

Abstract
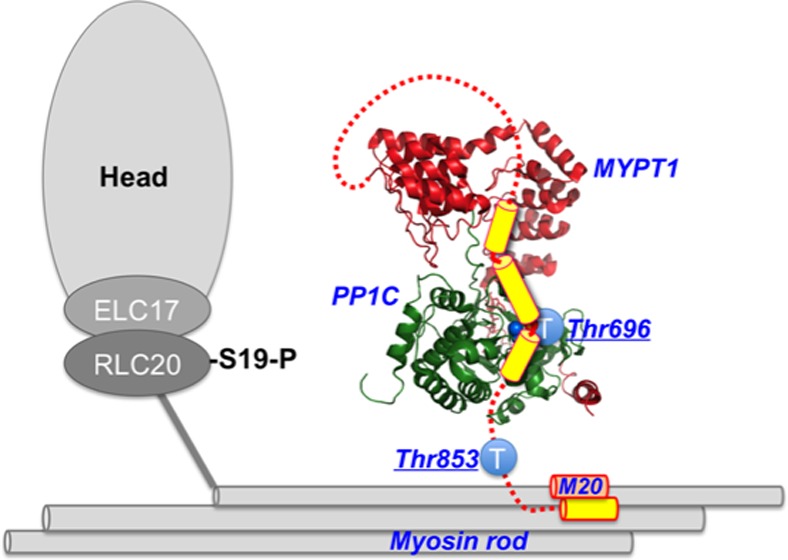
The myosin light chain phosphatase (MLCP) is a cytoskeleton-associated protein phosphatase-1 (PP1) holoenzyme and a RhoA/ROCK effector, regulating cytoskeletal reorganization. ROCK-induced phosphorylation of the MLCP regulatory subunit (MYPT1) at two sites, Thr696 and Thr853, suppresses the activity, although little is known about the difference in the role. Here, we developed a new method for the preparation of the recombinant human MLCP complex and determined the molecular and cellular basis of inhibitory phosphorylation. The recombinant MLCP partially purified from mammalian cell lysates retained characteristics of the native enzyme, such that it was fully active without Mn2+ and sensitive to PP1 inhibitor compounds. Selective thio-phosphorylation of MYPT1 at Thr696 with ROCK inhibited the MLCP activity 30%, whereas the Thr853 thio-phosphorylation did not alter the phosphatase activity. Interference with the docking of phospho-Thr696 at the active site weakened the inhibition, suggesting selective autoinhibition induced by phospho-Thr696. Both Thr696 and Thr853 sites underwent autodephosphorylation. Compared with that of Thr853, phosphorylation of Thr696 was more stable, and it facilitated Thr853 phosphorylation. Endogenous MYPT1 at Thr696 was spontaneously phosphorylated in quiescent human leiomyosarcoma cells. Serum stimulation of the cells resulted in dissociation of MYPT1 from myosin and PP1C in parallel with an increase in the level of Thr853 phosphorylation. The C-terminal domain of human MYPT1(495–1030) was responsible for the binding to the N-terminal portion of myosin light meromyosin. The spontaneous phosphorylation at Thr696 may adjust the basal activity of cellular MLCP and affect the temporal phosphorylation at Thr853 that is synchronized with myosin targeting.
Dynamic reorganization of the cytoskeleton is a fundamental process in cell motility. RhoA/ROCK signaling plays a dominant role in the regulation of cytoskeletal reorganization by inducing spatiotemporal phosphorylation of cytoskeletal proteins and their regulatory elements. Phosphorylation of myosin II regulatory light chains (MLC20) determines myosin motor activity, the affinity for actin filaments, and self-assembly in cells. The MLC20 phosphatase (MLCP) holoenzyme, consisting of the protein phosphatase-1 catalytic subunit δ (also named β) isoform (PP1C), myosin-targeting subunit MYPT1, and an accessory subunit M20,1−3 is a downstream effector of the RhoA/ROCK signaling axis.4 This trimeric PP1 holoenzyme also dephosphorylates other proteins, such as ERM, adducin, tau, merlin, and Rb, mediating RhoA signaling in the regulation of various cellular actions.5
Lines of evidence suggest that the MYPT1 regulatory subunit plays a central role in the spatiotemporal regulation of cellular MLCP (reviewed in refs (5) and (6)). The N-terminal 300-residue structural domain of MYPT1 forms a platform for the allosteric interaction with PP1C, and the interaction defines the substrate specificity and the sensitivity toward the endogenous inhibitor protein CPI-17.7−10 MYPT1 tethers PP1C to myosin filaments through interaction of the MYPT1 N-terminal domain with myosin subfragment-2 and/or the MYPT1 C-terminal 300-residue domain with the myosin rod domain.11,12 MYPT1 possesses multiple phosphorylation sites that negatively and positively regulate the cellular activity of MLCP.13−15 In permeabilized smooth muscle tissues, G-protein activation induces MLCP inhibition in parallel with MYPT1 thio-phosphorylation.16 ROCK phosphorylates MYPT1 at Thr696 and Thr853 (Thr695 and Thr850, respectively, in chicken MYPT1), and the phosphorylation suppresses MLCP activity.17,18 In addition, multiple kinases, such as a MYPT1-associated kinase (ZIPK), ILK, and PAK, are capable of selectively phosphorylating Thr696 and inhibiting the activity.19−21 In smooth muscle tissues, Thr696 phosphorylation is relatively insensitive to stimulus, whereas the level of Thr853 phosphorylation increases in response to G-protein activation.22−24 To fully interpret the physiological data showing the differential phosphorylation of MYPT1 into the MLCP activity in mammalian cells, we must know the roles of two inhibitory phosphorylation sites of human MYPT1 in the regulation.
It should be noted that characterization of MLCP has been mostly conducted using native or recombinant avian MLCP enzymes. Inhibition of the chimeric MLCP complex consisting of a full-length avian MYPT1 purified from bacteria lysates and isolated avian PP1C occurred upon phosphorylation at Thr696 but not Thr853.25 On the other hand, avian and human MYPT1 fragments that are phosphorylated at only Thr853 inhibit the activity of reconstituted or truncated enzymes.20,26,27 Phosphorylation of avian MYPT1 at Thr853 also interferes with the binding of the C-terminal segment to isolated myosin filaments,18 although cellular interaction between MLCP and myosin filaments is not fully understood. Thus, two inhibitory phosphorylation sites may play distinct roles in the regulation of cellular MLCP. The C-terminal domain of human MYPT1 possesses an extra 25-residue segment with a coiled-coil motif, in addition to the conserved Leu zipper (LZ) domain in chicken MYPT1, which is necessary for cGMP-induced smooth muscle relaxation and the binding to myosin filaments and M20.7,28 The difference in the structure of the C-terminal domain between species possibly affects MLCP regulation through Thr696 and Thr853 phosphorylation.
Unlike the native enzyme, PP1C expressed in bacteria requires a nonphysiological concentration of Mn2+ for activity and lacks sensitivity to specific inhibitor compounds.29,30 Therefore, the methodology for preparing the recombinant human MLCP holoenzyme that restores the function of the native complex is needed. Here, we successfully overexpressed the recombinant MLCP holoenzyme in mammalian cells. The phosphorylation-dependent inhibition of the MLCP holoenzyme was characterized using the holoenzyme partially purified from the mammalian cell lysates. The activity of the recombinant MLCP was independent of Mn2+ ion and inhibited via phosphorylation by ROCK. Systematic mutational analysis revealed distinguishable roles of two phosphorylation sites, Thr696 and Thr853, in the regulation of MLCP.
