

Abstract
Cardiac L-type calcium channel (CaV1.2), calmodulin (CaM), and Ca2+/calmodulin-dependent protein kinase II (CaMKII) form the CaV1.2/CaM/CaMKII signaling pathway, which plays an important role in maintaining intracellular Ca2+ homeostasis. The roles of CaM and CaMKII in the regulation of CaV1.2 in Ca2+-dependent inactivation and facilitation have been reported; however, alterations in this signaling pathway in the heart after myocardial ischemia (MI) had not been well characterized. In this study, we investigated the dynamic changes in CaV1.2, CaM, and CaMKII mRNA and protein expression levels in the left ventricles of the heart following MI in rats. The MI model was induced by ligating the left anterior descending coronary artery; the rats were divided into the following five groups: the 6 h post-MI group (MI-6h), 24 h post-MI group (MI-24h), 1 week post-MI group (MI-1w), 2 weeks post-MI group (MI-2w), and the sham group. The mRNA levels were measured by quantitative real-time polymerase chain reaction and the protein expression was determined by western blotting and immunohistochemistry. There were no observable differences in the CaV1.2 mRNA and protein levels at the early stages of MI, but these levels decreased at MI-2w. Both the mRNA and protein levels of CaM increased at MI-6h, peaked at MI-24h, and then reduced to normal levels at MI-2w. CaMKII mRNA and protein levels decreased at MI-6h and reached their lowest level at MI-24h. Taken together, these data demonstrate that there are dynamic changes in the CaV1.2/CaM/CaMKII signaling pathway following MI injuries, which suggests that different therapeutic regimens should be used at different time points after MI injuries.
Introduction
Myocardial ischemia (MI) is a complex pathophysiological process that involves various factors and pathways; one generally accepted initiation mechanism is Ca2+ overload. The voltage-dependent L-type Ca2+ channel (LTCC) is the major route for Ca2+ to enter cells. It had to control diverse physiological functions, including gene transcription, rhythmic firing, synaptic transmission, hormone secretion, and excitation-contraction coupling (Parkash, 2008). Four members of the CaV1 family (CaV1.1–CaV1.4) are present in the LTCC; CaV1.2 is mainly present in cardiomyocytes (Davies et al., 2007; Yang et al., 2011). In addition to physiological functions, changes in density or functions of the LTCC have been implicated in a variety of cardiovascular diseases, including atrial fibrillation (Grandi et al., 2011), heart failure (He et al., 2001), and ischemic heart disease (Aggarwal and Boyden, 1995). LTCC antagonists were widely used to treat cardiovascular diseases for decades. For example, they are frequently used to treat and manage ischemic heart disease. Additionally, these antagonists have been used as antiarrhythmic drugs (class IV) for supraventricular arrhythmias and to reduce blood pressure in patients with hypertension in order to decrease the risks of a major cardiovascular event (Fujino et al., 2005).
To date, many studies have elucidated the regulatory mechanisms of the LTCC. The functions of the LTCC are tightly controlled by protein kinase A (PKA) through phosphorylation mediated by β-adrenergic receptor and protein kinase C (PKC) through different signaling mechanisms based on different isoforms (Kamp and Hell, 2000). The LTCC is also regulated by calmodulin (CaM) and Ca2+/calmodulin-dependent protein kinase II (CaMKII), which induce dramatic changes in Ca2+ signaling. Our previous studies showed that CaM and CaMKII play distinct roles in CaV1.2 regulations in which the CaMKII phosphorylation site is slowly dephosphorylated during run-down of the channels; the effects of CaM are attenuated over time. Phosphorylation by CaMKII facilitates and prolongs the effects of CaM on the channel, and both CaMKII and CaM are required to reverse the run-down of the LTCC (Hao et al., 2008, 2009).
CaM is the primary intracellular Ca2+-binding protein and consists of two lobes with two Ca2+-binding sites in each lobe. CaM changes its conformation when Ca2+ is bound; therefore, it has been considered a Ca2+ sensor in various Ca2+ signaling cascades. CaMKII contains both Ca2+- and CaM-binding sites that are essential for signal transduction. CaMKII is activated through elevations in intracellular Ca2+ levels and, in turn, adjusts Ca2+ homeostasis in many ways. Autophosphorylation of CaMKII could activate inhibitory phosphorylation sites, which prevents further activation of CaMKII. Recent studies have highlighted the roles of CaM and CaMKII in the regulation of CaV1.2, especially in Ca2+-dependent inactivation (CDI) and Ca2+-dependent facilitation (CDF). CaM is necessary and could possibly be sufficient for the mediation of CDF and CDI in the LTCC, whereas CaMKII only affects their time courses. Thus, CaM is a key molecule in bifurcating the Ca2+ signal to CDF or CDI, and CaMKII plays a modulatory role in both pathways (Nie et al., 2007). However, it cannot be predicted how CaV1.2, CaM, and CaMKII change in the left ventricular myocardium during MI. Based on previous results, we hypothesize that the CaV1.2/CaM/CAMKII signaling pathway might be involved in myocardial impairments during MI.
