

Abstract
The twenty first amino acid, selenocysteine (Sec), is the only amino acid that is synthesized on its cognate transfer RNA (tRNASec) in all domains of life. The multistep pathway involves O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase (SepSecS), an enzyme that catalyzes the terminal chemical reaction during which the phosphoseryl–tRNASec intermediate is converted into selenocysteinyl-tRNASec. The SepSecS architecture and the mode of tRNASec recognition have been recently determined at atomic resolution. The crystal structure provided valuable insights that gave rise to mechanistic proposals that could not be validated because of the lack of appropriate molecular probes. To further improve our understanding of the mechanism of the biosynthesis of selenocysteine in general and the mechanism of SepSecS in particular, stable tRNASec substrates carrying aminoacyl moieties that mimic particular reaction intermediates are needed. Here, we report on the accurate synthesis of methylated, phosphorylated, and phosphonated serinyl-derived tRNASec mimics that contain a hydrolysis-resistant ribose 3′-amide linkage instead of the natural ester bond. The procedures introduced allow for efficient site-specific methylation and/or phosphorylation directly on the solid support utilized in the automated RNA synthesis. For the preparation of (S)-2-amino-4-phosphonobutyric acid–oligoribonucleotide conjugates, a separate solid support was generated. Furthermore, we developed a three-strand enzymatic ligation protocol to obtain the corresponding full-length tRNASec derivatives. Finally, we developed an electrophoretic mobility shift assay (EMSA) for rapid, qualitative characterization of the SepSecS-tRNA interactions. The novel tRNASec mimics are promising candidates for further elucidation of the biosynthesis of selenocysteine by X-ray crystallography and other biochemical approaches, and could be attractive for similar studies on other tRNA-dependent enzymes.
Keywords: amino acids, bioconjugation, RNA, RNA structures, solid-phase synthesis
Introduction
In humans, twenty five selenoproteins[1,2] have been shown to contain selenium in the form of a selenol group at the amino acid side chain of selenocysteine (Sec).[3] In all Sec-decoding organisms, the biosynthesis of Sec begins with the acylation of tRNASec by seryl-tRNA synthetase (SerRS) to form Ser-tRNASec.[4] The proposed mechanism in archaea and eukaryotes employs a phosphorylation step to generate the O-phosphoseryl-tRNASec (Sep-tRNASec) intermediate,[5,6] the substrate for the O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase (SepSecS), which catalyzes the tRNA-dependent Sep to Sec conversion (Figure 1).[7–9] The selenium donor for this transformation is selenophosphate, which is hydrolyzed subsequent to the nucleophilic attack.
Figure 1.

Key chemical transformations (1–3) during the biosynthesis of selenocysteine in humans mediated by the O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase (SepSecS; PLP=pyridoxal phosphate).
The aim of the present work was to develop an efficient synthesis of the serinyl-tRNASec derivatives that mimic intermediates of the biosynthesis of selenocysteine (Figure 2). The selected derivatives represent stable analogues and potential inhibitors at defined steps during the biosynthetic pathway, and hence, will act as promising compounds for high-resolution structural and mechanistic studies on the selenocysteine-handling enzymes (e.g., SerRS and SepSecS).
Figure 2.

Selection of target conjugates for phosphorylated, phosphonated, and methylated tRNASec mimics that carry a hydrolysis-resistant adenosine-3′-amide linkage. Anticodon loop replacement with the U1A protein recognition sequence is highlighted by dashed boxes. Potential sites for enzymatic ligation with T4 RNA ligase are indicated by blue bars.
From a synthetic point of view, our targets represent amino acid–RNA conjugates that are additionally methylated, phosphorylated, or phosphonated. The structural complexity of the amino acid–RNA and peptide–RNA conjugates, in particular the 3′-peptidyl-tRNA mimics, present a series of challenges for de novo synthesis.[10–21] The importance of tRNA conjugates in probing fundamental biological enzymatic mechanisms argues that the development of efficient synthetic protocols for such ligands is warranted.
We have recently introduced specifically functionalized polystyrene solid supports and demonstrated that standard Fmoc chemistry (Fmoc=9-fluorenylmethoxycarbonyl) for a single amino acid unit and short peptides, combined with phosphoramidite chemistry for RNA assemblies works very well.[10,12] Importantly, to fulfill the specific requirement of increased resistance towards hydrolytic cleavage, these conjugates were equipped with a ribose containing a 3′-amide linkage at the interface between the terminal adenosine-76 and the amino acid instead of a natural ester unit.
Results and Discussion
To achieve the synthesis of the phosphorylated, phosphonated, and/or methylated aminoacyl-RNA conjugates, we devised a flexible strategy that would allow for a combination of these modification patterns. Therefore, we considered two types of storable solid supports, one functionalized with either 3′-serinyl- or 3′-threonyl-adenosine that contains TBDMS-protected (TBDMS= tert-butyl dimethylsilyl) hydroxyl side chains (4 and 5) (Scheme 1), and the other one functionalized with adenosine that carries a masked phosphonate of the aminobutyric acid (Abu) moiety (10) (Scheme 2). These novel solid supports were readily accessible from the key precursor 3′-azido-3′-deoxyadenosine (1), for which we have previously introduced a straightforward synthesis.[10] Coupling of N-Fmoc-and O-TBDMS-protected L-serine or L-threonine under Staudinger–Vilarrasa conditions[16,17] furnished the amino acid linked building blocks 2 and 3 (Scheme 1). These derivatives were further transformed into pentafluorophenyl active esters by using adipic acid bis(pentafluorophenyl)ester to generate a tether that allowed coupling to amino-modified polystyrene yielding the desired 3′-aminoacylamino-3′-deoxyadenosine-functionalized solid supports 4 and 5.