Experimental Procedures
Preparation of Recombinant MLCP
The recombinant MLCP complex (MYPT1–PP1 dimer) was transiently expressed in COS1 cells. The cDNA fragment of Venus YFP (Vns)31 and the longest splicing variant of human MYPT132 were sequentially inserted at the HindIII–EcoRI site of the pCMV-FLAG-MAT expression vector (Sigma). Vns cDNA was a gift from A. Miyawaki (Riken) through a Material Transfer Agreement. The pHA3-PP1δ vector was cloned as described previously.32 The cDNA of the PP1δ isoform cloned from a pig cDNA library encodes an amino acid sequence identical to that of the human enzyme. COS1 cells (ATCC) were harvested in a 10 cm dish in the presence of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS). The cells in a 10 cm dish were transiently transfected for 24–36 h using FuGene HD or XtremeGene 9 (Roche) with each 5 μg of MYPT1 and PP1 vectors. A longer transfection did not increase the yield (data not shown). After the transfection, cells were rinsed with PBS and then homogenized using a Dounce homogenizer with 1 mL of extraction buffer [0.5 M NaCl, 20 mM Tris-HCl (pH 8.0), 0.1 mM EGTA, 5% glycerol, and 0.1% Tween 20] supplemented with 4 mM Pefabloc (PFB, Roche) and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP, Pierce). The lysates were incubated for 15 min on ice and clarified by centrifugation at 12000 rpm for 15 min (4 °C). Under these conditions, >80% of MYPT1 was recovered in the supernatant (data not shown). Talon Co2+ affinity resin slurry (100 μL) was added to the supernatant. After a 30 min incubation, the beads were collected and washed three times with extraction buffer (200 μL). The MLCP complex bound to the beads was released by stepwise elution with 100 μL of elution buffer [0.5 M imidazole-HCl (pH 7.0), including 0.1 M NaCl, 0.1 mM EGTA, 5% glycerol, 0.1% Tween 20, 4 mM PFB, and 0.5 mM TCEP]. The total protein concentration and the composition of each fraction were determined by a Bradford assay and immunoblotting, respectively. The elution fractions containing the MLCP complex were combined, dispensed into 50 μL aliquots, and stored at −80 °C until they were used. The preparation and activity of trimeric MLCP with the M20 subunit are shown in Figure S2 of the Supporting Information.
MLCP Assay
MLCP activity was determined as the amount of inorganic phosphate (Pi) released with the phospho-MLC20 peptide as the substrate, unless noted. We used the phosphopeptide to avoid allosteric effects caused by the multiple-site interaction with the myosin molecule. The phospho-MLC20 peptide {P-MLC20(3–26) mimicking human MLC20 residues 3–26 [KRAKAKTTKKRPQRAT(Sp)NVFAMFD]} was synthesized by LifeTein. The amount of released Pi was measured using a malachite green assay (BioMol Green kit, Enzo). The assay was performed in triplicate in a 96-well plate with a final volume of 50 μL. Conditions included 0.003–0.04 mg/mL recombinant MLCP in the presence of assay buffer [25 mM MOPS-NaOH (pH 7.0), 0.5 mM TCEP, 0.1 mM EDTA, 4 mM PFB, and 1 nM okadaic acid (OA)] at 30 °C. A small dose of OA was added to eliminate the activity of potential contaminants (PP2A/4/6) in the purified MLCP sample. The phosphatase reaction was initiated by the addition of 5 μL of 0.25 mM P-MLC20(3–26). After a 30 min incubation, 100 μL of BioMol Green reagent was added to each well to terminate the reaction. The plate was incubated at room temperature for 20–30 min, and the absorbance at 650 nm in each well was read by the plate reader. The amount of Pi was obtained using the OD value from the triplicate wells with a standard Pi solution. The MLCP assay with 32P-labeled MLC20 was conducted as described previously.33
Proximity Ligation Assay (PLA)
The PLA was performed using a Duolink-II kit (Olink) following the manufacturer’s protocol. Human leiomyosarcoma cells [sk-LMS-1 (ATCC)] were seeded onto fibronectin-coated coverslips. Cells were harvested overnight in DMEM without FBS, stimulated for 1 h with 10% FBS and then fixed with 10% TCA (for phosphorylation) or 4% paraformaldehyde (for protein–protein interaction).34 TCA fixation was critical for preserving The853 phosphorylation (data not shown). After being fixed, cells were permeabilized with 0.1% Triton X-100 and subjected to PLA staining. PLA signals and nuclei were detected using confocal microscopy with Texas Red and DAPI filter sets, respectively.35 More than 12 images were captured in each specimen. Spots of the PLA signal above background were counted using a semiautomated script written in IP-Lab. The number of PLA spots divided by the number of nuclei was obtained from each image, and mean values ± the standard error of the mean (SEM) of the ratio was defined as PLA/cell.
Other Methods
Molecular cloning, immunoblotting, immunoprecipitation, and the S-tag pulldown assay were performed as described previously.10,32 Expression vectors for myosin heavy chain fragments were prepared using cDNA of human non-muscle myosin heavy chain, a gift from M. Takahashi (Hokkaido University, Sapporo, Japan), as a polymerase chain reaction template. The DNA sequence was verified at the Kimmel Cancer Center Cancer Genomic Core Laboratory (Thomas Jefferson University). Primary antibodies for GFP, pan-actin, HA, and myc were purchased from the Aves lab, Sigma-Aldrich, and the University of Virginia Core Facility. Antibodies for total-MYPT1, P-MYPT1(T696), and P-MYPT1(T853) were from Millipore. The specificity of the phospho-specific antibodies was verified using Ala-substituted MYPT1 (see Figure 2A). Thio-phosphorylation was assessed following the method reported by Allen et al. using p-nitrobenzyl mesylate (PNBM) and the antibody specific to the alkylated thiophosphate (Epitomics).36 Dot blotting was performed using a nitrocellulose membrane. The phosphorylation of MLCP was terminated by adding 2×RIPA buffer. Aliquot (2 μL) of the sample was spotted on the membrane and subjected to immunostaining, followed by densitometry for quantification. A Student’s t test assuming an equal distribution and nonlinear regression were conducted using Excel and Kaleidagraph, respectively.
Figure 2.

Phosphorylation-dependent inhibition of the recombinant MLCP complex. (A) Phosphorylation of recombinant MLCP. Untreated (left) or phosphorylated recombinant MLCP with MYPT1 wild type (WT) and T696A, T853A, and T696A/T853A (AA) mutants by ROCK (100 milliunits) (right) was subjected to immunoblotting using antibodies for total-MYPT1, phospho-Thr696, and phospho-Thr853. No phosphorylation was detected with untreated MLCP. (B) MLCP assay. Each MLCP preparation was thio-phosphorylated for 90 min at 30 °C with ROCK (100 milliunits) and 0.1 mM ATPγS prior to the MLCP assay. The relative activity of thio-phosphorylated MLCP (filled bar) was normalized against the untreated enzyme (empty bar) (left). The mean value ± SEM was obtained from triplicate assays with at least three independent thio-phosphorylated MLCP trials. An asterisk indicates p < 0.05 compared to the value with the untreated enzyme. The difference in activity between thio-phosphorylated and untreated enzymes was defined as inhibition (%) (right).
Results
Figure 1 shows a summary of the preparation and characterization of the recombinant MLCP complex. FLAG-Vns-MAT (fvm)-tagged ectopic full-length (FL) MYPT1 was transiently co-expressed for 24 h with HA-tagged PP1δ in COS1 cells and partially purified using Co2+-chelated TALON beads. The eluent was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting with anti-FLAG and anti-HA antibodies (Figure 1A). On the Coomassie-stained gel (Figure 1A, left), fvm-MYPT1(FL) was a major component in the eluent along with HA-PP1δ (arrowhead). A truncated form, fvm-MYPT1(1–300), including the PP1 docking site also formed a complex with HA-PP1δ (Figure 1A, 300). The unidentified polypeptides existed in both preparations, suggesting that these are due to nonspecific binding to resins, but not degradation from MYPT1 FL. In a typical preparation, the yield was approximately 0.1 mg of partially purified enzyme from COS1 lysates from a 10 cm dish. On the basis of the densitometric analysis of the Coomassie gel, the molar ratio of fvm-MYPT1 to HA-PP1δ was close to 2, indicating approximately 50% fvm-MYPT1 is bound with HA-PP1δ. The yield, purity, and molar ratio of the recombinant MLCP were consistent between preparations and insensitive to the mutations described below. We also tested other cell lines, such as HEK293, HeLa, and A7r5, although only COS1 cells were able to produce a significant amount of the active MLCP complex (data not shown). Because of the low recovery, we were unable to further purify the MLCP complex (data not shown).
