

Abstract
The BRAF protein kinase, a molecule in the RAS-RAF-MEK-ERK signaling pathway, is mutated to harbor elevated kinase activity in about 7% of human cancers, making it an important therapeutic target for inhibition. Several BRAF protein kinase inhibitors have been developed through high-throughout-screening in vitro, however many of these compounds suffer from a lack of suitable kinase specificity and other chemotherapeutic properties. In silico screening has evolved as a powerful complimentary approach to protein kinase inhibitor identification. Here we describe an in silico screen for BRAF inhibitors leading to the identification of a series of purine-2,6-dione analogues with IC50 values in the single digit micromolar range and with significant selectivity for BRAF over other representative protein kinases. The binding modes of these inhibitors to BRAF are analyzed through molecular docking to derive structure-activity relationships and to assist in the future development of more potent and specific BRAF inhibitors.
INTRODUCTION
The RAS-RAF-MEK-ERK (MAPK) signaling pathway plays a central role in transducing signals from extracellular growth factors to the nucleus to promote cell proliferation and survival. The MAPK pathway also represents a common pathway that is activated at aberrantly high levels in a variety of human cancers. RAF protein kinases are central players in the MAPK signal transduction pathway and have been shown to be critical for mediating cell proliferation, survival, and angiogenesis in various cancer models1. The RAF protein kinase family consists of three isoforms named: ARAF, c-RAF-1 and BRAF. Earlier functional studies on the RAF family focused on c-RAF-1 and these studies revealed that RAF kinases are tightly regulated and require multiple phosphorylation events from diverse upstream protein kinases to achieve kinase activation. The importance of BRAF activation was highlighted by a more recent study showing that it is mutated in approximately 7% of human cancer2, and most notably in melanoma (50–70%), ovarian (~35%), thyroid (~30%) and colorectal (~10%) cancers. Among the many activating BRAF mutations that were identified in human cancers, a single V600E mutation within the BRAF kinase domain accounts for over 90% of all these mutations and the BRAFV600E mutant protein was found to be 500-fold more active than the wild-type protein in vivo3. The oncogenic BRAFV600E bypasses the requirement for upstream regulation by phosphorylation resulting in a MAPK signaling pathway that is constitutively activated in the absence of extracellular growth factor signals. These data strongly implicate the BRAF protein kinase as an important target for developing small molecule inhibitors in the treatment of human cancers and melanoma in particular.
Towards the preparation of BRAF inhibitors, the general RAF protein kinase inhibitor, Sorafenib, was developed through high-throughput solution screening and approved by the FDA for the use in renal carcinoma4, although this compound failed to show therapeutic activity in the treatment of malignant melanoma. While several other BRAF inhibitors identified through high throughput solution screening are currently in various stages of development, the discovery of novel, potent and specific inhibitors against BRAF protein kinase remains a major challenge.