Scheme 1.
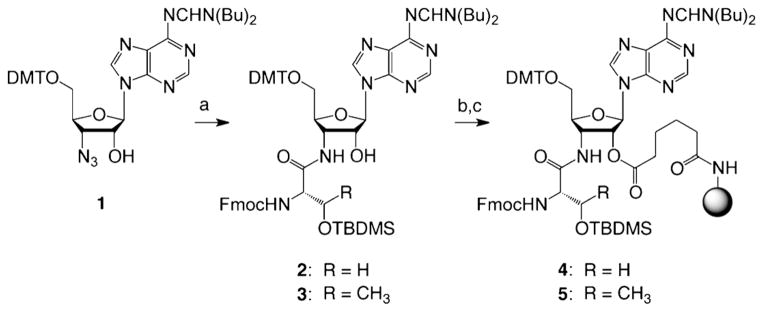
Synthesis of the 3′-serinyl- and 3′-threonylamino-3′-deoxyade-nosine-functionalized solid supports for RNA solid-phase synthesis. Reaction conditions: a) Fmoc-Ser(OTBDMS)-OBt (1.3 equiv, Bt =benzotriazol-1-yl) (alternatively, Thr), P(CH3)3 (2.2 equiv), THF, 0 °C to RT, 16 h, 89%. b) PfpOOC(CH2)4COOPfp (3.5 equiv, Pfp = pentafluorophenyl), 4-dimethylaminopyridine (DMAP, 1.1 equiv), DMF/pyridine (1:1), RT, 1 h, 71%. c) amino-functionalized support (GE Healthcare, Custom Primer Support 200 Amino, 3 equiv (w/w)), pyridine (2 equiv), DMF, RT, 22 h, average loading: 50 μmolg−1. DMTO=4,4′-dimethoxytritylchlorid.
Scheme 2.
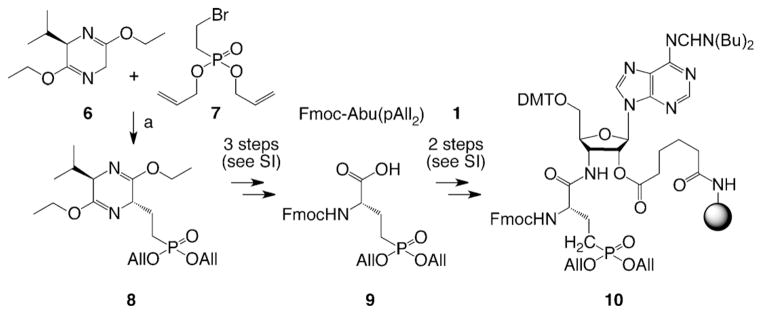
Synthesis of protected (S)-2-amino-4-(bis(allyloxy) phosphoryl) butanoic acid (All =allyl) and the 3′-amino-3′-deoxyadenosine-functionalized solid support. Reaction conditions: a) BuLi (2.5M in hexane, 1.0 equiv), compound 7 (0.9 equiv), diallyl(eth-2-enyl)phosphonate (elimination product of 7, 0.1 equiv), THF, −78°C to RT, 40 min, 85%. For details of chemical transformations of compound 8 to the solid support 10 and the synthesis of compounds 6 and 7 see the Supporting Information (SI).
The asymmetric synthesis of N-Fmoc- and O-allyl-protected (S)-2-amino-4-phosphonobutyric acid (9) that was required for the solid support 10, was optimized from an early report in the literature[22] and based on Schöllkopfs bislactim ether 6 and the bromoethylphosphonate ester 7 (Scheme 2). Importantly, although the hydrolysis of the bislactim ether function of 8 to the corresponding amino acid ester can be performed under very mild acidic conditions, enzymatic hydrolysis of the ester by using chymotrypsin had to be used to generate the free carboxylic acid moiety (see the Supporting Information). Of note, chymotrypsin is selective for L-configurated amino acid esters. The corresponding hydrolyzed amino acids are conveniently separated from unreacted D-configurated counterparts and isolated in high enantiomeric excess (ee) (see also Ref. [23]). After protection of the amino group, compound 9 was linked to the amino-modified polystyrene to yield the solid support 10, analogous to its serinyl and threonyl counterparts 4 and 5.