To date, changes in the CaV1.2/CaM/CaMKII signaling pathway following MI have not been well documented. Many clinical studies have suggested that treatment timing for myocardial infarction is vital for healing. Thus, this study was conducted to examine the dynamic changes in the CaV1.2/CaM/CaMKII signaling pathway in the left ventricular myocardium following MI in rats. Our current data demonstrated that the CaV1.2/CaM/CaMKII signaling pathway changes differently following MI at different stages, suggesting that the current therapeutic regimen should be modified for different time points following MI.
Materials and Methods
MI model preparation
Experiments were performed using Wistar rats weighing 250–300 g, supplied by the Laboratory Animal Centre of China Medical University (SCXK Liao, No. 2008-0005). All animals received humane care in compliance with the Principles of Laboratory Animal Care (NIH publication No. 85-23, revised 1985). All animal experiments were approved by the Animal Care and Use Committee of China Medical College.
Rats were anesthetized by intraperitoneal injection with 1% sodium pentobarbital (40 mg/kg) and subsequently placed in supine position. Three electrocardiogram (ECG) limb electrodes were then connected. Under sterile conditions, the rats' hearts were exposed with a 2-cm left lateral thoracotomy. The left anterior descending coronary artery was completely ligated with a 3/8 circular needle followed by a 5/0 silk thread through the shallow layer of the myocardium. To make it completely consistent, the ligation procedure should be finished in a fixed position with similar strength. If the ligation was not firm, then the rat would be excluded from the test group. The heart was subsequently put back into the thoracic cavity, and the chest wall was sewn up. Induction of MI was successful when ST-segment elevation continued for 30 min (Zhao et al., 2009). The percentage for the infarct was measured by the quantitative histochemical staining method (Harnarayan et al., 1970). The ratio of the infarct area to total heart area was 38.36%±8.59%. Specimens were collected from the infarct area of left ventricular anterior wall after ligation of coronary artery.
Experimental animals were randomly divided into four groups: 6 h post-MI group (MI-6h), 24 h post-MI group (MI-24h), 1 week post-MI group (MI-1w), and 2 weeks post-MI group (MI-2w). The sham group rats underwent the same procedure except for the ligation process. Infection after operation was prevented through the use of the antibiotic penicillin at 100,000 U/day (i.p. injection for 3 days). The rats were killed at similar times and their blood was collected from the abdominal aorta; then, the heart was immediately removed and the infarcted left ventricular tissues were harvested at different time points after the operation.
Lactate dehydrogenase determination
Lactate dehydrogenase (LDH) release in the blood serum was evaluated at different time points after MI. LDH levels were determined by a colorimetric method according to the manufacturer's instructions.
Hematoxylin-eosin staining
Rats were sacrificed at the designated time points and their hearts were quickly harvested from the thoracic cavity and immersed in paraformaldehyde for 48 h. Fixed tissues were embedded in paraffin and 5-μm-thick histological sections were prepared. The sections were mounted onto slides and stained with hematoxylin and eosin (HE). To avoid air oxidation and stain fading, the sections were mounted with gum for microscopic examination and photography. The morphological features of myocardial tissue were observed under a light microscope for analysis of myocardium injuries.
Quantitative real-time polymerase chain reaction analysis of Cav1.2, CaM, and CamKII mRNA expression
Total RNA was extracted from ∼50 mg of myocardial tissue samples using Trizol reagent (Invitrogen); 0.5 μg of total RNA was used for first-strand cDNA synthesis with the RT reagent kit (TaKaRa). The reaction conditions were followed based on the instructions in the manufacturer's protocols: 30°C for 10 min, 42°C for 30 min, 99°C for 5 min, and 5°C for 5 min. Then, quantitative real-time polymerase chain reaction (qRT-PCR) was performed after RT with dilutions of cDNA template with SYBR premix Ex Taq II (TaKaRa). The reaction conditions were as follows: 95°C for 30 s, 95°C for 5 s, and 60°C for 30 s, 40 cycles. The following primer sequences were used: Cacna1c, sense 5′-gatgcaagacgctatgggctatga g-3′ and antisense 5′-gcatgctcatgt-ttcggggtttgt c-3′; Cam, sense 5′-atggctgaccagctg ac-3′ and antisense 5′-ctttgcagtcatcat c-3′; Camk2, sense 5′-ggttcaccgacgagtatc ag-3′ and antisense 5′-gtcttcaaagagttcgcc ac-3′; and Gapdh, sense 5′-caacgaccccttcattga cc-3′ and antisense 5′-gaagacgccagtagactc ca-3′. Each primer pair produced a single band after amplification with a single melting curve. Standard curves of the purpose gene were generated with reverse-transcribed RNA from the heart tissue and analyzed using quintupled serial dilutions. Concentrations were determined by comparing the Ct values of the samples to the standard curves. The results have been expressed as relative expression compared with the Gapdh expression.