Due to the cost-inefficient and time-consuming process of conventional drug discovery over the last decade, high throughput virtual screening (HTVS) has emerged as an attractive and complementary approach to traditional high-throughout solution screening (HTS). HTVS typically depends on the availability of a high resolution crystal structure of the protein target as a template for computational screening. Over the years, HTVS has been applied to the successful identifications of biologically active molecules against targets such as HIV-1 protease, thymidylate synthase, influenza hemagglutinin, and parasitic proteases 5, 6. The availability of the crystal structure of the BRAF protein kinase3, 7 now provides an opportunity to utilize the HTVS strategy to identify BRAF inhibitors. Here we described a HTVS approach to identify a series of novel inhibitors against the BRAF protein kinase. Specifically, we screened about 90,000 low molecular weight compounds from a publicly available small molecule database using the HTVS approach and after two generations of optimization from a primary inhibitor lead, we developed a novel series of compounds with IC50 values in the single digit micromolar range against BRAF kinase. We also show that these compounds display selectivity for the inhibition of BRAF over other representative protein kinases and we employ molecular docking of these compounds bound to BRAF to derive structure-activity relationships. These results establish that virtual screening is an effective strategy for the discovery of potent and selective BRAF inhibitors and we provide a novel lead scaffold for future design of more potent and specific BRAFWT and BRAFV600E protein kinase inhibitors.
RESULTS AND DISCUSSION
High throughout virtual screening procedure
In this procedure, residues within a distance of 6 Å around the BRAF inhibitor, compound 50 (SB590885)7 were isolated for constructing a grid for screening using the program DOCK4.08. This grid was large enough to include every residue of the BRAF kinase ATP binding pocket. For creation of a library of compounds for screening we selected a public database containing a large number of small molecule compounds that would be available for subsequent solution screening at nominal cost. To this end, we selected the SPECS database that contained about 300,000 small molecule compounds. To further refine the database to include the compounds that were likely to be soluble in aqueous solution to allow for a follow-up solution assay, we filtered the database for compounds with a logS value of greater than - 4, resulting in a database of about 90,000 small molecule compounds. To efficiently screen these compounds within a reasonable time we initially used DOCK4.08 as the primary molecular docking program, a docking program that had already been successfully used for the identification of inhibitors for the HIV-1 protease, thymidylate synthase, influenza hemagglutinin, and parasitic proteases5. As a pilot study to test the effectiveness of our docking procedure, we included 2 known BRAF inhibitors, compounds 50 and 51 (BAY439006)3 as positive controls for this docking procedure in an initial trial run with 998 randomly selected compounds from the SPECS database. As expected, these two inhibitors were placed as the top two scoring compounds in this docking procedure. We then carried out docking against the remaining compounds and the top 5,000 hits generated from the energy scoring function of DOCK4.0 were docked using three other docking programs employing different scoring functions; XSCORE (version 1.2.1) for calculating a binding score for a given protein-ligand complex structure9, SLIDE (version 2.3.1) for calculation of hydrogen bonds and the hydrophobic complementarity while considering the flexibility of both protein and ligand10, 11 and AutoDock3.0 for calculating the free energy of binding12. Specifically, the program XScore was first performed on the top 5,000 candidate compounds that were generated from the program DOCK4.0. The top 2,000 compounds from the program XScore were then selected for reevaluation using the SLIDE scoring function. The top 783 componds from SLIDE were finally evaluated according to free energy of binding with the program AutoDock3.0. According to their binding modes, free energy scores and scaffold diversity, 20 compounds from 10 manually classified groups were selected (Table S1) for BRAF inhibition solution studies using an ELISA-based activity assay system.