With the functionalized supports 4, 5, and 10 in hand, we next explored the methylation of their α-amino group. For solid-phase peptide synthesis, methods for selective N-methylation of a specific amino acid have already been reported.[24–27] The most efficient solid-phase procedure, first described by Miller and Scanlan,[24] is a three-step sequence involving amine activation by an o-nitrobenzenesulfonyl group (o-NBS), followed by direct alkylation or Mitsunobu reaction on the activated nitrogen and then removal of the sulfonamide group. Recently, this strategy has been further optimized by Kessler and coworkers.[28,29] For our structurally more complex adenosine-containing supports, selective N-methylation of the 3′-aminoacylamino moiety was achieved by an analogous three-step procedure (Scheme 3). Reaction with o-NBS-Cl transformed the primary amine into the corresponding o-nitrobenzenesulfonamide that was deprotonated with a sterically hindered base, 7-methyl-1,5,7- triazabicyclo[4.4.0]dec-5-ene (MTBD), and subsequently methylated by using methyl p-nitrobenzenesulfonate. Cleavage of the o-NBS group proceeded with 2-mercaptoethanol. Protection of the N-methylamino group with Fmoc was required to block this group from undesired branching during RNA chain assembly by using standard phosphoramidite chemistry. The reaction sequence was efficient, and automated RNA synthesis by using the methylated supports 11–13 provided the crude aminoacyl-oligoribonucleotides of high quality after standard deprotection with methylamine (30% in ethanol/water), followed by tetrabutylammonium fluoride (TBAF) in tetrahydrofuran. Analysis by anion-exchange HPLC demonstrated that the major peak of the crude products represented the desired N-methylated conjugate that was further purified by HPLC and analyzed by LC-ESI mass spectrometry as exemplified in Figure 3A. Importantly, when the solid support 10 was applied, palladium-mediated cleavage of the allyl groups to liberate the phosphonate moiety was conducted in apolar solvent as the first deprotection step of the bioconjugate, prior to methylamine and subsequent fluoride treatments (see the Supporting Information).
Scheme 3.

Selective N-methylation of the 3′-amino-3′-deoxyadenosine-functionalized solid support 4, 5, and 10. Reaction conditions: a) piperidine (20 v/v% in CH3CN), RT, 15 min. b) o-NBS-Cl, sym-collidine, CH2Cl2, RT, 2 h. c) 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene, methyl p-nitrobenzenesulfonate, DMF, RT, 30 min; d) HSCH2CH2OH, 1,8-diazabicyclo[5,4,0]undec-7-ene, DMF, RT, 30 min; e) Fmoc N-hydroxysuccinimide ester, H2O/dioxane (1:1), RT, 14 h.
Figure 3.

Examples for the synthesis of the 3′-aminoacylamino-RNA conjugates based on the solid supports 11 and 14–16. Anion-exchange HPLC profiles of the crude and the purified (insets) conjugates C1, C2, C7, and C8 (A–D, upper panels) and LC-ESI mass spectra of purified products (A–D, lower panels). Anion-exchange chromatography conditions: Dionex DNAPac PA-100 (4×250 mm) column; temperature: 60°C; flow rate: 1 mLmin−1; eluent A: 25 mM Tris-HCl (pH 8.0, Tris=tris(hydroxymethyl)aminomethane), 6M urea; eluent B: 25 mM Tris-HCl (pH 8.0), 6M urea, 500 mM NaClO4; gradient: 0–35% B in A within 30 min; UV detection at λ=260 nm.
Comparable to the N-methylation, the phosphorylation procedure of the 3′-serinyl- and 3′-threonyl-RNA conjugates preceded oligoribonucleotide assembly directly on the adenosine supports 4 and 5 (Scheme 4). After manual deprotection of the O-TBDMS group by rinsing the support with a solution of TBAF and acetic acid in THF in a frit-fitted syringe, followed by washing and drying, it was transferred onto a column for automated solid-phase synthesis. Phosphorylation was accomplished by a modified coupling cycle without initial detritylation by using the bis(2-cyanoethoxy) (diisopropy1amino) phosphine reagent[30,36] and benzylthiotetrazol for activation, followed by oxidation with iodine/ THF/water. The resulting phosphorylated serine side chain was appropriately protected to immediately continue with RNA strand assembly. Deprotection of the conjugate was performed according to standard procedures (Figure 3B and C).
Scheme 4.
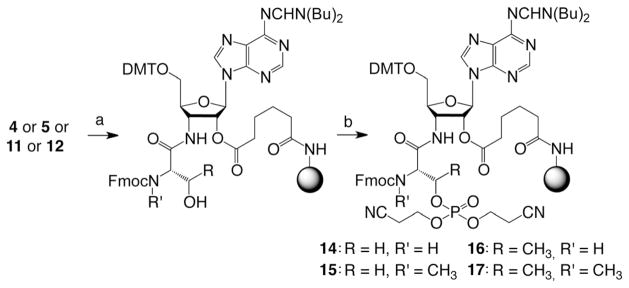
Selective O-phosphorylation of the 3′-amino-3′-deoxyadenosine-functionalized solid support 4, 5, 11, and 12. Reaction conditions: a) TBAF3·H2O (1.0 M), acetic acid (0.5 M), THF, RT, 30 min. b) i) 0.1 M (iPr)NP(OCH2CH2CN)2 (130 μL), 0.3M 5-benzylthio-1H-tetrazole (360 μL), CH3CN; ii) capping (1.2 min, Cap A/Cap B = 1:1) with Cap A: 0.2M phenoxyacetic anhydride in THF, Cap B: 0.2M N-methylimidazole and 0.2M sym-collidine in THF; oxidation (1.0 min) with I2 (0.2 M) in THF/pyridine/H2O (35:10:5).
