Oct 22
- Wu N, Wang LS, Tan DCW, Moochhala SM, Yang YY. Mathematical modeling and in vitro study of controlled drug release via a highly swellable and dissoluble polymer matrix: polyethylene oxide with high molecular weights. J Control Release. 2005;102:569–81. doi: 10.1016/j.jconrel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Mahalingam R, Jasti B, Birudaraj R, Stefanidis D, Killion R, Alfredson T, et al. Evaluation of polyethylene oxide compacts as gastroretentive delivery systems. AAPS PharmSciTech. 2009;10:98–103. doi: 10.1208/s12249-008-9182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman KC. A critical review of gastric retentive controlled drug delivery. Pharm Dev Technol. 2007;12:1–10. doi: 10.1080/10837450601168680. [DOI] [PubMed] [Google Scholar]
- Klausner EA, Lavy E, Friedman M, Hoffman A. Expandable gastroretentive dosage forms. J Control Release. 2003;90:143–62. doi: 10.1016/s0168-3659(03)00203-7. [DOI] [PubMed] [Google Scholar]
- Werle M, Bernkop SA. Thiolated chitosans: useful excipients for oral drug delivery. J Pharm Pharmacol. 2008;60:273–81. doi: 10.1211/jpp.60.3.3001. [DOI] [PubMed] [Google Scholar]
- Chambin O, Jannin V, Champion D, Chevalier C, Rochat-Gonthier MH, Pourcelot Y. Influence of cryogenic grinding on properties of a self-emulsifying formulation. Int J Pharm. 2004;278:79–89. doi: 10.1016/j.ijpharm.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72. doi: 10.1023/a:1016268311867. [DOI] [PubMed] [Google Scholar]
- Li HT, Hardy RJ, Gu XC. Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS PharmSciTech. 2008;9:437–43. doi: 10.1208/s12249-008-9060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CJ. Effects of drug solubility, drug loading, and polymer molecular weight on drug release from Polyox® tablets. Drug Dev Ind Pharm. 1998;24:645–51. doi: 10.3109/03639049809082366. [DOI] [PubMed] [Google Scholar]
- Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polyglycolized glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106:15–23. [Google Scholar]
- Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AA, et al. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J Pharm Sci. 1998;87:164–9. doi: 10.1021/js970300n. [DOI] [PubMed] [Google Scholar]
- Fischl MA, Richman DD, Flexner C, Para MF, Jaubrich R, Karim A, et al. Phase I/II study of the toxicity, pharmacokinetics, and activity of the HIV protease inhibitor SC-52151. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15:28–34. doi: 10.1097/00042560-199705010-00005. [DOI] [PubMed] [Google Scholar]