Figure 1.

Characterization of the recombinant MLCP complex. Full-length (FL) FLAG-Vns-MAT (fvm)-tagged MYPT1 and fragment 1–300 (300) were co-expressed with 3×HA-PP1C (δ isoform) and partially purified as described in Experimental Procedures. Each sample was subjected to SDS–PAGE, and the proteins were detected by Coomassie staining (A, left) and immunoblotting with anti-FLAG and anti-HA antibodies (A, right). The arrowhead indicates the location of 3×HA-PP1C detected with the anti-HA antibody. The MLCP complex was subjected to an MLCP assay in the presence of MnCl2, okadaic acid (OA), and microcystin LR (MC) with P-MLC20(3–26) as a substrate (B). Bar C indicates the activity without additives, which was set to 100%. An asterisk indicates p < 0.05 (n = 3), compared with bar C.
A litmus test of the recombinant PP1 preparation is the sensitivity to Mn2+. MLCP activity was determined by measuring the release of phosphate from a phospho-peptide mimicking MLC20(3–26:P-Ser19) as a substrate using the Malachite Green method. The recombinant fvm-MLCP(FL) complex dephosphorylated the P-MLC20 peptide in the absence of Mn2+. Under this condition [25 μM P-MLC20(3–26)], the specific activity of fvm-MLCP(FL) was ∼200 nmol min–1 mg–1. The MLCP activity was unchanged by the addition of 0.1 mM MnCl2 but decreased to 59% in the presence of 1 mM MnCl2 (Figure 1B). Okadaic acid (OA) and microcystin LR (MC) are potent inhibitor compounds for PP1 and PP2A. The addition of 10 nM okadaic acid, which inhibits PP2A, had no effect on MLCP activity, while the addition of 1 μM okadaic acid and 10 nM MC, which inhibits both PP1 and PP2A, substantially decreases MLCP activity to 23 and 12%, respectively (Figure 1B). These results suggest that the Mn2+ and inhibitor sensitivity of the recombinant MLCP is indistinguishable from that of the native MLCP enzyme. Furthermore, the recombinant MLCP did not hydrolyze p-nitrophenyl phosphate (PNPP), a synthetic substrate dephosphorylated by Mn2+-dependent PP1 (data not shown).29
Figure 2 shows the phosphorylation-dependent inhibition of the recombinant MLCP. No phosphorylation at Thr696 or Thr853 was detected in the MLCP preparations, including the fvm-MYPT1 wild type (WT), T696A, T853A, and T696A/T853A (AA) (Figure 2A, left). Probably, it is due to autodephosphorylation (see Figure 4). The incubation with recombinant ROCK (100 milliunits) yielded phosphorylation of mutant MYPT1s at Thr696 or Thr853 at a level equivalent to that of WT. Under the same condition, the MLCP complex was thio-phosphorylated with ROCK and ATPγS and subjected to the phosphatase assay (Figure 2B). Thio-phosphorylation caused a decrease in the activity of WT MLCP to 29%. Substitution of Thr696 with Ala eliminated the phosphorylation-dependent inhibition, whereas the extent of inhibition of T853A MLCP was indistinguishable from that of WT MLCP (Figure 2B). Neither AA MLCP nor the MLCP consisting of MYPT1(1–300) was sensitive to the thio-phosphorylation. Further addition of ROCK and/or prolonged thiophosphorylation did not enhance the inhibitory potency (data not shown). These results suggest that two inhibitory phosphorylation sites of human MYPT1 play distinguishable roles in the regulation of MLCP. As shown in Figure S1A of the Supporting Information, there are two Pro residues in the region prior to Thr853 that corresponds to the middle of an α-helix prior to Thr696 (B-helix) (Figure S1B of the Supporting Information),37 although both regions are enriched with basic residues.9 The difference in the structure may confer specific roles on the two inhibitory phosphorylation sites. We also tested whether the M20 subunit plays a role in the regulation using the recombinant trimeric MLCP (Figure S2 of the Supporting Information). Neither the specific activity nor the extent of phosphorylation-dependent inhibition was distinguishable between the dimeric and trimeric holoenzymes. Therefore, we characterized only the MYPT1–PP1 dimeric complex.
Figure 4.
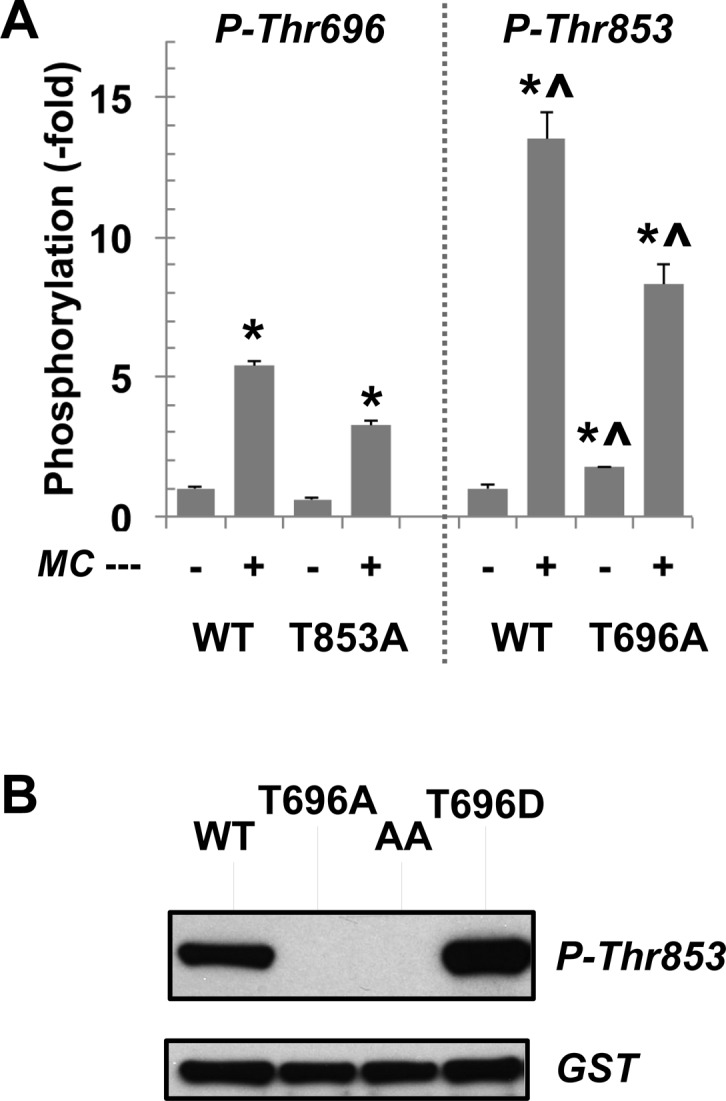
Phosphorylation of MYPT1 by ROCK. (A) WT, T696A, and T853A MLCPs were phosphorylated for 30 min at 30 °C with ROCK (36 milliunits) in the absence or presence of 1 μM MC-LR and subjected to immuno-dot blotting using antibodies for total, P-T696, and P-T853 MYPT1. Potential contamination of PP2A was eliminated with OA (10 nM) in the mixture. The mean value ± SEM of staining density of phospho-Thr696 or Thr853 vs total MYPT1 was obtained from triplicate assays. An asterisk and a carrot indicate p < 0.05 against the value without MCLR and the value of P-Thr696, respectively. (B) GST-tagged MYPT1 fragment (residues 654–880) phosphorylated for 30 min at 30 °C with ROCK (36 milliunits) and subjected to immunoblotting. Representative data from duplicate assays are shown.