Furthermore, our approach allowed the combination of methylation and phosphorylation to achieve both modification patterns on the same conjugate, as exemplified for conjugate C8 in Figure 3D. This diversity enables the synthesis of derivatives that could potentially interfere with discrete steps in the reaction cascade of the biosynthesis of selenocysteine. A selection of hydrolysis-resistant tRNASec mimics synthesized during the course of this study, ranging from short tetranucleotides (C2 and C3) to acceptor stem mimics (C6, C7, and C9) to 3′-tRNASec fragments (C4, C5, and C8) is shown in Table 1.
Table 1.
Selection of 3′-aminoacylamino oligoribonucleotides.
| Sequence 5′-RNA-3′-NH-aa(modification/s) | Mwcalcd | Mwobs[a] | Yield[b] [nmol] | |
|---|---|---|---|---|
| C1 | pGCG CCA-Ser(NαCH3) | 2053.3 | 2052.9 | 140 |
| C2 | pGCC A-Ser(p) | 1388.9 | 1388.7 | 77 |
| C3 | pGCC A-Thr(p) | 1402.9 | 1402.6 | 74 |
| C4 | pUCC ACC UUU CGG GCG CCA-Ser(p) | 5889.5 | 5889.5 | 129 |
| C5 | pUCC ACC UUU CGG GCG CCA-Abu(p) | 6193.7 | 6194.3 | 25 |
| C6 | GCC CGG AUG UUC GCU UUC GGG CGC CA-Ser(p) | 8453.1 | 8453.1 | 34 |
| C7 | GCC CGG AUG UUC GCU UUC GGG CGC CA-Thr(p) | 8466.1 | 8467.0 | 58 |
| C8 | pUCC ACC UUU CGG GCG CCA-Ser(p/NαCH3) | 5903.5 | 5903.4 | 69 |
| C9 | GCC CGG AUG UUC GCU UUC GGG CGC CA-Ser(p/NαCH3) | 8467.1 | 8467.4 | 23 |
Measured by LC-ESI mass spectrometry.
Yield of purified, isolated RNA.
To generate full-length tRNASec mimics that comprise the modified 3′-termini, we elaborated an efficient enzymatic ligation strategy. A previously introduced concept for ligative tRNA synthesis utilizes 3′-fragments that are 18 nt long and 5′-fragments with a size of 58 nt.[11,31] The ligation site was positioned within the TΨC loop to allow proper annealing of the binary pre-ligation complex required for T4 RNA ligase.
However, when this strategy is applied onto tRNASec, the required 5′-fragment is 71 nt long. This is primarily because of the enlarged variable loop of tRNASec. We therefore decided to test a three-strand one-pot approach with an additional site for ligation located in the anticodon loop (Figure 4A).
Figure 4.

Optimized enzymatic ligation of phosphonated, methylated, and/or phosphorylated aminoacylamino-tRNASec by using three synthetic fragments and T4 RNA ligase, exemplified for Abu(p)-tRNASec. A) Concept for enzymatic ligation of tRNAs with a large variable arm. B) HPLC profiles following the ligation progress. C) HPLC profile (top) and LC-ESI mass spectrum (bottom) of the purified tRNA derivative. For conditions see the Supporting Information.
Annealing of the ternary pre-ligation complex by using the 38, 33 and 19 nt long strands was efficient and gave high yields for the double ligation by using T4 RNA ligase (Figure 4B). The RNA was separated by phenol/chloroform extraction from the ligation mixture and purified by anion-exchange chromatography. The expected molecular weight of the isolated product was confirmed by LC-ESI mass spectrometry (Figure 4C). By using this approach, tRNASec mimics with various modification patterns and sequence variations were obtained (Table 2).
Table 2.
Enzymatic ligation of selected tRNASec variants.
| tRNA mimic[a] | Mwcalcd | Mwobs[d] | Yield[e] [nmol] |
|---|---|---|---|
| Abu(p)-tRNASec (90 nt)[b] | 29095 | 29096 | 1.0 |
| Abu(p)-tRNAU1ASec (93 nt)[c] | 30028 | 30028 | 0.5 |
| Ser(p/NαCH3)-tRNASec (93 nt)[c] | 30044 | 30043 | 4.0 |
For sequences see Figure 2.
Ligation of 38, 33, and 19 nt long fragments.
Ligation of 39, 35, and 19 nt long fragments.
Measured by LC-ESI mass spectrometry.
Yield of purified, isolated tRNA.
To test the biological relevance of the novel tRNASec mimics, we employed an electrophoretic mobility shift assay (EMSA). The binding capacity to SepSecS of each of the ligands was evaluated in a qualitative and rapid fashion (Figure 5). Under our experimental conditions, the binding to SepSecS of the unacylated tRNASec, which was used in the earlier structural study,[8] and the chemically modified tRNASec mimics was indistinguishable. tRNASec and the mimics run slightly faster than the 100 bp DNA fragment in the DNA ladder (Figure 5, lanes 7–14).