Western blot analysis of CaV1.2, CaM, and CaMKII protein expression
Proteins were extracted from ∼100 mg of myocardial tissue samples followed by tissue homogenization and sonification in RIPA lysis buffer with a cocktail of proteinase inhibitors. Two well-known phosphatase inhibitors, NaF and Na3VO4, were used in this study. Total protein levels were determined using a Bradford protein assay kit. Aliquots of protein extracts from the samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to the polyvinylidene difluoride membrane (Millipore) by electrophoresis. Membranes were blocked with 5% non-fat milk blocking buffer in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature and incubated with the indicated primary antibodies overnight at 4°C. The following primary antibodies were prepared according to manufacturer's instructions: mouse anti-CaM (1:500; Santa Cruz), mouse anti-CaMKII (1:500; Santa Cruz), rabbit anti-p-CaMKII (1:500; Santa Cruz), and rabbit anti-CaV1.2 (1:500; Sigma). Unbound antibodies were removed by repeated washing in TBST. Horseradish peroxidase-conjugated sheep anti-rabbit or sheep anti-mouse immunoglobulin IgG (1:5000) was used as the secondary antibody; the membranes were incubated for 2 h at room temperature. The immunoreactive proteins were detected by enhanced chemiluminescence. The optical density of each protein band was analyzed with the Fluor Chem V2.0 software.
Immunohistochemistry analysis of CaV1.2, CaM, and CaMKII proteins
The left ventricles were immersed in 4% paraformaldehyde for 48 h and embedded in paraffin blocks. Five-micrometer sections were cut, deparaffinized, rehydrated, and incubated in 3% H2O2 at 37°C for 25 min. Slides were treated with citric acid buffer at 95°C for 10 min, and non-specific binding was blocked using goat serum in PBS buffer at 37°C for 30 min. The sections were then incubated with primary antibodies against rat CaM, CaMKII, or CaV1.2 overnight at 4°C. Slides were then incubated with biotinylated secondary antibody at 37°C for 30 min. The antibody complex was detected with 30-min incubation in a streptavidin-biotin-horseradish peroxidase complex at 37°C. Color was developed with diaminobenzidine, which generated a brown reaction byproduct. Slides were counterstained with hematoxylin for 5 min, and mounted with gum. The preparations were examined under a light microscope.
Statistical analysis
The data are presented as mean±standard error. Statistical analysis was performed with the SPSS13.0 statistical software followed by ANOVA; p<0.05 was considered statistically significant for differences between the sham and experimental groups.
Results
Pathological changes of MI in the left ventricular myocardium
ECGs were recorded before and after coronary artery ligation and successful infarction was confirmed by ST-segment elevation (Fig. 1A). A 4.5-fold increase in LDH released into the serum was also observed at MI-6h, compared with that of the sham group; levels returned to normal at MI-24h (Fig. 1B). Variable degrees of myocardial injury at MI-24h and MI-2w were observed through HE staining (Fig. 1C). In the sham group, the myocardial cells were well arranged, with clear cellular nuclei, and the cytoplasm was uniformly stained. In MI-24h group, acute heart injury characterized with neutrophil infiltration, interstitial edema, and partial rupture was observed. It was revealed that there was marked increase of neutrophil infiltration and hyperplastic fibroblast infiltration in the MI-2w heart tissues.
FIG. 1.

Pathologic changes in myocardial ischemia (MI) rats. Change in electrocardiograms (ECGs) of MI rats. (A) Normal ECG was detected in normal rats (upper panel) and a marked ST-segment elevation was detected in MI rats (lower panel). Note: arrow shows the ST-segment elevation. (B) Levels of lactate dehydrogenase (LDH) released in rat serum during MI (**p<0.01). (C) Representative slides showing hematoxylin-eosin staining in the left ventricular myocardium of rats (400×). The interstitial edema, neutrophil infiltration, and myocardial disorganization were observed at MI-24h (shown as arrows). Increased neutrophil infiltration and hyperplastic fibroblast infiltration were observed at MI-2w (shown as arrows).
Pathological changes differed between the early and later stages of MI injury. In the early stages, ST-segment elevation and increased LDH release were apparent, and HE staining showed acute myocardium injury. In the later stages, LDH release approached normal levels, and HE staining showed chronic myocardium injury. Therefore, the pathological changes differed between different stages of MI injury.
Messenger RNA and protein expression of CaV1.2, CaM, and CaMKII in the left ventricular myocardium of MI rats
Four different time points were selected to evaluate the changes in mRNA and protein levels after MI. The mRNA levels of CaV1.2 were evaluated by qRT-PCR (Fig. 2A); CaV1.2 mRNA levels in the left ventricle of MI rats did not change in MI-6h or MI-24h group, but were lower at MI-2w than the levels of the sham group (0.65±0.05 vs. 1.00±0.04, respectively; p<0.05). Next, changes in CaM mRNA and protein levels after MI were evaluated. CaM mRNA expression in the left ventricles of MI rats was significantly higher than that in the sham group at MI-6h (2.44±0.10 vs. 1.00±0.06, respectively; p<0.01) and peaked at MI-24h (3.11±0.10 vs. 1.00±0.06, respectively; p<0.01); levels were similar to those of the sham group at MI-2w (1.35±0.14 vs. 1.00±0.06, respectively; Fig. 2B). Since phospho-CaMKII has been shown to regulate the activity of CaV1.2, we later examined the expression of CaMKII and phospho-CaMKII in the hearts of rats with MI. Our data showed that CaMKII decreased significantly at MI-6h and MI-24h. CaMKII mRNA expression in the left ventricles of MI rats was significantly lower than that of the sham group at MI-6h (0.28±0.07 vs. 1.00±0.08, respectively; p<0.01); it reached its lowest level at MI-24h (0.11±0.09 vs. 1.00±0.08, respectively; p<0.01) and then recovered to 60% of the sham levels at MI-2w (0.60±0.04 vs. 1.00±0.08, respectively; p<0.01; Fig. 2C).