GST-tagged MYPT1 fragments, including either phosphorylated Thr696 or Thr853, is capable of docking at the active site and inhibiting purified MLCP, suggesting an autoinhibition mechanism for the regulation.27 We tested the autoinhibition model using the recombinant MLCP complex. As shown in Figure 3A, thio-phosphorylation of T853A MLCP at Thr696 reduced the Vm value from 0.32 to 0.15 nmol min–1 mg–1 and the Km value from 12.3 to 4.7 μM, indicating noncompetitive inhibition, in disagreement with the autoinhibition model, including competition at the active site. The autoinhibition model was further tested using the antibody for phospho-MYPT1(Thr696) (Figure 3B). To avoid interference with the Malachite Green assay by components in the antibody solution, 32P-labeled MLC20 was used as a substrate for the assay. Incubation of WT MLCP with ROCK and ATP reduced the activity to 60% (Figure 3B). Addition of the anti-P-MYPT1(Thr696) antibody after the phosphorylation reversed the phosphorylation-induced inhibition. Thus, masking of phospho-Thr696 is sufficient for the attenuation of inhibition. In the sequence alignment (Figure S1B of the Supporting Information), acidic and basic residue clusters, DEE and RK, prior to MYPT1 Thr696, are highly conserved between species (human, fruit fly, and chicken) as well as other MYPT1 family members, such as MYPT2 and MBS85. If phospho-Thr696 docks at the active site, the position of the basic cluster, RK, is predicted to be in the proximity of an acidic cluster that exists at the edge of the PP1C active site groove, based on X-ray crystal structure data.9 To test this possibility, the MLCP complexes including two versions of Gln-substituted MYPT1, RK > QQ (“RK”) and DEE > QQQ (“DEE”), were subjected to the thio-phosphorylation-dependent inhibition assay (Figure 3C). Compared with that of the WT, relative phosphorylation levels of DEE and RK MYPT1 at Thr696 were 1.0 ± 0.04 and 1.4 ± 0.27, respectively (n = 3). Thio-phosphorylation of DEE MLCP yielded a moderate decrease in activity to 72% (Figure 3C). On the other hand, the RK mutation eliminated the thio-phosphorylation-dependent inhibition of MLCP, suggesting an importance of the basic cluster prior to phospho-Thr696 in the inhibition, in agreement with the autoinhibition model. Therefore, we propose that the direct binding of phospho-Thr696 is a cause of the inhibition of human MLCP.
Figure 3.

Autoinhibition of MLCP through Thr696 phosphorylation. (A) Activity of thiophospho-MLCP. Specific activity of untreated T853A MLCP (U-853A) and T853A MLCP thio-phosphorylated at Thr696 (tP-853A) determined using P-MLC20(3–26) at the indicated concentration. Vm and Km values were obtained by curve fitting (n = 3). (B) Interference with autoinhibition by the anti-P-T696-MYPT1 antibody. WT MLCP was preincubated for 90 min at 30 °C under the indicated conditions in the absence or presence of ROCK (100 milliunits) and then subjected to the MLCP assay using 32P-labeled MLC20 (1 μM). The anti-P-T696 antibody was added 30 min prior to the assay (n = 3). (C) Thio-phosphorylation-dependent inhibition of MLCP activity. Untreated and thio-phosphorylated WT, DEE > QQQ (DEE), and RK > QQ (RK) MLCPs were subjected to the MLCP assay using P-MLC20(3–26), and the relative activities against untreated enzymes are shown. p < 0.05 (n = 6). The number indicates the relative extent of thio-phosphorylation of MYPT1, which was determined by immuno-dot blotting using PNBM and the antibody for the alkylated thiophosphate, as described in Experimental Procedures.
The inhibition of MLCP induced by the phosphorylation was less potent, compared with thio-phosphorylation (Figures 2B and 3B). We asked if phospho-Thr696 and Thr853 are dephosphorylated in the recombinant MLCP (“auto-dephosphorylation”). WT, T696A, and T853A MLCP complexes were phosphorylated with a submaximal amount of recombinant ROCK in the absence and presence of microcystin, and the specific phosphorylation at Thr696 and Thr853 was assessed using immunostaining (Figure 4A). The inhibition of phosphatase activity with microcystin resulted in a 5-fold increase in the level of phosphorylation at Thr696, which indicates the extent of autodephosphorylation at the inhibitory phospho-Thr696 (Figure 4A, left). The level of phosphorylation of Thr853 was more sensitive to microcystin, which elevated it 13-fold, suggesting a higher turnover rate of the phosphorylation at Thr853, compared with the rate of that at Thr696. In the T696A MLCP, phosphorylation of Thr853 was less sensitive to microcystin, compared with that of WT MLCP (Figure 4A). The T853A mutation also moderately reduced the level of phosphorylation at Thr696. Thus, each phosphorylation at Thr696 and Thr853 may stabilize the phosphorylation at the other site. A synergy between two phosphorylation sites was determined using the GST-MYPT1(654–880) fragment that does not possess the phosphatase activity. Recombinant ROCK failed to phosphorylate the T696A protein at Thr853, whereas the phosphorylation at Thr853 was enhanced by phospho-mimicking substitution at Thr696 (Figure 4B). These results suggest communication between two inhibitory phosphorylation sites of MYPT1. For an unknown reason, we failed to produce stable GST-MYPT1(654–880) protein with a substitution of Thr853 with Ala or Asp.
Figure 5 shows the phosphorylation of endogenous MYPT1 and the interaction with myosin filaments in human leiomyosarcoma. The level of phosphorylation of MYPT1 at Thr696 was spontaneously high in the quiescent cells and unchanged upon serum stimulation (Figure 5A, top, and Figure S3 of the Supporting Information). This spontaneous phosphorylation at Thr696 is potentially attributed to the slower autodephosphorylation (Figure 4A). On the other hand, the level of phosphorylation of MYPT1 at Thr853 and the level of diphosphorylation of MLC20 at Thr18/Ser19 were low under quiescent conditions and elevated in response to serum stimulation (Figure 5A and Figure S3 of the Supporting Information). ROCK inhibition blocked the increase in the level of phosphorylation (Figure 5A and Figure S3 of the Supporting Information). A parallel assay for Thr853 phosphorylation and dissociation of MYPT1 from myosin filaments was performed using the proximity ligation assay (PLA) (Figure 5B,C). Cells were fixed with 4% paraformaldehyde and subjected to the PLA, except for the assay that detected MYPT1 phosphorylation, in which 10% TCA was used as a fixative to preserve the phosphorylation. The PLA signals and the number of nuclei in each image were counted under a fluorescence microscope (Figure 5B). The relative PLA signal was determined using the anti-total MYPT1 antibody paired with anti-P-MYPT1(Thr853), anti-MLC20, and anti-PP1δ antibodies and preimmune IgG as a control (Ctl) that indicates the background signal (Figure 5C). The magnitude of the PLA signal between P-MYPT1(Thr853) and total-MYPT1 was increased upon serum stimulation, indicating an increased level of Thr853 phosphorylation (Figure 5B,C). In parallel, the magnitude of the PLA signal between anti-total MYPT1 and MLC20 was significantly reduced upon stimulation, indicating an increase in the distance between MYPT1 and MLC20. In addition, the magnitude of the PLA signal against PP1δ was also reduced in parallel to Thr853 phosphorylation (Figure 5C). These results suggest that phosphorylation of MYPT1 at Thr853 is sensitive to ROCK-induced signaling and is synchronized with the disassembly of the myosin–MYPT1–PP1δ complex in cells.