Figure 5.

Synthetic tRNASec derivatives bind to SepSecS. The electrophoretic mobility shift assay was performed by using 6% native polyacrylamide gel and the gel was stained by ethidium bromide. SepSecS forms a binary complex with transcribed tRNASec (lane 8), and tRNASec mimics (lanes 10, 12, and 14), whereas it does not bind DNA (lane 4) or pre-mi-RNA-21 (lane 6). N-m-Sep-tRNASec is an equivalent annotation to Ser(p/NαCH3)-tRNASec (see Table 1).
After incubation with SepSecS prior to native gel electrophoresis, a significant decrease in the electrophoretic mobility of both the unacylated tRNASec and the tRNASec mimics was observed (Figure 5, lanes 8, 10, 12, and 14), which is consistent with the formation of the high-affinity SepSecS-tRNASec complex. That the slower species represents the binary complex was confirmed by Coomassie staining, which revealed the presence of SepSecS in the band as well (Figure S1 in the Supporting Information, lanes 8, 10, 12, and 14). In the absence of tRNASec, SepSecS does not enter the native gel, but it stays in the loading well (Figure S1 in the Supporting Information, lanes 2, 4, and 6). However, the enzyme migrates faster once in complex with tRNA, which is consistent with the increase of the overall negative charge in the ribonucleoprotein complex compared to SepSecS alone (Figure S1 in the Supporting Information). Moreover, the titration experiment shows that the mimics saturate the tRNA-binding sites on SepSecS to the comparable level as the unacylated tRNASec (Figure S2 in the Supporting Information), suggesting that the nature and stoichiometry of the binary complex is likely to be the same regardless of the tRNASec derivative used. This also argues that the mimics adopt the same fold as the in vitro transcribed tRNASec. Finally, control experiments with either the DNA fragment (Figure 5, lanes 3 and 4) or the human pre-miRNA-21 oligonucleotide (Figure 5, lanes 5 and 6) support the notion that the interaction of SepSecS with tRNASec and tRNASec mimics is specific and that the mimics developed herein are suitable for structural studies.
Conclusion
From a methodological point of view and compared to previously published procedures to generate aminoacyl–tRNA and peptidyl–tRNA conjugates (reviewed in Ref. [32] and [33], see also Ref. [34] and [35]), the challenge here was the design of a versatile approach to N-methylation, O-phosphorylation, and phosphonation of amino acid side chains of such bioconjugates. By introducing robust protocols for direct methylation of the α-amino group, as well as the phosphorylation directly on the aminoacyl-nucleoside-functionalized solid support, this goal has been successfully accomplished for aminoacyl–tRNA conjugates required here. Consequently, the same approach could, in principle, be extended to methylated and/or phosphorylated peptidyl–tRNA conjugates. Also, the serine analogue Abu(p), introduced as a functional part of an adenosine-modified solid support, could be used as an amino acid building block (Fmoc-Abu(pAll2)) for incorporation at internal positions of the peptidyl moiety.[22] Finally, our work included the development of an efficient one-pot, three-strand tRNA ligation protocol for tRNASec, which is generally applicable to other tRNA species containing a long variable loop.
We have prepared structurally complex tRNASec mimics that carry modified amino acid moieties, which may, interfere with discrete steps in the biosynthetic cycle of seleno-cysteine. Because the aminoacyl moieties are attached to tRNASec through a hydrolysis-resistant linkage, the new mimics are attractive for high-resolution crystallographic studies of the unexplored catalytic mechanism of SepSecS and interactions between the active site of SepSecS and the 5′-end of Sep-tRNASec. Finally, a similar approach could be utilized in studying other tRNA-dependent enzymes including the ribosome.
Experimental Section
For the synthesis of the functionalized solid supports 4, 5, and 10 as well as amino acid derivative 9 see the Supporting Information.
Nα-Methylation on solid support
Fmoc deprotection
The solid supports 4, 5, or 10 (30 mg) were suspended in acetonitrile for 1 min, followed by treatment with piperidine (20% solution in acetonitrile, 2 mL) for 10 min, and again for 5 min with fresh solution. Then, the beads were washed with acetonitrile and dried under vacuum.
NBS activation
The deprotected supports were suspended in a solution of 2-nitrobenzenesulfonyl chloride (150 mg, 0.68 mmol) and sym-collidine (98 μL, 0.72 mmol) in CH2Cl2 (1 mL) and agitated for 2 h at room temperature. The beads were washed with CH2Cl2 and dried.
N-Methylation
The beads were swelled in DMF and subsequently treated with a solution of MTBD (10 μL, 0.07 mmol) and methyl-4-nitrobenzene sulfonate (18 mg, 0.08 mmol) in DMF (1 mL) for 30 min. The solution was discharged and the solid support was washed with DMF for direct use in the next step.