In vitro analysis of BRAF inhibitors identified through virtual screening
Eighteen virtual screening hits (compounds 1–18) shown in Table S1 and Figure S1a were assayed for BRAF activity at an inhibitor concentration of 100 μM using an ELISA-based MEK phosphorylation assay. From this initial screen, only compound 1 reduced BRAF kinase activity, to about 80% of wild-type activity, and a subsequent measurement of the dose-response inhibition curve of compound 1 against BRAF produced an IC50 value of 29 μM (Figure 1c).
Figure 1.
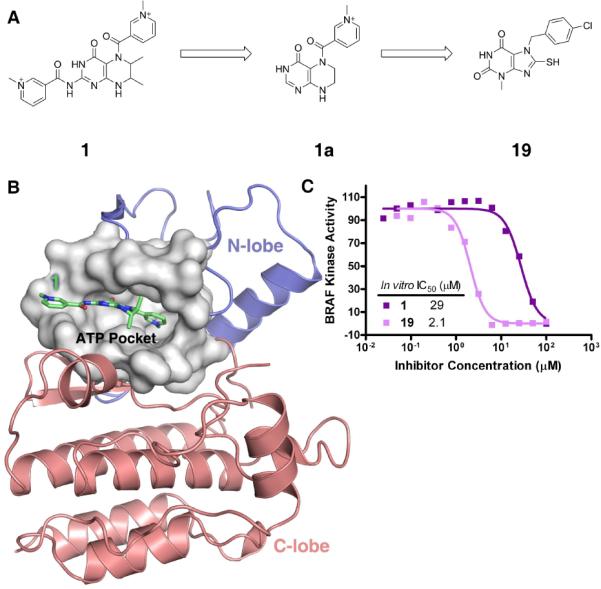
Identification of Compound 1 and 19 as BRAF inhibitors: (a) Molecular structures of compound 1, symmetry extracted scaffold 1a and compound 19; (b) The binding mode of compound 1 in the active site of the BRAF protein kinase. The surface representation is colored white to show all ATP pocket residues within 8 Å from compound 1. The N-lobe and C-lobe of the BRAF kinase domain are colored blue and red, respectively; (c) Dose response curve of BRAF kinase inhibition by compounds 1 (purple) and 19 (pink) using an in vitro BRAF ELISA kinase assay;
Development of second generation BRAF inhibitors
Upon close examination of the molecular structure of compound 1, we noted that the hexahydropteridine portion of the molecule contained two symmetrical methylpyridinium groups at opposite ends suggesting that the hexahydropteridine portion and only one of the two methylpyridinium groups might be employed for BRAF inhibition (Figure 1a). In order to obtain more direct insights into the binding mode of the compound 1 to BRAF, we analyzed its docked conformation within the BRAF active site (Figure 1b). This docking result revealed that one of the methylpyridinium groups and the hexahydropteridine portion of the molecule formed interactions with the BRAF active site through extensive hydrophobic interactions with BRAF active site residues Trp463, Val471, Leu514, Trp531 and Phe583. In contrast, the second methylpyridinium group was pointing outside of the BRAF active site, making minimal interactions with the protein. Based on this observation, we hypothesized that the inhibitory activity of compound 1 was largely due to the hexahydropteridine moiety combined with only one of the two methylpyridinium groups of compound 1. To test this hypothesis, we derived a new scaffold, named compound 1a (Figure 1a) consisting of only the hexahydropteridine and methylpyridinium groups as a query to search the SPECS database for compounds with similar scaffolds. From this approach, compound 19 was identified and tested using the BRAF ELISA assay for inhibitory activity against BRAF. Consistent with our hypothesis, compound 19, which has a purine-2,6-dione scaffold similar to our query structure was indeed a relatively potent BRAF inhibitor, showing a 90% reduction of BRAF activity at an inhibitor concentration of 50 μM. A dose response inhibition curve of compound 19 against BRAF produced an IC50 value of 2.1 μM (Figure 1c and Table 1).
Table 1.
Molecular structures of compounds that show inhibitory activity against BRAFWT.
| Compound | Structure | IC50 for BRAFWT (μM) |
|---|---|---|
| 1 |