FIG. 2.
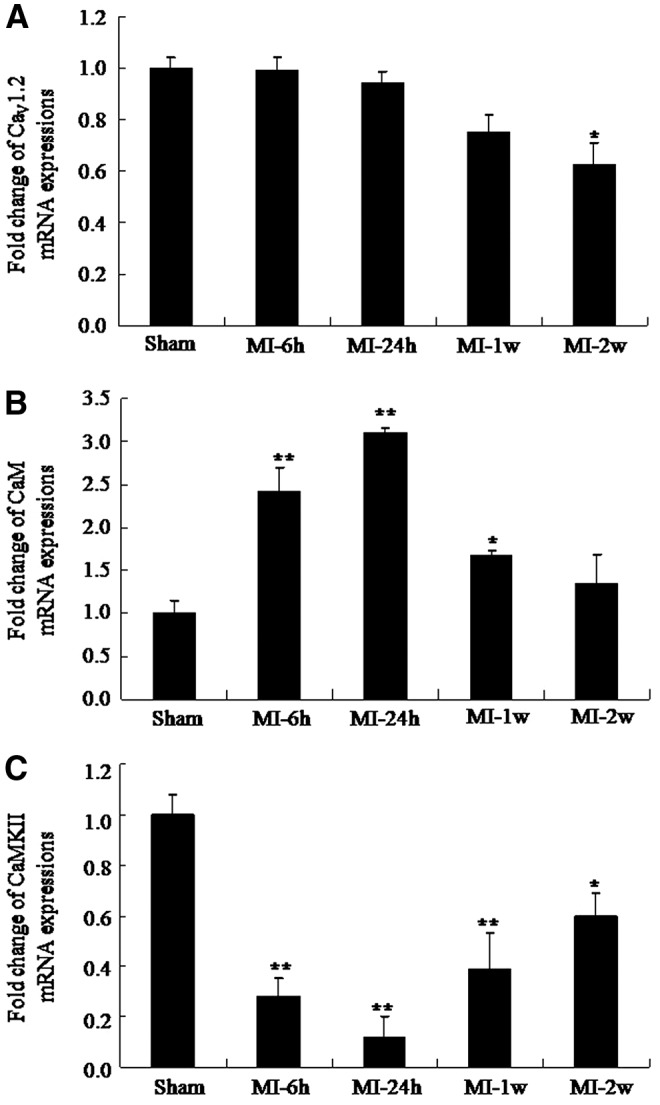
Dynamic change in Cacna1c, Cam, and Camk2 mRNA expression in the left ventricular tissue of MI rats. (A) Cacna1c mRNA levels (mean±standard error [SE], n=6, *p<0.05 compared with sham group). (B) Cam mRNA levels (mean±SE, n=6, *p<0.05 **p<0.01 compared with sham group). (C) Camk2 mRNA levels (mean±SE, n=6, *p<0.05 **p<0.01 compared with sham group).
Similarly, western blot results showed that protein levels did not change at MI-6h and MI-24h, but that the levels decreased at MI-2w (0.43±0.17 vs. 1.00±0.12, respectively; p<0.05; Fig. 3A, B). These findings indicate that CaV1.2 mRNA and protein levels post-MI did not change in the early stages, but markedly decreased during the later stages of MI injury. Protein levels of CaM were also higher than that of the sham group at MI-6h (2.10±0.50 vs. 1.00±0.38, respectively; p<0.05) and peaked at MI-24h (3.06±0.65 vs. 1.00±0.38; p<0.01); the levels were similar to those of the sham group at MI-2w (0.72±0.39 vs. 1.00±0.38, respectively; Fig. 3A, C). The protein levels of CaMKII and phospho-CaMKII were significantly lower than the sham group levels at MI-24h (0.41±0.03 vs. 1.00±0.04 and 0.37±0.08 vs. 1.00±0.02, respectively; p<0.05) and then recovered to 72% and 66% at MI-2w compared with sham levels (0.72±0.01 vs. 1.00±0.04 and 0.66±0.07 vs. 1.00±0.02, respectively; Fig. 3A, D, E). However, we found no differences between phospho-CaMKII and total CaMKII levels (p>0.05). The GAPDH signal confirms that unequal loading of the samples did not account for the differences observed.