NBS cleavage
The beads were incubated with a solution of 2-mercaptoethanol (18 μL, 0.26 mmol) and 1,5-diazabicyclo[5.4.0]undec-5-ene (DBU) (16 μL, 0.11 mmol) in DMF (1 mL) for 30 min. During that time, the solution turned yellow. The beads were washed and a second treatment with 2-mercaptoethanol (9 μL, 0.13 mmol) and DBU (8 μL, 0.06 mmol) in DMF (1 mL) for 1 min showed no color reaction and confirmed that cleavage was complete. The beads were washed with DMF and acetonitrile, and finally dried under vacuum.
Fmoc protection
Sodium carbonate (100 mg) was dissolved in H2O/dioxane (1:1, 2 mL), followed by addition of the beads. After 15 min, Fmoc N-hydroxysuccinimide ester (3 mg, 0.08 mmol) was added and the suspension was agitated overnight at room temperature. The beads were washed with water, acetonitrile, and CH2Cl2 and dried under vacuum.
O-Phosphorylation on solid support
TBDMS deprotection
After swelling of the solid support 4, 5, 11, or 12 (30 mg) in THF (1 mL) for 10 min, it was treated with a solution of TBAF·3H2O (1.0 M) and acetic acid (0.5 M) in THF (1 mL) for 30 min at room temperature. Then, the beads were washed with THF and CH2Cl2 and dried.
O-Phosphorylation
Phosphorylation was performed in an automated manner on an oligonucleotide synthesizer by using bis(2-cyanoethyl)-(N,N-diisopropyl)phosphoramidite[29,35] and following a standard synthesis coupling cycle that lacked the detritylation step. For details see “RNA synthesis”.
RNA synthesis
All oligonucleotides were synthesized on an ABI 392 Nucleic Acid Synthesizer following standard synthesis protocols. Detritylation (120 s): dichloroacetic acid/1,2-dichloroethane (4:96); coupling (120 s): phosphoramidites (0.1M in acetonitrile, 130 μL) were activated with benzylthiotetrazole (0.3M in acetonitrile, 180 μL); capping (2×10 s, Cap A/Cap B=1:1): Cap A: phenoxyacetic anhydride (0.2M in THF), Cap B: N-methyl imidazole (0.2M), sym-collidine (0.2) in THF; oxidation (20 s): I2 (0.2 M) in THF/pyridine/H2O (35:10:5). Amidites, benzylthiotetrazole, and capping solutions were dried over activated molecular sieves (4 Å) overnight.
Deprotection of 3′-aminoacylamino-RNA conjugates
Allyl deprotection
Conjugates synthesized on solid support 10 were treated with a solution of N-methyl morpholine (37 μL, 0.34 mmol) and acetic acid (37 μL, 65 mmol) in chloroform (1 mL). After addition of tetrakis(triphenylphosphine) palladium(0) (12 mg, 0.01 mmol), the suspension was agitated for 5 h at room temperature. Subsequently, the solid support was collected on a Büchner funnel, washed with chloroform and dried under vacuum.
Acyl deprotection and cleavage from the solid support
For conjugates synthesized on solid support 4, 5, 11, and 12 and for conjugates synthesized on solid support 10 after allyl deprotection (see above), the beads were transferred into an Eppendorf tube and equal volumes of methylamine in ethanol (8M, 0.5 mL) and methylamine in H2O (40%, 0.5 mL) were added. After 6 h shaking at room temperature the supernatant was filtered and evaporated to dryness.
2′-O-TOM deprotection
The obtained residue was treated with TBAF·3H2O in THF (1 M, 1 mL) overnight at room temperature. The reaction was quenched by the addition of triethylammonium acetate (TEAA) (1M, pH 7.4, 1 mL). After reducing the volume of the solution, it was applied on a size-exclusion chromatography column (GE Healthcare, HiPre 26/10 Desalting, 2.6×10 cm, Sephadex G25). By eluating with H2O, the conjugate-containing fractions were collected, evaporated to dryness, and the residue was dissolved in H2O (1 mL). Analysis of the crude products was performed by anion-exchange chromatography on a Dionex DNAPac PA-100 column (4×250 mm) at 60°C. Flow rate: 1 mLmin−1; eluent A: 25 mM Tris·HCl (pH 8.0), 6M urea; eluent B: 25 mM Tris·HCl (pH 8.0), 0.5M NaClO4, 6 M urea; gradient: 0–60% B in A within 45 min or 0–40% B in A within 30 min for short sequences up to fifteen nucleotides, UV detection at λ=260 nm.
Purification of 3′-aminoacylamino-RNA conjugates
The crude RNA products were purified on a semipreparative Dionex DNAPac PA-100 column (9×250 mm) at 60°C with flow rate of 2 mLmin−1 (for eluents see above). Fractions containing the conjugate were loaded on a C18 SepPak Plus cartridge (Waters/Millipore), washed with 0.1–0.15M (Et3NH)+HCO3−, H2O, and eluted with H2O/CH3CN (1:1). Conjugate-containing fractions were evaporated to dryness and dissolved in H2O (1 mL). The quality of the purified conjugate was analyzed by analytical anion-exchange chromatography (for conditions see above). The molecular weight of all synthesized RNAs was confirmed by LC-ESI mass spectrometry (see the Supporting Information). Yields were determined by UV photometrical analysis of conjugate solutions.