|
29.42 |
| 19 |

|
2.10 |
| 22 |

|
30.11 |
| 23 |

|
10.91 |
| 24 |

|
1.72 |
| 25 |

|
11.08 |
| 26 |

|
4.60 |
| 27 |

|
57.71 |
| 28 |

|
3.75 |
| 29 |

|
37.48 |
| 30 |

|
17.36 |
| 31 |

|
>50 |
| 32 |

|
57.89 |
| 33 |

|
11.36 |
Based on the positive results obtained with compound 19, we used it as a query scaffold and identified compounds 22–41 (Table 1 and Figure S1c) as close analogues of compound 19. From these compounds, we identified compound 24 (Figure 2a), which showed inhibition against BRAF with an IC50 value of 1.7 μM (Figure 2b and Table 1). Taken together, our in vitro assay results established the compound 24 as the most potent BRAF inhibitors within this library of compound 19 analogues.
Figure 2.

Identification of compound 24 as a third generation BRAF specific inhibitor. (a) Molecular structure of compound 24; (b) Dose response curve of compound 24 inhibition against the BRAF kinase; (c) Kinase profiling of compound 24 against GSK3β, Pim1, PAK1 and PAK4 protein kinases and PI3Kγ lipid kinase.
Selectivity of compound 24 for BRAF
In order to probe the kinase selectivity of compound 24 against BRAF over other kinases, we selected 6 kinases, PI3Kγ, GSK3β, Pim1, MST1, PAK1 and PAK4 which represent 4 major kinase families; lipid kinases and CMGC, CAMK, and STE protein kinases. As shown in Figure 2c, compound 24 did not show significant inhibition against GSK3β, Pim1, MST1, PAK1 and PAK4 at an inhibitor concentration of 100 μM and it only showed PI3Kγ inhibition with an IC50 value of 25 μM. Our data establishes compound 24 as a BRAF inhibitor with a more than 50-fold selectivity over other representative protein kinases and more than a 12-fold selectivity over PI3K lipid kinases. Taken together, our kinase profiling data suggested that the HTVS approach described here can lead to the identification of low micromolar BRAF inhibitors with significant kinase inhibitor selectivity.
Molecular modeling of BRAF inhibitors
To understand the mode of inhibition of compounds 19 and 24 to BRAF, we docked these compounds into the active site of BRAF using AutoDock 3.0. As shown in Figure 4a, compound 19 is modeled to bind in the ATP binding pocket of BRAF such that one of the carbonyl oxygen atoms of the purine-2,6-dione ring forms a hydrogen bond with the amide nitrogen of residue Cys532 in the hinge region between the N-lobe and C-lobe of the BRAF kinase domain. In addition, the model suggests that extensive hydrophobic interactions are formed between compound 19 and the ATP binding pocket of BRAF kinase. In particular, residues Ile463, Ser466, Val471, Ala481, Val482, Lys483, Leu514, Thr529, Trp531, Phe583 and Phe595 are predicted to make van der Waals contacts with compound 19.
Figure 4.

Future directions for designing BRAF inhibitors with greater potency and specificity. (a) Base scaffold with three possible positions for modification; (b) Corresponding position of three modification sites within the ATP binding pocket of the BRAF protein kinase. Surface contours corresponding to the P-loop, Act. Loop (Activation loop) and Cat. loop (Catalytical loop) are color coded yellow, cyan and red, respectively.
In contrast to the compound 19, compound 24 is modeled to bind to the BRAF active site in a different orientation, apparently due to the bulky phenyl group derivatization on the maleimide nitrogen of the purine-2,6-dione ring (Figure 3b). Nonetheless, this phenyl group is predicted to bind essentially the same hydrophobic pocket as targeted by the chlorophenyl moiety of compound 19. In addition to extensive hydrophobic interactions that are predicted to form between BRAF and compound 24 with residues Ile463, Val471, Ala481, Ile527, Lys483, Leu514, Thr529, Trp531, Cys532 and Phe583, compound 24 also is modeled to make two additional hydrogen bonding interactions with Thr529 and Gly534 of BRAF. In particular, the thio-ethanol group of compound 24 forms hydrogen bonds with the amide carbonyl of Gly534 and the maleimide carbonyl of compound 24 also interacts with the side chain of Thr529 by forming a potential hydrogen bond. This model can also explain why compound 24 is a more potent inhibitor than compound 23 (Table 1).
Figure 3.