FIG. 3.

Dynamic change in CaV1.2, calmodulin (CaM), calmodulin-dependent protein kinase II (CaMKII), and phospho-CaMKII protein expression in the left ventricular tissue of MI rats. (A) Representative protein bands of CaV1.2, CaM, CaMKII, and phospho-CaMKII in MI rats. (B) CaV1.2 protein levels (mean±SE, n=6, *p<0.05 compared with sham group). (C) CaM protein levels (mean±SE, n=6, *p<0.05 **p<0.01 compared with sham group). (D, E) CaMKII and phospho-CaMKII protein levels (mean±SE, n=6, *p<0.05).
Immunohistochemical staining for CaM showed that more scattered brown color dye within the cytoplasm was observed at MI-24h compared with that for the sham group (Fig. 4A). These results showed that CaM levels increased significantly at the early stages, but gradually recovered to normal levels at the later stages of MI. The levels of CaM mRNA and protein that differed at different time points after MI injury would influence CaV1.2 in different ways throughout the course of MI. Immunostaining for phospho-CaMKII showed that there was lesser brown color dye within the cytoplasm at MI-24h compared with that for the sham group (Fig. 4B). These results suggested that the decreased expression of CaMKII and phospho-CaMKII at early stages of MI was, at least in part, a negative feedback mechanism to reduce Ca2+ overload induced by increased CaM levels.
FIG. 4.

Expression of CaM and phospho-CaMKII in the left ventricular myocardium of rats (400×). (A) Very slim-stained CaM in cell membrane and cytoplasm in sham while increased staining in the cytoplasm regions was observed in the MI-24h. (B) Immunohistochemical expression of phospho-CaMKII showed slim stain in the cytoplasmic region in the MI-24h group.
Discussion
The present study demonstrated that CaV1.2 mRNA and protein levels did not change at the early stages of MI (MI-6h and MI-24h), but decreased significantly at the later stages (MI-1w and MI-2w) in rats. Consistent with our data, the Ca2+ channel α1c subunit expression decreased dramatically in the ventricle in ischemic hypertrophic myocardium at 6 weeks after MI (Huang et al., 2008; Goonasekera et al., 2012). In the failing mouse heart, the investigators consistently observed a significant reduction in α1c protein after 8 weeks of pressure overload stimulation (Goonasekera et al., 2012). Since CaV1.2 levels decreased at the later stages of MI, LTCC blockers have either no effect or can negatively affect the survival and cardiovascular events at this time point in clinical trials (Mahe et al., 2003). An increase in cytoplasmic Ca2+ concentration in myocardial cells with hypoxia has been reported (Song et al., 2005; Liu et al., 2009), and intracellular Ca2+ overload is an important cause for damage and dysfunction in the myocardium (Tang et al., 2011; Dhalla et al., 2012). As CaV1.2 is involved in the major route for Ca2+ to enter into myocardial cells, the decreased expression of CaV1.2 and the overload of intracellular Ca2+ seemed to be paradoxical, which suggests that intracellular Ca2+ overload of MI may be related to other regulatory proteins of CaV1.2, such as Na+/Ca2+ exchanger, Na+/H+ exchanger, RyR, and SERCA (Zima and Blatter, 2006; Vittone et al., 2008). CaMKII may regulate a variety of Ca2+-dependent proteins, such as LTCC, intracellular Ca2+ release ryanodine receptor channels, and phospholamban (PLB). LTCC opening is primarily initiated by cell membrane depolarization, but opening probability is also regulated by phosphorylation and this is the mechanism by which CaMKII increases Ca2+ entry. CaMKII phosphorylation causes LTCCs to enter a high-activity gating mode (Dzhura et al., 2000). Ryanodine receptor 2 (RYR2) was a CaMKII-regulated protein that was involved in the regulation of the intranuclear Ca2+ concentration. Currie et al. (2004) have reported that CaMKII may promote activation of RYR2 of the sarcoplasmic reticulum (SR), thereby increasing RyR2 Ca2+ sensitivity and spontaneous release of Ca2+ from internal stores. The PLB was also defined as a CaMKII-regulated phosphoprotein (Sag et al., 2007). CaMKII could cause increased PLB phosphorylation at T17, which might help preserve SR Ca2+ and make PLB unable to inhibit SERCA2a, thereby increasing Ca2+ reuptake into intracellular stores (Schwoerer et al., 2008). In this study, our findings revealed that there was a significant reduction of CaMKII levels in the acute phase of ischemia, which might decrease the activation of LTCC and RYR2 and intracellular Ca2+ concentration. Phospho-CaMKII may promote activation of RYR2 of the SR, thereby increasing RyR2 Ca2+ sensitivity and spontaneous release of Ca2+ from internal storage (Currie et al., 2004). Further, phospho-CaMKII could cause increased PLB phosphorylation at T17, which might help preserve SR Ca2+, and disabled PLB's ability to inhibit SERCA2a, thereby increasing Ca2+ reuptake into intracellular stores (Schwoerer et al., 2008). In this study, our findings revealed a significant reduction of CaMKII levels in the acute phase of ischemia, which might decrease the activation of LTCC and RYR2 and intracellular Ca2+ concentration.