Figure 5.
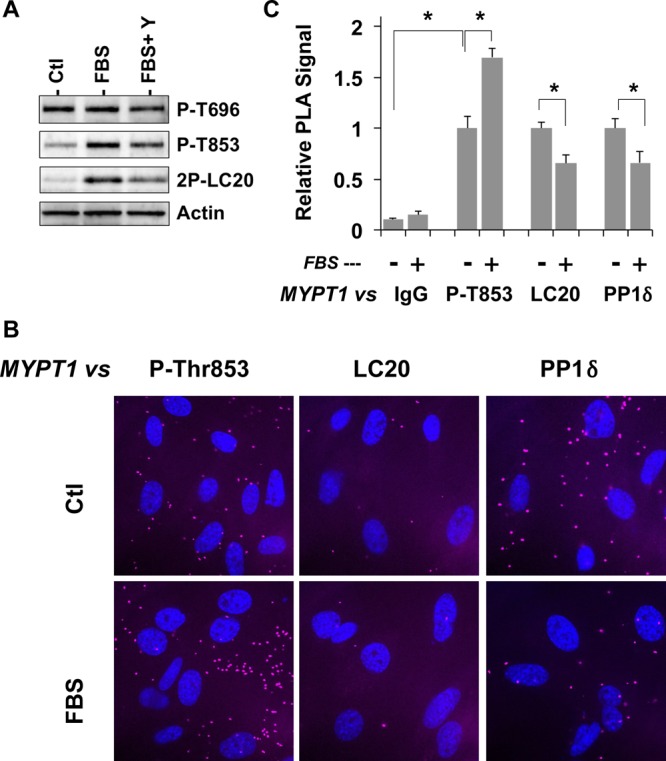
Phosphorylation and regulation of MLCP in leiomyosarcoma. (A) Cellular phosphorylation of MYPT1. Quiescent leiomyosarcoma cells were stimulated for 1 h with 10% FBS in the absence and presence of 10 μM Y27632, fixed with 10% TCA, and subjected to immunoblotting. 2P-MLC20 denotes MLC20 diphosphorylated at Thr18 and Ser19. (B and C) PLA analysis. Quiescent and FBS-stimulated cells on coverslips were fixed with 10% TCA (for P-Thr853) or 4% paraformaldehyde (MLC20 and PP1δ) and subjected to the PLA using anti-total MYPT1 paired with preimmune IgG (control), anti-P-MYPT1(T853), anti-MLC20, and anti-PP1δ. Magenta and blue portions are the PLA signal and the nucleus, respectively (B). Numbers of PLA spots and nuclei were obtained in each image field. The mean value ± SEM of the ratio of the number of PLA spots to the number of nuclei was obtained from 12–43 image fields in two independent assays (C). Asterisks denote p < 0.05 vs the quiescent condition.
N-Terminal and C-terminal domains of chicken MYPT1 have been reported to bind to heavy meromyosin (HMM) and rod domains (Figure 6A), respectively, although little is known about the contact regions of human MYPT1. S tag fusion peptides of three regions in the myosin rod domain, S-2, the N-terminal portion of light meromyosin (LMM-N), and the assembly competence domain (ACD), a minimal region required for filament formation, were expressed in COS1 cells and used for the S pull-down assay with the MLCP complex, including myc-tagged human MYPT-1 (Figure 6B,C). The myc-MYPT1 subunit of the MLCP complex in the cell lysates was coprecipitated only with S tag LMM-N, but not with S-2 or the ACD domain (Figure 6B). Next, the pull-down assay was conducted using S-S-2 and -LMM-N beads with cell lysates including myc-MYPT1(1–498) (N) and myc-MYPT1(495–1030) (C) (Figure 6C). Only the C-terminal segment of MYPT1 was coprecipitated with S-LMM-N beads (Figure 6C, right). Neither myc-MYPT1-N nor myc-MYPT1-C was coprecipitated with S-S-2 (Figure 6C, left). In a manner consistent with the pull-down assay data, only myc-MYPT1-C was coprecipitated with the cytoskeletal fraction in the cell lysates (Figure 6D). Thus, the C-terminal domain of human MYPT1 docks at the LMM-N domain. As shown in Figure S4 of the Supporting Information, human MYPT1(934–1030), including both Leu zipper (LZ) and coiled-coil (CC) domains, was predominantly found in the cytoskeletal fraction, suggesting that the C-terminal helical domain of MYPT1 is responsible for the interaction with the helical LMM subdomain.
Figure 6.

Interaction of MYPT1 with myosin. (A) Structure of myosin II heavy chain. Abbreviations: S-1, subfragment-1; S-2, subfragment-2; LMM-N, N-terminal portion of light meromyosin; ACD, assembly competent domain. (B and C) Binding of recombinant MLCP to S-tagged myosin fragments. S-tagged S-2, LMM-N, and ACD were expressed in COS1 cells and captured with S-protein affinity beads. The beads were mixed with cell lysates, including the complex of myc-MYPT1 with 3×HA-PP1C (B) or myc-MYPT1(1–498) with 3×HA-PP1δ and myc-MYPT1(495–1030) with 3×HA-M20 (C). The proteins bound to the beads were analyzed by immunoblotting. (D) Subfractionation of cells expressing recombinant MLCP. myc-MYPT1(1–498) (“N”) and myc-MYPT1(495–1030) (“C”) were co-expressed in COS1 cells with 3×HA-PP1C (H124N) and 3×HA-M20, respectively. Cells were lysed with buffer [10 mM HEPES (pH 7.4) with 0.2 M NaCl, 5 mM Mg(OAc)2, 2 mM EGTA, and 0.1% Triton X-100] and subjected to airfuge for 30 min at 80000 rpm. S and P denote the cytoplasm and cytoskeletal fractions, respectively.
Discussion
In this work, we established a new method for the preparation of the recombinant PP1 holoenzyme and revealed distinguishable roles of MYPT1 phosphorylation at two inhibitory phosphorylation sites in the regulation of human MLCP. Recently, we have proposed the autoinhibition of MLCP in response to phosphorylation at either Thr696 or Thr853, based on the data that the MYPT1 fragments phosphorylated at Thr696 or Thr853 are capable of inhibiting purified MLCP and the endogenous MLCP in smooth muscle fibers.27 In the recombinant MLCP complex, only Thr696 phosphorylation is responsible for the inhibition of MLCP. The inhibition is attenuated by substitution with Gln or the neutralizing antibody, suggesting the direct docking of phospho-Thr696 at the active site. Phospho-Thr696 is autodephosphorylated at a rate slower than that of phospho-Thr853. The difference in the stability of the phospho-ester likely confers the function of each phosphorylation. It should be noted that the apparent Vm value of MLCP activity decreases in response to Thr696 thiophosphorylation, implying an allosteric inhibition in the Michaelis–Menten model. The thiophosphorylation-dependent reduction of Vm is also shown in the previous report with the native MLCP and the reconstituted enzyme.13,25 We presume that the high affinity and the proximity of the thiophosphorylated intrinsic autoinhibitory domain to the active site result in stable docking of thiophospho-Thr696, reducing the number of accessible active sites (decreasing [E]total). Under the circumstances, the reversible competitive inhibition model of the Michaelis–Menten equation is not applicable. This stabilized autoinhibitory interaction of phospho-Thr696 with the active site must be necessary for the potent regulation of MLCP in cells, where the enzyme is anchored to the substrate, resulting in an apparent high [S] value.7,12
Accumulating lines of evidence show the spontaneous MYPT1 phosphorylation at Thr696 in resting smooth muscle22−24 and quiescent cells, including human leiomyosarcoma in this work. The slow autodephosphorylation and the basal kinase activity at Thr696 likely contribute to the elevated levels of phosphorylation and autoinhibition under the resting condition. The spontaneous autoinhibition of MYPT1 by Thr696 phosphorylation suggests that cellular MLCP activity is at least partially suppressed when the holoenzyme is synthesized in cells. MLCP autoinhibition is necessary for maintaining the basal level of myosin phosphorylation and cytoskeleton stability. For example, when the ectopic MLCP lacking the autoinhibition site is introduced into cells by microinjection or transfection, the stress fibers are distorted because of hypo-phosphorylation of myosin.32,38 The data in this work also suggest that the phosphorylation at Thr696 facilitates the phosphorylation at Thr853. The priming effect of phospho-Thr696 to Thr853 phosphorylation is more evident in the MYPT1 fragment. Thus, the slow turnover of Thr696 phosphorylation is responsible for regulating the basal activity of MLCP as well as the sensitivity of Thr853 phosphorylation to the agonist stimulus. Although, in general, the spontaneous Thr696 phosphorylation is unchanged in smooth muscle tissues,22−24,39 moderate fluctuations have been reported in cell cultures25,26,40 and smooth muscle tissues under certain conditions.41−44 Purified kinases, such as ROCK, ZIPK, and ILK, are capable of phosphorylating MYPT1 at Thr696, but cellular kinases responsible for the phosphorylation remain unclear.6 ROCK is unlikely involved in Thr696 phosphorylation, because ROCK inhibition does not effectively reduce the level of Thr696 phosphorylation in leiomyosarcoma cells (this study) or others.45 ZIPK associated with MYPT1 is a strong candidate as a Thr696 kinase,19 although genetically modified ZIPK failed to phosphorylate MYPT1 at Thr696 in permeabilized smooth muscle strips.46 Identifying specific kinases responsible for basal and pathologic phosphorylation at Thr696 is needed for a full understanding of the autoinhibitory regulation of cellular MLCP.