Docking of compound 19, 24 and 50 in the BRAF active site. (a) Interactions of compound 19 with the BRAF protein kinase ATP binding pocket. (b) Interaction of compound 24 with the BRAF protein kinase ATP binding pocket. Hydrogen bonding interactions are represented as red dashed lines in both panel (a) and panel (b). (c) Comparison of binding modes of compounds 19 (pink), 24 (blue) and 50 (yellow) in the ATP binding pocket of the BRAF kinase. Surface contours corresponding to the P-loop, Act. loop (Activation loop) and Cat. loop (Catalytical loop) are color coded yellow, cyan and red, respectively.
Comparison with other BRAF inhibitors
Several BRAF inhibitors, identified through solution screening, have been described in the literature including a bis-aryl urea compound, BAY43-9006 (Sorafenib)13 and a triarylimidazole derivative called compound 507. Although these compounds bind more potently to BRAF than compounds 19 and 24 that are described here (Table S2), identified through in silico screening, there are some interesting similarities and differences, particularly with the compound 50, that might be exploited to improve the BRAF potency of compounds 19 and 24 for BRAF. In particular, a comparison of the 19 and 24 inhibitors with the compound 50, in complex with BRAF7 shows significant similarities in their BRAF binding modes (Figure 3c). Like the compound 50 inhibitor, the purine-2,6-dione ring of both compounds 19 and 24 intercalates in between residues F583 and W531 by making a π−π stacking interaction7. In addition, the chlorophenyl moiety of compound 19 and the phenyl group of compound 24 adopt a similar orientation as the indane-oxime group of the compound 50 within the BRAF binding pocket with the bulky group pointing towards the BRAF activation loop where it may play an important role in BRAF selectivity7, 14. When compared with the compound 50, the chlorophenyl group of compound 19 and the phenyl group of compound 24 occupy significantly less space within the BRAF active site than the corresponding indane-oxime group of the compound 50 (Figure 3c). Based on this observation, we propose that analogs of compounds 19 and 24 containing additional hydrophilic modifications in this position may mediate additional protein interaction and shape complementarity near the activation loop and this strategy would be likely to increase BRAF inhibitor potency and specificity (Figure 4).
Our SAR analysis also reveals that bulkier substitutions on the thiol moiety significantly reduce BRAF inhibitor potency. For example compounds 31 and 32 which contains a nitrophenyl-hydrazine group in this position shows a least a 20-fold reduction in BRAF inhibitor potency relative to compound 19 (Table 1). There are also other compounds with bulky derivatives at this position that show reduced inhibitory potency against BRAF (Table 1). These results demonstrate that compounds with bulky substitutions in this position are not favorable for BRAF inhibition.
Conclusion and future prospects
We have identified a series of purine-2,6-dione analogues as novel inhibitors against the BRAF kinase using the HTVS approach from a filtered small molecule SPECS compound database containing about 90,000 compounds. Among these newly identified purine-2,6-dione based inhibitors, compound 1 was found to inhibit the enzymatic activity of the BRAF protein kinase in a ELISA based MEK phosphorylation assay with an IC50 value at 29 μM. Based on the molecular docking of this compound bound to BRAF, a fragment scaffold (scaffold 1a in Figure 1a) was hypothesized to be the functional moiety for BRAF inhibition. This scaffold was extracted as a query for searching structural analogues of compound 1 for testing their inhibition in an in vitro solution screen. The BRAF ELISA assay results established compound 19 from this library of analogues as a potent BRAF inhibitor with an IC50 value at 2.1 μM against BRAF. Based on the structure of this new inhibitor lead of compound 19, 12 compounds out of 20 compound 19 analogues were identified as low micro-molar inhibitors of BRAF kinase. The best inhibitor, compound 24 inhibits the kinase activity of BRAF with an IC50 value of 1.7 μM. We also demonstrated selectivity of compound 24 for BRAF over other representative protein and lipid kinases in our kinase profiling studies. Furthermore, the SAR studies of inhibitor analogues and docking studies of compounds 19 and 24 