The cyclic adenosine monophosphate (cAMP)–dependent PKA, a multifunctional enzyme that could be modulated by intracellular Ca2+ concentrations, may phosphorylate several proteins, such as LTCC and PLB and RyR2. The phosphorylation of LTCC causes the Ca2+ influx and a Ca2+-induced Ca2+ release, thereby increasing intracellular Ca2+ concentrations. The phosphorylation of PLB accelerates Ca2+ uptake into the SR. The phosphorylation of RyR2 may increase RyR2 Ca2+ sensitivity and the spontaneous release of Ca2+ from internal storage (Leineweber et al., 2006). Previous studies demonstrated that β-adrenoceptor is generally activated during MI (Schomig et al., 1995; Ungerer et al., 1996). Noradrenaline binds to β-adrenoceptor to activate stimulating adenylate cyclase G protein (Gs). Then, adenylate cyclase is activated by Gs, which turns adenosine triphosphate (ATP) to cAMP. The cAMP in turn activates PKA and increases intracellular Ca2+ concentrations, thereby activating CaM/CaMKII (Lu et al., 2012). In this study, the results indicated that the reduction of CaMKII levels is a negative feedback regulator of cellular Ca2+ overload, which could reduce the extent of ischemic myocardial injury.
In our study, CaM mRNA and protein levels considerably increased at the early stages of MI and recovered to normal levels at the later stages in rats with MI, which suggested that CaM changed in different modalities during MI to regulate CaV1.2 in different ways. Our previous study has shown that the run-down of CaV1.2 activity is reversed by CaM and ATP (Xu et al., 2004). The direct binding of CaM with the IQ motif of the Ca2+-channel α1-subunit has been reported to be involved in the molecular mechanism by which it exerts its Ca2+-dependent effects, namely, CDI and CDF. Based on our previous and current findings, we propose that, under the condition of MI, the relative increase in CaM in cardiomyocytes at the early stages may facilitate CaV1.2 channel activities through CDF and enhance Ca2+ inflow into cells, while increased [Ca2+] may result in CDI to avoid Ca2+ overload and myocardial damage. Consistent with our results, inhibition of CaM was found to be essential for cardioprotection in the ischemic heart (Afanas'ev et al., 2006; Fukunaga et al., 2006). In this study, decreased CaV1.2 expression and Ca2+ overload can be explained by the increased CaM expression. Therefore, our findings have established a link between increasing CaM and MI, and have defined an essential role of CDF due to CaM.
In this current study, CaMKII and phospho-CaMKII mRNA and protein levels had considerably decreased at the early stages of MI in rats and inclined to return to normal levels at the later stages, which shows the dynamic changes during MI and regulation of CaV1.2 in different ways. CaMKII is activated through increases in intracellular Ca2+/CaM, which in turn maintains Ca2+ homeostasis. In our previous study, CaM and CaMKII had distinct roles in the reversal of run-down; in that, CaM served to activate the channel and CaMKII-mediated phosphorylation facilitated the effects of CaM. Dzhura et al. (2000) reported that the single LTCC activity could be facilitated by CaM in inside-out patches, but only in the presence of CaMKII. The changes in the expression of CaMKII and CaM may be related to transcription initiation and mRNA accumulation. Further, altered degradation of proteins may also be involved in the expression of CaMKII and CaM. Here, we propose another possible mechanism in which decreased levels of CaMKII and phosphor-CaMKII may serve as a negative feedback to regulate CDF that was caused by CaM to avoid Ca2+ influx through the LTCC in MI. The CDF and CDI are two continuous processes accompanied by an increase in cellular Ca2+. The CDF was first induced and then followed by an increase in Ca2+ channel open probability; then, the CDI immediately appeared. However, the excess of CDF might lead to Ca2+ overload and inhibit the occurrence of CDI (Pitt et al., 2001). Uemura has suggested a molecular role of CaMKII in the ischemic heart, which appeared to be characteristic features reversible and immediate translocation of substantial amounts of CaMKII, and reduced autophosphorylation (Uemura et al., 2002). In addition, the dynamic changes in CaMKII and phospho-CaMKII can also regulate other ion channels of cardiomyocytes, such as potassium and sodium channels (Mori et al., 2000; El-Haou et al., 2009), which have been shown to be involved in Ca2+ homeostasis in cells.
In conclusion, this study is the first, to our knowledge, to show dynamic changes in the CaV1.2/CaM/CaMKII signaling pathway in rats with MI. The most interesting finding was that the expression of CaV1.2 did not change at early stages, but decreased during later stages of MI. CaM, an important regulator protein of the LTCC, was upregulated at the early stages of MI. In contrast, CaMKII was downregulated at the early stages of MI. Thus, it would be reasonable to conclude that decreased expression of CaMKII might serve as a negative feedback to inhibit Ca2+ overload. The CaV1.2/CaM/CaMKII signaling pathway is a promising therapeutic target for preventing or alleviating cardiac function after MI; we propose that current clinical therapeutic regimens should be modified depending on the time point after MI.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (30870907, 31071004, and 81100108) and the Scientific Research Project of the Education Department of Liaoning Province (L2010572). The authors would like to acknowledge the reviewers for their helpful comments on this article.