Enzymatic ligation of tRNASec
Equimolar amounts of three chemically synthesized tRNA fragments (see Figure 2 for sequence, Figure 4 for 5′-phosphate modification, and Table 2) were dissolved in water (2/3 of the final total reaction volume, final RNA concentration of 40 μM for each strand). One tenth of the final reaction volume of 10× ligation buffer (10 mM adenosintriphosphate (ATP), 500 mM 4-(2-hydroxyethyl)-1-piperazineehtanesulfonic acid (HEPES)-NaOH, 100 mM MgCl2, and 100 mM dithiothreitol (DTT)) was added. The solution was heated to 90°C for 5 min and then allowed to cool to room temperature within 3 h. Then, bovine serum albumin (BSA) stock solution (1 mgmL−1) was added (Fermentas). The reaction mixture was vortexed and centrifuged before it was treated with T4 RNA ligase (0.4 UμL−1, Fermentas, 10 UμL−1 in storage buffer). Final concentrations: 40 μM for each RNA strand, 1 mM ATP, 50 mM HEPES-NaOH (pH 8.0), 10 mM MgCl2, 10 mM DTT, 0.1 mgmL−1 BSA, 0.4 UμL−1 T4 RNA ligase. The solution was incubated at 37°C overnight, afterwards it was extracted twice with an equal volume of a phenol/chloroform/isoamyl alcohol solution (25:24:1, v/v/v) and twice with an equal volume of chloroform/isoamyl alcohol solution (24:1, v/v). All organic layers were rewashed once with an equal volume of H2O. The aqueous layers were combined and reduced in vacuo to approximately 50% (v/v) before purification. Monitoring of the ligation reaction, purification, and analysis of the 3′-modifed full-length tRNASec were performed by anion-exchange chromatography as described above.
EMSA binding assay
The ability of the tRNASec mimics to bind to human SepSecS was assayed by using DNA retardation gels. Each of the tRNASec mimics (125–500 ng) was mixed with SepSecS (1 μg) in TGS buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 5% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), 10 μM PLP), and the mixture was incubated 30 min at 23°C. After incubation, High-Density TBE sample buffer was added to a final volume of 10 μL and the samples were subjected to electrophoresis in 6% DNA retardation gels (Invitrogen), in TBE buffer (44.5 mM Tris base, 44.5 mM boric acid, 1 mM EDTA, pH 8.3) at 100 mV for 75 min. The samples containing in vitro transcribed human tRNASec served as a positive control, whereas samples containing either 171 bp DNA fragment (polymerase chain reaction (PCR) product of the template used for in vitro transcription of tRNASec) or 60 nt long human pre-miRNA-21 (5′-UAG CUU AUC AGA CUG AUG UUG ACU GUU GAA UCU CAU GGC AAC ACC AGU CGA UGG GCU GUC-3′) served as negative controls. The molar ratio between a nucleic acid and SepSecS was kept at 2:4. For the titration experiment, increasing amounts of tRNASec and mimics were incubated with a constant amount of SepSecS yielding molar ratios of 1:4, 2:4, and 4:4. All calculations were based on the SepSecS monomer concentration. Human tRNASec and SepSecS were purified according to the previously published procedure.[8] Nucleic acids (tRNASec mimics, tRNASec, DNA, and pre-miRNA-21) were visualized by ethidium bromide staining, whereas Coomassie staining was used to visualize SepSecS.
Supplementary Material
Acknowledgments
This work was funded by the Austrian Science Foundation (FWF) grants P21641 and I1040 (R.M.) and by Grant GM097042 from the National Institute of General Medical Sciences (to M.S.). The authors thank Prof. Zain Paroo (University of Illinois at Chicago) for providing human pre-miRNA-21.
Footnotes
Sec=selenocysteine.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201302188.
Contributor Information
Prof. Miljan Simonović, Email: msimon5@uic.edu.
Prof. Dr. Ronald Micura, Email: ronald.micura@uibk.ac.at.