into the BRAF active site provides important pharmacophore clues for future inhibitor optimization to increase BRAF inhibitor potency and selectivity (Figure 4). In particular, compounds with modifications that extend the R1 position of the phenyl group with a long aliphatic chain with a hydrophilic terminal end might allow the compound analog to reach deeper into the BRAF binding pocket formed by the activation loop and the αC helix such that the hydrophilic extension interacts with residue Asp594 of BRAF (compound 28 in Table 1 and Figure S2). The significance of this modification is underscored when considering the fact that this pocket of the BRAFV600E oncogenic mutant protein may differ drastically from that of the BRAFWT protein due to the different conformations of the activation loop between BRAFWT and BRAFV600E 7, 14. Indeed, we have shown that compounds 19 and 24 do not distinguish between BRAFWT and BRAFV600E (data not shown) pointing to the potential importance of modifying the R1 position to obtain BRAF inhibitors with isoform and oncogene specificity. Derivatization of the thiol group at the R2 position may also increase inhibitor binding potency for BRAF, although large derivatizations in this position may prevent an R1 derivatization off the phenyl group from extending into the site near the activation loop. We therefore suggest that the derivatization of the R2 position with a relatively small substituent may be more favorable when combined with modifications in the R1 position. Indeed, the SAR analysis reveals that substitutions of a bulky group in the R2 position are not well tolerated when there is another bulky substitution in the R1 position (compound 27 in Table 1 and compounds 34, 35, 36 in Figure S1c). In the R3 position, the substitution of a bulky group such as a phenyl will likely change the binding conformation of the inhibitor in the BRAF ATP binding pocket, while modifications of the R3 position to a hydrophilic group would be in position to mediate hydrogen bonding interactions with protein pocket residues.
Taken together, our study demonstrates that an efficient and cost-effective virtual screening procedure can be used to identify potent BRAF inhibitors that also show considerable specificity for BRAF over other protein and lipid kinases. Moreover, the promising results obtained in this study will serve as an excellent platform for developing BRAF inhibitors with even greater potency and specificity for further development as therapeutic agents.
EXPERIMENTAL SECTION
High throughput virtual screening (HTVS)
The X-ray crystal structure of the BRAF kinase domain in an active configuration7 (PDB accession code: 2FB8) was used as the target structure in this approach. The modified small molecular database containing ~90,000 molecules for virtual screening was generated as a SPECS subset from the Zinc15 with a predicted solubility filter (logS > −4). The virtual screening was performed on a 4-processor Linux workstation and the National Science Foundation Terascale Computing System at the Pittsburgh Supercomputing Center (http://www.psc.edu). A heuristic docking and consensus scoring strategy was used to evaluate the results of the virtual screening. Specifically, DOCK4.0 was performed as the primary screening tool using the active site of the BRAF/compound 50 crystal structure with the inhibitor excluded from the coordinates (PDB accession code: 2FB8) as a receptor for compound binding. Residues around the compound 50 at a radius of 6 Å were isolated for constructing the grid for docking screening. This radius was large enough to include all the residues involved in inhibitor binding. During the docking procedure, Kollman-all-atom charges were assigned to the protein, and Geisterger-Hückel charges assigned to the small molecules in the SPECS database. In the docking search, the conformational flexibility of the molecules from the database was considered. Thirty configurations per ligand building cycle and 50 maximum anchor orientations were used in the anchor-first docking algorithm. All docked configurations were energy minimized using 100 maximum iterations and 1 minimization cycle. Prior to screening the target molecular database, this docking protocol was tested by screening a small molecule database containing 1,000 compounds consisting of two known BRAF inhibitors (Compounds 50 and 51) and 998 randomly selected low molecular