Disclosure Statement
The authors have declared that no competing interests exist.
References
- Afanas'ev S.A., Kozlov B.N., Korovin N.V., and Shipulin V.M. (2006). Inhibition of calmodulin prevents spasms in autologous arterial bypass grafts during surgical treatment of ischemic heart disease. Fiziol Cheloveka 32, 38–41 [PubMed] [Google Scholar]
- Aggarwal R., and Boyden P.A. (1995). Diminished Ca2+ and Ba2+ currents in myocytes surviving in the epicardial border zone of the 5-day infarcted canine heart. Circ Res 77, 1180–1191 [DOI] [PubMed] [Google Scholar]
- Currie S., Loughrey C.M., Craig M.A., and Smith G.L. (2004). Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biocheml J 377, 357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A., Hendrich J., Van Minh A.T., Wratten J., Douglas L., and Dolphin A.C. (2007). Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci 28, 220–228 [DOI] [PubMed] [Google Scholar]
- Dhalla N.S., Rangi S., Babick A.P., Zieroth S., and Elimban V. (2012). Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev 17, 671–681 [DOI] [PubMed] [Google Scholar]
- Dzhura I., Wu Y., Colbran R.J., Balser J.R., and Anderson M.E. (2000). Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol 2, 173–177 [DOI] [PubMed] [Google Scholar]
- El-Haou S., Balse E., Neyroud N., Dilanian G., Gavillet B., Abriel H., Coulombe A., Jeromin A., and Hatem S.N. (2009). Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res 104, 758–769 [DOI] [PubMed] [Google Scholar]
- Fujino T., Hasebe N., and Kikuchi K. (2005). Evidence-based usefulness of Ca antagonists and ACEIs and ARBs for the primary and secondary prevention of major cardiovascular and renal events in patients with hypertension. Clin Calcium 15, 1695–1708 [PubMed] [Google Scholar]
- Fukunaga K., Han F., Shioda N., Moriguchi S., Kasahara J., and Shirasaki Y. (2006). DY-9760e, a novel calmodulin inhibitor, exhibits cardioprotective effects in the ischemic heart. Cardiovasc Drug Rev 24, 88–100 [DOI] [PubMed] [Google Scholar]
- Goonasekera S.A., Hammer K., Auger-Messier M., Bodi I., Chen X., Zhang H., Reiken S., Elrod J.W., Correll R.N., York A.J., Sargent M.A., Hofmann F., Moosmang S., Marks A.R., Houser S.R., Bers D.M., and Molkentin J.D. (2012). Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J Clin Invest 122, 280–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi E., Pandit S.V., Voigt N., Workman A.J., Dobrev D., Jalife J., and Bers D.M. (2011). Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res 109, 1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L.Y., Wang W.Y., Minobe E., Han D.Y., Xu J.J., Kameyama A., and Kameyama M. (2009). The distinct roles of calmodulin and calmodulin kinase II in the reversal of run-down of L-type Ca2+ channels in guinea-pig ventricular myocytes. J Pharmacol Sci 111, 416–425 [DOI] [PubMed] [Google Scholar]
- Hao L.Y., Xu J.J., Minobe E., Kameyama A., and Kameyama M. (2008). Calmodulin kinase II activation is required for the maintenance of basal activity of L-type Ca2+ channels in guinea-pig ventricular myocytes. J Pharmacol Sci 108, 290–300 [DOI] [PubMed] [Google Scholar]
- Harnarayan C., Bennett M.A., Pentecost B.L., and Brewer D.B. (1970). Quantitative study of infarcted myocardium in cardiogenic shock. Br Heart J 32, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Conklin M.W., Foell J.D., Wolff M.R., Haworth R.A., Coronado R., and Kamp T.J. (2001). Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res 49, 298–307 [DOI] [PubMed] [Google Scholar]
- Huang K., Huang D., Fu S., Yang C., and Liao Y. (2008). Abnormal calcium “sparks” in cardiomyocytes of post-myocardial infarction heart. J Huazhong Univ Sci Technolog Med Sci 28, 401–408 [DOI] [PubMed] [Google Scholar]
- Kamp T.J., and Hell J.W. (2000). Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res 87, 1095–1102 [DOI] [PubMed] [Google Scholar]
- Leineweber K., Bohm M., and Heusch G. (2006). Cyclic adenosine monophosphate in acute myocardial infarction with heart failure: slayer or savior? Circulation 114, 365–367 [DOI] [PubMed] [Google Scholar]
- Liu X.H., Chen P.F., Pan L.L., Silva R.D., and Zhu Y.Z. (2009). 4-Guanidino-n-butyl syringate (Leonurine, SCM 198) protects H9c2 rat ventricular cells from hypoxia-induced apoptosis. J Cardiovasc Pharmacol 54, 437–444 [DOI] [PubMed] [Google Scholar]