References
- 1.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt RL, Simonović M. Croat Med J. 2012;53:535–550. doi: 10.3325/cmj.2012.53.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cone JE, Del Rio RM, Davis JN, Stadtman TC. Proc Natl Acad Sci USA. 1976;73:2659–2663. doi: 10.1073/pnas.73.8.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambrogelly A, Palioura S, Söll D. Nat Chem Biol. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 5.Chiba S, Itoh Y, Sekine S, Yokoyama S. Mol Cell. 2010;39:410–420. doi: 10.1016/j.molcel.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Sherrer RL, O’Donoghue P, Söll D. Nucleic Acids Res. 2008;36:1247–1259. doi: 10.1093/nar/gkm1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J, Palioura S, Salazar JC, Su D, O’Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Söll D. Proc Natl Acad Sci USA. 2006;103:18923–18927. doi: 10.1073/pnas.0609703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palioura S, Sherrer RL, Steitz TA, Söll D, Simonović M. Science. 2009;325:321–325. doi: 10.1126/science.1173755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. PLoS Biol. 2007;5:e4. doi: 10.1371/journal.pbio.0050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moroder H, Steger J, Graber D, Fauster K, Trappl K, Marquez V, Polacek N, Wilson DN, Micura R. Angew Chem. 2009;121:4116–4120. doi: 10.1002/anie.200900939. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:4056–4060. doi: 10.1002/anie.200900939. [DOI] [PubMed] [Google Scholar]
- 11.Graber D, Moroder H, Steger J, Trappl K, Polacek N, Micura R. Nucl Acids Res. 2010;38:6796–6802. doi: 10.1093/nar/gkq508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steger J, Graber D, Moroder H, Geiermann A-S, Aigner M, Micura R. Angew Chem. 2010;122:7632–7634. doi: 10.1002/anie.201003424. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:7470–7472. doi: 10.1002/anie.201003424. [DOI] [PubMed] [Google Scholar]
- 13.Steger J, Micura R. Bioorg Med Chem. 2011;19:5167–5174. doi: 10.1016/j.bmc.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiermann AS, Polacek N, Micura R. J Am Chem Soc. 2011;133:19068–19071. doi: 10.1021/ja209053b. [DOI] [PubMed] [Google Scholar]
- 15.Geiermann AS, Micura R. ChemBioChem. 2012;13:1742–1745. doi: 10.1002/cbic.201200368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapuis H, Strazewski P. Tetrahedron. 2006;62:12108–12115. [Google Scholar]
- 17.Terenzi S, Biala E, Nguyen-Trung NQ, Strazewski P. Angew Chem. 2003;115:3015–3018. doi: 10.1002/anie.200350926. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:2909–2912. doi: 10.1002/anie.200350926. [DOI] [PubMed] [Google Scholar]
- 18.Chemama M, Fonvielle M, Arthur M, Valéry JM, Ethève-Quelquejeu M. Chem Eur J. 2009;15:1929–1938. doi: 10.1002/chem.200801563. [DOI] [PubMed] [Google Scholar]
- 19.Chemama M, Fonvielle M, Lecerf M, Mellal D, Fief H, Arthur M, Valéry J-M, Ethève-Quelquejeu M. Curr Protoc Nucl Acids Chem. 2011;Chapter 4(Unit 4.44) doi: 10.1002/0471142700.nc0444s44. [DOI] [PubMed] [Google Scholar]
- 20.Okuda K, Seila AC, Strobel SA. Tetrahedron. 2004;60:12101–12112. [Google Scholar]
- 21.Schmeing TM, Huang KS, Strobel SA, Steitz TA. Mol Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Shapiro G, Buechler D, Ojea V, Pombo-Villar E, Ruiz M, Weber HP. Tetrahedron Lett. 1993;34:6255–6258. [Google Scholar]
- 23.Marczak E, Lipkowski AW, Czerwinski K, Kazimierczak J. In: Peptide Science—Present and Future. Shimonishi Y, editor. Kluwer Academic; Dordrecht: 2002. pp. 779–781. [Google Scholar]
- 24.Miller SC, Scanlan TS. J Am Chem Soc. 1997;119:2301–2302. [Google Scholar]
- 25.Reichwein JF, Liskamp MJ. Tetrahedron Lett. 1998;39:1243–1246. [Google Scholar]
- 26.Yang L, Chiu K. Tetrahedron Lett. 1997;38:7307–7310. [Google Scholar]
- 27.Laplante C, Hall DG. Org Lett. 2001;3:1487–1490. doi: 10.1021/ol015803l. [DOI] [PubMed] [Google Scholar]
- 28.Biron E, Chatterjee J, Kessler H. J Pept Sci. 2006;12:213–219. doi: 10.1002/psc.711. [DOI] [PubMed] [Google Scholar]
- 29.Chatterjee J, Laufer B, Kessler H. Nat Protoc. 2012;7:432–444. doi: 10.1038/nprot.2011.450. [DOI] [PubMed] [Google Scholar]
- 30.Bannwarth W, Trzeciak A. Helv Chim Acta. 1987;70:175–186. [Google Scholar]
- 31.Graber D, Trappl K, Steger J, Geiermann AS, Rigger L, Moroder H, Polacek N, Micura R. Methods Mol Biol. 2012;848:201–213. doi: 10.1007/978-1-61779-545-9_13. [DOI] [PubMed] [Google Scholar]
- 32.Lu K, Duan QP, Ma L, Zhao DX. Bioconjugate Chem. 2010;21:187–202. doi: 10.1021/bc900158s. [DOI] [PubMed] [Google Scholar]
- 33.Lönnberg H. Bioconjugate Chem. 2009;20:1065–1094. doi: 10.1021/bc800406a. [DOI] [PubMed] [Google Scholar]
- 34.Diezmann F, Eberhard H, Seitz O. Biopolymers. 2010;94:397–404. doi: 10.1002/bip.21440. [DOI] [PubMed] [Google Scholar]
- 35.Aviñó A, Ocampo SM, Caminal C, Perales JC, Eritja R. Mol Diversity. 2009;13:287–293. doi: 10.1007/s11030-009-9110-7. [DOI] [PubMed] [Google Scholar]
- 36.Uhlmann E, Engels J. Tetrahedron Lett. 1986;27:1023–1026. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.