weight compounds from the SPECS database, and this control experiment produced the two known BRAF inhibitors as the top scoring ligands. Following this control experiment, the target database was screened and the top 5,000 molecules were taken as the hit list for further analysis. These molecules were re-ranked using, in sucession, XSCORE (version 1.2.1)9, the dockingand-scoring mode of SLIDE (version 2.3.1)10, 11 and AutoDock3.0. Based on the results of these scoring functions, the molecules were ranked and the top 200 molecules were extracted and carefully considered for their receptor binding and scaffold diversity. Finally, 18 available candidate compounds from different scaffolds were purchased for in vitro assay.
Molecular docking by Autodock 3.0
The molecular docking program AutoDock 3.016, was used for the automated molecular docking simulations. The docking scheme is summarized as follows. First, the BRAF receptor molecule was checked for polar hydrogen and assigned for partial atomic charges. The PDBQS file was created and the atomic solvation parameters were also assigned. The torsion angles of the docked ligands that could be sampled during the molecular docking procedure were defined to permit a conformation search for ligands during the molecular docking process. Second, a 3D search grid was created using the AutoGrid algorithm17 to evaluate the binding energies between the ligands and the BRAF receptor. The receptor was embedded in the 3D grid and a probe atom was placed at each grid point. The affinity and electrostatic potential grid was calculated for various types of atoms in the ligands. The energetic configuration of a particular ligand was determined using tri-linear interpolation of affinity values and electrostatic interaction of the eight grid points around each atom of the ligand. Third, a series of the docking parameters were defined. The atom types, generations and the run numbers for the LGA algorithm were properly assigned according to the requirement of the Amber force field. The number of generations, energy evaluations, and docking runs were set to 370,000, 1,500,000, and 20, respectively. The atomic charges were assigned as Kollman all-atom for the BRAF receptor and Gasteiger-Marsili18 was used for the ligands. Finally, the docked complexes of ligand-receptor were selected according to the criteria of interacting energy combined with geometrical matching quality. These complexes were used as the starting conformation for further energetic minimization and geometrical optimization before the final binding models were generated.
Cloning, protein expression and purification
The full length human BRAF clone was kindly provided by Dr. Richard Marais (Institute of Cancer Research, United Kingdom) and the full length mouse p50cdc37 clone was provided by Dr. Wade Harper (Harvard Medical School). The human BRAF kinase domain (residues 433–726) with an N-terminal purification tag (MDRGSH6GS) and full length mouse p50cdc37 were PCR-amplified and subcloned into the pFastBac Dual vector. The BRAF kinase domain was expressed and purified essentially as previously described 3.
In vitro BRAF kinase assay
Recombinantly expressed GST-MEK diluted in TTBS buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.05% TWEEN-20) to 50 μg/mL was bound in 100 μL volume to the wells of a 96-well glutathione coated plate (Pierce Biotechnology). 1 μL of compound with 2× serial dilutions in 100% DMSO stock were added in a mixture of 50 μL 50 mM HEPES pH 7.0 with 25 ng of BRAF kinase. This mixture was incubated at room temperature for 1 hour before it was added into the GST-MEK treated wells of the 96-well plate. An additional 50 μL of phosphorylation buffer (50 mM HEPES pH 7.0, 200 mM NaCl, 10 mM MgCl2, 200 μM ATP) was added into this mixture to start the kinase reaction at 37°C for 30 min with intermittent shaking. The kinase reaction was stopped by extensive washing with TTBS buffer and a 1:5000 dilution of Anti-phospho-MEK1 (Ser218/222)/MEK2 (Ser222/226) monoclonal antibody (Millipore) in TTBS was subsequently added into the wells and incubated for 1 hour with shaking. Goat Anti-Rabbit IgG (H+L)-HRP Conjugate (BioRad Laboratories) in a 1:5000 dilution was finally added into the wells to incubate at room temperature with shaking. Finally, the SuperSignal ELISA Pico Chemiluminescent Substrate (Pierce Biotechnology) was added into the wells. The luminescence signal was recorded with a luminescence filter by a Wallac 1420 luminometer (PerkinElmer). These data were processed and IC50 values were derived from sigmoidal dose response curve fitting by GraphPad Prism 4.0.