Trinkle-Mulcahy et al. originally reported G-protein-induced incorporation of [35S]thiophosphate into MYPT1 in parallel with force development and MLCP inhibition using partially permeabilized portal vein smooth muscle strips.16 Later, two phosphorylation sites of MYPT1 at Thr696 and Thr853 leading to MLCP inhibition were identified using purified ROCK.17,18 It becomes evident that Thr853 is enhanced in response to G-protein activation in cells.22,23 Unlike the recombinant proteins purified from bacterial lysates,26 the activity of the recombinant MLCP complex prepared using mammalian cells is insensitive to Thr853 phosphorylation. Instead, PLA analysis shows that increases in the distance of MYPT1 from myosin and PP1C parallel the increase in the level of Thr853 phosphorylation, suggesting that Thr853 phosphorylation is indicative of disassembly of the myosin–MLCP complex in cell cultures. Lines of evidence support the disassembly of the MLCP–myosin filament. Gong et al. showed that treatment with arachidonic acid causes dissociation of MYPT1 from PP1C.47 Velasco et al. reported that the phosphorylation of isolated chicken MYPT1 at Thr850 (corresponding to human Thr853) interferes the binding to myosin filaments.18 Agonist-induced redistribution of MYPT1 and PP1C was reported in smooth muscle cells.48 Also, fluorescence photobleaching analysis suggests the high mobility of GFP-tagged PP1C in living cells, suggesting a reversible association–dissociation cycle of the cellular PP1 holoenzyme.49 Thus, disassembly of the holoenzyme induced by G-protein-induced Thr853 phosphorylation is a possible cause of the suppression of MLCP activity in cells. Because Thr853 phosphorylation is not sufficient for disassembly of our recombinant MLCP (data not shown), we presume that multiple pathways in ROCK signaling are involved in the regulation of MLCP–myosin complex stability in cells.
There are more than 100 PP1 regulatory subunits that confer specific functions to cellular PP1 catalytic subunits.50 There have been limitations in characterizing cellular PP1 signaling, because classical inhibitor compounds for PP1 cannot distinguish between PP1 holoenzymes in cells. Recently, guanabenz was shown to lead to the dissociation of PP1C from a regulatory subunit, GADD34, and treatment with the compound enhances cell viability under ER stress through the inhibition of the GADD34–PP1C complex.51 Independently, we developed inhibitor compounds using MLCP purified from aorta extracts. The lead compound induces apoptosis of prostate cancer cells, suggesting the potential for anticancer therapy, although the limitation of the production of the native enzyme has hampered further characterization and refinement of the compound.33 PP1C has been prepared using expression systems with Escherichia coli and Sf9 cells.29,30 These preparations produce a sufficient amount of recombinant PP1C but yielded the enzyme that did not retain native PP1C function. For high-yield preparation, a high concentration of Mn2+ ion (1 mM) is necessary for bacterial culture and the solutions for purification.29 This is beyond the physiologic concentration of the ion, which has been estimated to be <20–50 nmol/g in tissues and 200 and 20 nmol/L in blood and serum, respectively.52 The recombinant PP1C activity is Mn2+-dependent, unlike the native enzyme. In addition, the PP1C preparation is not phospho-Ser/Thr-specific, dephosphorylating phospho-Tyr and PNPP, which are not hydrolyzed by native PP1C. PP1C expressed in Sf9 cells, a eukaryotic insect cell culture, also displays partial Mn2+-dependent activity like bacterial PP1C. Importantly, all bacterial preparations of PP1C displayed altered interactions with the regulatory subunits.30 For example, E. coli PP1C is relatively insensitive to inhibition by thiophospho-DARPP-32 or thiophosphoro-inhibitor-1, endogenous PP1C inhibitors, or regulation by PP1 regulatory subunits, such as spinophilin and PNUTS. This has significantly limited the study of PP1C–regulatory subunit interaction. In our mammalian expression system, the recombinant MLCP retains native function, such as Mn2+-independent activity and inhibition by okadaic acid, and lacks activity toward the PNPP substrate (data not shown). It is possible that the co-expression of PP1C and the regulatory subunit contributes to the correct folding and physiological metal binding of PP1C through multiple interactions. We predict that the COS1 expression method and the Malachite Green assay with the phosphopeptide can be applied for other PP1 holoenzymes that retain physiological function and for high-throughput assay formats. The new preparation system for the PP1 holoenzymes opens fresh avenues for further investigation of mechanisms underlying cellular PP1 regulation, as well as the development of tools for therapeutic intervention against diseases.
Acknowledgments
We thank Professor Shin-ya Ohki (Japan Advanced Institutes for Science and Technology) for valuable suggestions.