- Lu Y., Zheng Y., Liu X., Liang X., Ngai S., Li T., and Zhang W. (2012). Metabolomic profiles of myocardial ischemia under treatment with salvianolic acid B. Chin Med 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe I., Chassany O., Grenard A.S., Caulin C., and Bergmann J.F. (2003). Defining the role of calcium channel antagonists in heart failure due to systolic dysfunction. Am J Cardiovasc Drugs 3, 33–41 [DOI] [PubMed] [Google Scholar]
- Mori M., Konno T., Ozawa T., Murata M., Imoto K., and Nagayama K. (2000). Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry 39, 1316–1323 [DOI] [PubMed] [Google Scholar]
- Nie H.G., Hao L.Y., Xu J.J., Minobe E., Kameyama A., and Kameyama M. (2007). Distinct roles of CaM and Ca2+/CaM -dependent protein kinase II in Ca2+ -dependent facilitation and inactivation of cardiac L-type Ca2+ channels. J Physiol Sci 57, 167–173 [DOI] [PubMed] [Google Scholar]
- Parkash J. (2008). Inflammatory cytokine signaling in insulin producing beta-cells enhances the colocalization correlation coefficient between L-type voltage-dependent calcium channel and calcium-sensing receptor. Int J Mol Med 22, 155–163 [PubMed] [Google Scholar]
- Pitt G.S., Zuhlke R.D., Hudmon A., Schulman H., Reuter H., and Tsien R.W. (2001). Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 276, 30794–30802 [DOI] [PubMed] [Google Scholar]
- Sag C.M., Dybkova N., Neef S., and Maier L.S. (2007). Effects on recovery during acidosis in cardiac myocytes overexpressing CaMKII. J Mol Cell Cardiol 43, 696–709 [DOI] [PubMed] [Google Scholar]
- Schomig A., Richardt G., and Kurz T. (1995). Sympatho-adrenergic activation of the ischemic myocardium and its arrhythmogenic impact. Herz 20, 169–186 [PubMed] [Google Scholar]
- Schwoerer A.P., Neuber C., Schmechel A., Melnychenko I., Mearini G., Boknik P., Kirchhefer U., Schmitz W., Ehmke H., Eschenhagen T., and El-Armouche A. (2008). Mechanical unloading of the rat heart involves marked changes in the protein kinase-phosphatase balance. J Mol Cell Cardiol 45, 846–852 [DOI] [PubMed] [Google Scholar]
- Song L.S., Pi Y., Kim S.J., Yatani A., Guatimosim S., Kudej R.K., Zhang Q., Cheng H., Hittinger L., Ghaleh B., Vatner D.E., Lederer W.J., and Vatner S.F. (2005). Paradoxical cellular Ca2+ signaling in severe but compensated canine left ventricular hypertrophy. Circ Res 97, 457–464 [DOI] [PubMed] [Google Scholar]
- Tang H., Viola H.M., Filipovska A., and Hool L.C. (2011). Cav1.2 calcium channel is glutathionylated during oxidative stress in guinea pig and ischemic human heart. Free Radic Biol Med 51, 1501–1511 [DOI] [PubMed] [Google Scholar]
- Uemura A., Naito Y., and Matsubara T. (2002). Dynamics of Ca2+/calmodulin-dependent protein kinase II following acute myocardial ischemia-translocation and autophosphorylation. Biochem Biophys Res Commun 297, 997–1002 [DOI] [PubMed] [Google Scholar]
- Ungerer M., Kessebohm K., Kronsbein K., Lohse M.J., and Richardt G. (1996). Activation of beta-adrenergic receptor kinase during myocardial ischemia. Circ Res 79, 455–460 [DOI] [PubMed] [Google Scholar]
- Vittone L., Mundina-Weilenmann C., and Mattiazzi A. (2008). Phospholamban phosphorylation by CaMKII under pathophysiological conditions. Front Biosci 13, 5988–6005 [DOI] [PubMed] [Google Scholar]
- Xu J.J., Hao L.Y., Kameyama A., and Kameyama M. (2004). Calmodulin reverses rundown of L-type Ca2+ channels in guinea pig ventricular myocytes. Am J Physiol Cell Physiol 287, C1717–C1724 [DOI] [PubMed] [Google Scholar]
- Yang L., Katchman A., Morrow J.P., Doshi D., and Marx S.O. (2011). Cardiac L-type calcium channel (Cav1.2) associates with gamma subunits. FASEB J 25, 928–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.J., Wang J.H., Fu B., Ma M.X., Li B.X., Huang Q., and Yang B.F. (2009). Effects of 3-aminobenzamide on expressions of poly (ADP ribose) polymerase and apoptosis inducing factor in cardiomyocytes of rats with acute myocardial infarction. Chin Med J (Engl) 122, 1322–1327 [PubMed] [Google Scholar]
- Zima A.V., and Blatter L.A. (2006). Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71, 310–321 [DOI] [PubMed] [Google Scholar]