Supplementary Material
Acknowledgement
We thank Dr. M.Y. Zheng from Shanghai Institute of Materia Medica, Chinese Academy of Sciences for kindly providing the ZlogS program for the solubility prediction on the small molecules in the SPECS database. We also thank Dr. Ruchi Anand for carrying out MST1 kinase assay and Jasna Maksimoska for carrying out GSK3β, Pim1, PAK1 and PAK4 kinase assays. We thank Dr. Richard Marais for providing the full length human BRAF clone and Dr. Wade Harper for providing the full length mouse clone. We thank the Wistar Protein Expression facility for preparing recombinant proteins. This work was supported by an NIH grants to R.M. (CA 94165)
Abbreviations
- RAS
small G protein Ras
- RAF
RAF proto-oncogene serine/threonine-protein kinase
- MEK
dual specificity mitogen-activated protein kinase kinase
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- PI3Kγ
phosphatidyl-inositol-3-kinase gamma isoform
- GSK3β
glycogen synthase kinase 3 beta isoform
- Pim1
proto-oncogene serine/threonine-protein kinase Pim 1
- MST1
STE20-like kinase MST1
- PAK
p21 activated kinase
- CMGC
CDK/MAPK/GSK3/CDK-like protein kinase family
- CAMK
calcium/calmodulin dependent kinase family
- STE
ste20 like protein kinase family.
Footnotes
Supporting Information Available: 1H NMR spectra for all compounds described, PI3Kγ, GSK3β, MST1, Pim1, PAK1 and PAK4 kinase assay procedures, a consensus ranking of 18 initial hits from HTVS, a comparison of compound 24 with conventional BRAF kinase inhibitors and additional references. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther. 2005;4(4):677–685. doi: 10.1158/1535-7163.MCT-04-0297. [DOI] [PubMed] [Google Scholar]
- 2.Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6(4):313–9. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 3.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad T, Eisen T. Kinase inhibition with BAY 43-9006 in renal cell carcinoma. Clin Cancer Res. 2004;10(18 Pt 2):6388S–92S. doi: 10.1158/1078-0432.CCR-040028. [DOI] [PubMed] [Google Scholar]
- 5.Vangrevelinghe E, Zimmermann K, Schoepfer J, Portmann R, Fabbro D, Furet P. Discovery of a Potent and Selective Protein Kinase CK2 Inhibitor by High-Throughput Docking. J. Med. Chem. 2003;46(13):2656–2662. doi: 10.1021/jm030827e. [DOI] [PubMed] [Google Scholar]
- 6.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proceedings of the National Academy of Sciences %R 10.1073/pnas.0609757104. 2007;104(18):7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, Kumar R, Rusnak DW, Takle AK, Wilson DM, Hugger E, Wang L, Karreth F, Lougheed JC, Lee J, Chau D, Stout TJ, May EW, Rominger CM, Schaber MD, Luo L, Lakdawala AS, Adams JL, Contractor RG, Smalley KS, Herlyn M, Morrissey MM, Tuveson DA, Huang PS. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66(23):11100–5. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 8.Kuntz ID, Meng EC, Shoichet BK. Structure-based molecular design. Acc. Chem. Res. 1994;27:117–23. [Google Scholar]
- 9.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46(12):2287–303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 10.Zavodszky MI, Sanschagrin PC, Korde RS, Kuhn LA. Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening. J Comput Aided Mol Des. 2002;16(12):883–902. doi: 10.1023/a:1023866311551. [DOI] [PubMed] [Google Scholar]
- 11.Schnecke V, Swanson CA, Getzoff ED, Tainer JA, Kuhn LA. Screening a peptidyl database for potential ligands to proteins with side-chain flexibility. Proteins. 1998;33(1):74–87. [PubMed] [Google Scholar]
- 12.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9(1):1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 14.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris GM, Goodsell DS, Huey R, Hart WE, Halliday RS, Belew RK, Olson AJ. Autodock Version 3.0.3. The Scripps Research Institute, Molecular Graphics Laboratory, Department of Molecular Biology. 1999. [Google Scholar]
- 17.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J. Comput. Chem. 1999;19:1639–1662. [Google Scholar]
- 18.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity-A rapid access to atomic charges. Tetrahedron. 1980;36:3219–28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.