Supporting Information Available
Sequence alignment and three-dimensional structure of human MYPT1 phosphorylation sites (Figure S1), effects of the M20 subunit on MLCP activity (Figure S2), densitometric analysis of MYPT1 phosphorylation in LMS cells (Figure S3), and subcellular fractionation with the lysates of cells expressing the MYPT1 fragments (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK088905, the Brandywine Valley Hemophilia Foundation, Pennsylvania C.U.R.E. (to M.E.), and the Institutional Program for Young Researcher Overseas Visits from the Japan Society for the Promotion of Science (to A.N.). Research in this publication includes work conducted by the Kimmel Cancer Center Genomics Facility, supported in part by National Cancer Institute Grant P30-CA56036.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alessi D.; Macdougall L. K.; Sola M. M.; Ikebe M.; Cohen P. (1992) The Control of Protein Phosphatase-1 by Targetting Subunits. Eur. J. Biochem. 210, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Shirazi A.; Iizuka K.; Fadden P.; Mosse C.; Somlyo A. P.; Somlyo A. V.; Haystead T. A. (1994) Purification and Characterization of the Mammalian Myosin Light Chain Phosphatase Holoenzyme. The Differential Effects of the Holoenzyme and Its Subunits on Smooth Muscle. J. Biol. Chem. 269, 31598–31606. [PubMed] [Google Scholar]
- Shimizu H.; Ito M.; Miyahara M.; Ichikawa K.; Okubo S.; Konishi T.; Naka M.; Tanaka T.; Hirano K.; Hartshorne D. J.; et al. (1994) Characterization of the Myosin-Binding Subunit of Smooth Muscle Myosin Phosphatase. J. Biol. Chem. 269, 30407–33011. [PubMed] [Google Scholar]
- Hartshorne D. J.; Ito M.; Erdodi F. (2004) Role of Protein Phosphatase Type 1 in Contractile Functions: Myosin Phosphatase. J. Biol. Chem. 279, 37211–37214. [DOI] [PubMed] [Google Scholar]
- Matsumura F.; Hartshorne D. J. (2008) Myosin Phosphatase Target Subunit: Many Roles in Cell Function. Biochem. Biophys. Res. Commun. 369, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassie M. E.; Moffat L. D.; Walsh M. P.; MacDonald J. A. (2011) The Myosin Phosphatase Targeting Protein (Mypt) Family: A Regulated Mechanism for Achieving Substrate Specificity of the Catalytic Subunit of Protein Phosphatase Type 1δ. Arch. Biochem. Biophys. 510, 147–159. [DOI] [PubMed] [Google Scholar]
- Johnson D.; Cohen P.; Chen M. X.; Chen Y. H.; Cohen P. T. (1997) Identification of the Regions on the M110 Subunit of Protein Phosphatase 1m That Interact with the M21 Subunit and with Myosin. Eur. J. Biochem. 244, 931–999. [DOI] [PubMed] [Google Scholar]
- Hirano K.; Phan B. C.; Hartshorne D. J. (1997) Interactions of the Subunits of Smooth Muscle Myosin Phosphatase. J. Biol. Chem. 272, 3683–3688. [DOI] [PubMed] [Google Scholar]
- Terrak M.; Kerff F.; Langsetmo K.; Tao T.; Dominguez R. (2004) Structural Basis of Protein Phosphatase 1 Regulation. Nature 429, 780–784. [DOI] [PubMed] [Google Scholar]
- Eto M.; Kitazawa T.; Brautigan D. L. (2004) Phosphoprotein Inhibitor Cpi-17 Specificity Depends on Allosteric Regulation of Protein Phosphatase-1 by Regulatory Subunits. Proc. Natl. Acad. Sci. U.S.A. 101, 8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui T.; Inagaki M.; Ikebe M. (1992) Purification and Characterization of Smooth Muscle Myosin-Associated Phosphatase from Chicken Gizzards. J. Biol. Chem. 267, 16727–16735. [PubMed] [Google Scholar]
- Ichikawa K.; Hirano K.; Ito M.; Tanaka J.; Nakano T.; Hartshorne D. J. (1996) Interactions and Properties of Smooth Muscle Myosin Phosphatase. Biochemistry 35, 6313–6620. [DOI] [PubMed] [Google Scholar]
- Ichikawa K.; Ito M.; Hartshorne D. J. (1996) Phosphorylation of the Large Subunit of Myosin Phosphatase and Inhibition of Phosphatase Activity. J. Biol. Chem. 271, 4733–4440. [DOI] [PubMed] [Google Scholar]
- Yuen S.; Ogut O.; Brozovich F. V. (2011) Mypt1 Protein Isoforms Are Differentially Phosphorylated by Protein Kinase G. J. Biol. Chem. 286, 37274–37279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassie M. E.; Sutherland C.; Ulke-Lemee A.; Chappellaz M.; Kiss E.; Walsh M. P.; MacDonald J. A. (2012) Cross-Talk between Rho-Associated Kinase and Cyclic Nucleotide-Dependent Kinase Signaling Pathways in the Regulation of Smooth Muscle Myosin Light Chain Phosphatase. J. Biol. Chem. 287, 36356–36369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L.; Ichikawa K.; Hartshorne D. J.; Siegman M. J.; Butler T. (1995) Thiophosphorylation of the 130-Kda Subunit Is Associated with a Decreased Activity of Myosin Light Chain Phosphatase in a-Toxin-Permeabilized Smooth Muscle. J. Biol. Chem. 270, 18191–18194. [DOI] [PubMed] [Google Scholar]
- Kawano Y.; Fukata Y.; Oshiro N.; Amano M.; Nakamura T.; Ito M.; Matsumura F.; Inagaki M.; Kaibuchi K. (1999) Phosphorylation of Myosin-Binding Subunit (Mbs) of Myosin Phosphatase by Rho-Kinase in Vivo. J. Cell Biol. 147, 1023–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco G.; Armstrong C.; Morrice N.; Frame S.; Cohen P. (2002) Phosphorylation of the Regulatory Subunit of Smooth Muscle Protein Phosphatase 1m at Thr850 Induces Its Dissociation from Myosin. FEBS Lett. 527, 101–104. [DOI] [PubMed] [Google Scholar]
- MacDonald J. A.; Borman M. A.; Muranyi A.; Somlyo A. V.; Hartshorne D. J.; Haystead T. A. (2001) Identification of the Endogenous Smooth Muscle Myosin Phosphatase-Associated Kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi A.; MacDonald J. A.; Deng J. T.; Wilson D. P.; Haystead T. A.; Walsh M. P.; Erdodi F.; Kiss E.; Wu Y.; Hartshorne D. J. (2002) Phosphorylation of the Myosin Phosphatase Target Subunit by Integrin-Linked Kinase. Biochem. J. 366, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa N.; Koga Y.; Ikebe M. (2002) Phosphorylation of Cpi17 and Myosin Binding Subunit of Type 1 Protein Phosphatase by P21-Activated Kinase. Biochem. Biophys. Res. Commun. 297, 773–778. [DOI] [PubMed] [Google Scholar]
- Kitazawa T.; Eto M.; Woodsome T. P.; Khalequzzaman M. (2003) Phosphorylation of the Myosin Phosphatase Targeting Subunit and Cpi-17 During Ca2+ Sensitization in Rabbit Smooth Muscle. J. Physiol. 546, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiro N.; Koga Y.; Ikebe M. (2003) Agonist-Induced Changes in the Phosphorylation of the Myosin-Binding Subunit of Myosin Light Chain Phosphatase and Cpi17, Two Regulatory Factors of Myosin Light Chain Phosphatase, in Smooth Muscle. Biochem. J. 369, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. P.; Susnjar M.; Kiss E.; Sutherland C.; Walsh M. P. (2005) Thromboxane A2-Induced Contraction of Rat Caudal Arterial Smooth Muscle Involves Activation of Ca2+ Entry and Ca2+ Sensitization: Rho-Associated Kinase-Mediated Phosphorylation of Mypt1 at Thr-855, but Not Thr-697. Biochem. J. 389, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Ito M.; Ichikawa K.; Isaka N.; Nishikawa M.; Hartshorne D. J.; Nakano T. (1999) Inhibitory Phosphorylation Site for Rho-Associated Kinase on Smooth Muscle Myosin Phosphatase. J. Biol. Chem. 274, 37385–33790. [DOI] [PubMed] [Google Scholar]
- Muranyi A.; Derkach D.; Erdodi F.; Kiss A.; Ito M.; Hartshorne D. J. (2005) Phosphorylation of Thr695 and Thr850 on the Myosin Phosphatase Target Subunit: Inhibitory Effects and Occurrence in A7r5 Cells. FEBS Lett. 579, 6611–6615. [DOI] [PubMed] [Google Scholar]
- Khromov A.; Choudhury N.; Stevenson A. S.; Somlyo A. V.; Eto M. (2009) Phosphorylation-Dependent Autoinhibition of Myosin Light Chain Phosphatase Accounts for Ca2+ Sensitization Force of Smooth Muscle Contraction. J. Biol. Chem. 284, 21569–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q. Q.; Fisher S. A.; Brozovich F. V. (2004) Unzipping the Role of Myosin Light Chain Phosphatase in Smooth Muscle Cell Relaxation. J. Biol. Chem. 279, 597–603. [DOI] [PubMed] [Google Scholar]
- Zhang A. J.; Bai G.; Deans-Zirattu S.; Browner M. F.; Lee E. Y. (1992) Expression of the Catalytic Subunit of Phosphorylase Phosphatase (Protein Phosphatase-1) in Escherichia coli. J. Biol. Chem. 267, 1484–1190. [PubMed] [Google Scholar]
- Watanabe T.; da Cruz e Silva E. F.; Huang H. B.; Starkova N.; Kwon Y. G.; Horiuchi A.; Greengard P.; Nairn A. C. (2003) Preparation and Characterization of Recombinant Protein Phosphatase 1. Methods Enzymol. 366, 321–338. [DOI] [PubMed] [Google Scholar]
- Nagai T.; Ibata K.; Park E. S.; Kubota M.; Mikoshiba K.; Miyawaki A. (2002) A Variant of Yellow Fluorescent Protein with Fast and Efficient Maturation for Cell-Biological Applications. Nat. Biotechnol. 20, 87–90. [DOI] [PubMed] [Google Scholar]
- Eto M.; Kirkbride J. A.; Brautigan D. L. (2005) Assembly of Mypt1 with Protein Phosphatase-1 in Fibroblasts Redirects Localization and Reorganizes the Actin Cytoskeleton. Cell Motil. Cytoskeleton 62, 100–109. [DOI] [PubMed] [Google Scholar]
- Grindrod S.; Suy S.; Fallen S.; Eto M.; Toretsky J.; Brown M. L. (2011) Effects of a Fluorescent Myosin Light Chain Phosphatase Inhibitor on Prostate Cancer Cells. Front. Oncol. 1, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K.; Yonemura S.; Matsui T.; Tsukita S. (1999) Immunofluorescence Detection of Exrin/Radixin/Moesin (Erm) Proteins with Their Carboxyl-Terminal Threonine Phosphorylated in Cultured Cells and Tissues. Application of a Novel Fixation Protocol Using Trichloroacetic Acid (Tca) as a Fixative. J. Cell Sci. 112, 1149–1158. [DOI] [PubMed] [Google Scholar]
- Kim J. I.; Young G. D.; Jin L.; Somlyo A. V.; Eto M. (2009) Expression of Cpi-17 in Smooth Muscle During Embryonic Development and in Neointimal Lesion Formation. Histochem. Cell Biol. 132, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. J.; Li M.; Brinkworth C. S.; Paulson J. L.; Wang D.; Hubner A.; Chou W. H.; Davis R. J.; Burlingame A. L.; Messing R. O.; Katayama C. D.; Hedrick S. M.; Shokat K. M. (2007) A Semisynthetic Epitope for Kinase Substrates. Nat. Methods 4, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S.; Iwaoka R.; Eto M.; Ohki S. Y. (2009) Solution Structure of the Inhibitory Phosphorylation Domain of Myosin Phosphatase Targeting Subunit 1. Proteins 77, 732–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsukawa G.; Yamakita Y.; Yamashiro S.; Hartshorne D. J.; Sasaki Y.; Matsumura F. (2000) Distinct Roles of Rock (Rho-Kinase) and Mlck in Spatial Regulation of Mlc Phosphorylation for Assembly of Stress Fibers and Focal Adhesions in 3t3 Fibroblasts. J. Cell Biol. 150, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Yazbi A. F.; Johnson R. P.; Walsh E. J.; Takeya K.; Walsh M. P.; Cole W. C. (2010) Pressure-Dependent Contribution of Rho Kinase-Mediated Calcium Sensitization in Serotonin-Evoked Vasoconstriction of Rat Cerebral Arteries. J. Physiol. 588, 1747–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko T.; Ito M.; Kureishi Y.; Okamoto R.; Moriki N.; Onishi K.; Isaka N.; Hartshorne D. J.; Nakano T. (2003) Activation of Rhoa and Inhibition of Myosin Phosphatase as Important Components in Hypertension in Vascular Smooth Muscle. Circ. Res. 92, 411–418. [DOI] [PubMed] [Google Scholar]
- Sakurada S.; Takuwa N.; Sugimoto N.; Wang Y.; Seto M.; Sasaki Y.; Takuwa Y. (2003) Ca2+-Dependent Activation of Rho and Rho Kinase in Membrane Depolarization-Induced and Receptor Stimulation-Induced Vascular Smooth Muscle Contraction. Circ. Res. 93, 548–556. [DOI] [PubMed] [Google Scholar]
- Jin L.; Ying Z.; Webb R. C. (2004) Activation of Rho/Rho Kinase Signaling Pathway by Reactive Oxygen Species in Rat Aorta. Am. J. Physiol. 287, H1495–H1500. [DOI] [PubMed] [Google Scholar]
- Lubomirov L. T.; Reimann K.; Metzler D.; Hasse V.; Stehle R.; Ito M.; Hartshorne D. J.; Gagov H.; Pfitzer G.; Schubert R. (2006) Urocortin-Induced Decrease in Ca2+ Sensitivity of Contraction in Mouse Tail Arteries Is Attributable to Camp-Dependent Dephosphorylation of Mypt1 and Activation of Myosin Light Chain Phosphatase. Circ. Res. 98, 1159–1167. [DOI] [PubMed] [Google Scholar]
- Bhetwal B. P.; Sanders K. M.; An C.; Trappanese D. M.; Moreland R. S.; Perrino B. A. (2013) Ca2+ Sensitization Pathways Accessed by Cholinergic Neurotransmission in the Murine Gastric Fundus. J. Physiol. 591, 2971–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsome T. P.; Polzin A.; Kitazawa K.; Eto M.; Kitazawa T. (2006) Agonist- and Depolarization-Induced Signals for Myosin Light Chain Phosphorylation and Force Generation of Cultured Vascular Smooth Muscle Cells. J. Cell Sci. 119, 1769–1780. [DOI] [PubMed] [Google Scholar]
- Moffat L. D.; Brown S. B.; Grassie M. E.; Ulke-Lemee A.; Williamson L. M.; Walsh M. P.; MacDonald J. A. (2011) Chemical Genetics of Zipper-Interacting Protein Kinase Reveal Myosin Light Chain as a Bona Fide Substrate in Permeabilized Arterial Smooth Muscle. J. Biol. Chem. 286, 36978–36991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong M. C.; Fuglsang A.; Alessi D.; Kobayashi S.; Cohen P.; Somlyo A. V.; Somlyo A. P. (1992) Arachidonic Acid Inhibits Myosin Light Chain Phosphatase and Sensitizes Smooth Muscle to Calcium. J. Biol. Chem. 267, 21492–21498. [PubMed] [Google Scholar]
- Shin H. M.; Je H. D.; Gallant C.; Tao T. C.; Hartshorne D. J.; Ito M.; Morgan K. G. (2002) Differential Association and Localization of Myosin Phosphatase Subunits During Agonist-Induced Signal Transduction in Smooth Muscle. Circ. Res. 90, 546–553. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L.; Sleeman J. E.; Lamond A. I. (2001) Dynamic Targeting of Protein Phosphatase 1 within the Nuclei of Living Mammalian Cells. J. Cell Sci. 114, 4219–4228. [DOI] [PubMed] [Google Scholar]
- Bollen M.; Peti W.; Ragusa M. J.; Beullens M. (2011) The Extended Pp1 Toolkit: Designed to Create Specificity. Trends Biochem. Sci. 35, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaytler P.; Harding H. P.; Ron D.; Bertolotti A. (2011) Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 332, 91–94. [DOI] [PubMed] [Google Scholar]
- Keen C. L.; Ensunsa J. L.; Clegg M. S. (2000) Manganese Metabolism in Animals and Humans Including the Toxicity of Manganese. Met. Ions Biol. Syst. 37, 89–121. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.