

Abstract
Adaptor protein Grb2 binds phosphotyrosines in the epidermal growth factor (EGF) receptor (EGFR) and thereby links receptor activation to intracellular signaling cascades. Here, we investigated how recruitment of Grb2 to EGFR is affected by the spatial organization and quaternary state of activated EGFR. We used the techniques of image correlation spectroscopy (ICS) and lifetime-detected Förster resonance energy transfer (also known as FLIM-based FRET or FLIM–FRET) to measure ligand-induced receptor clustering and Grb2 binding to activated EGFR in BaF/3 cells. BaF/3 cells were stably transfected with fluorescently labeled forms of Grb2 (Grb2–mRFP) and EGFR (EGFR–eGFP). Following stimulation of the cells with EGF, we detected nanometer-scale association of Grb2–mRFP with EGFR–eGFP clusters, which contained, on average, 4 ± 1 copies of EGFR–eGFP per cluster. In contrast, the pool of EGFR–eGFP without Grb2–mRFP had an average cluster size of 1 ± 0.3 EGFR molecules per punctum. In the absence of EGF, there was no association between EGFR–eGFP and Grb2–mRFP. To interpret these data, we extended our recently developed model for EGFR activation, which considers EGFR oligomerization up to tetramers, to include recruitment of Grb2 to phosphorylated EGFR. The extended model, with adjustment of one new parameter (the ratio of the Grb2 and EGFR copy numbers), is consistent with a cluster size distribution where 2% of EGFR monomers, 5% of EGFR dimers, <1% of EGFR trimers, and 94% of EGFR tetramers are associated with Grb2. Together, our experimental and modeling results further implicate tetrameric EGFR as the key signaling unit and call into question the widely held view that dimeric EGFR is the predominant signaling unit.
The epidermal growth factor (EGF) receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases.1,2 The EGFR signaling network contributes to a number of processes important to cancer development and progression, including cell proliferation, angiogenesis, and metastatic spread. EGFR overexpression and truncation3 have been observed in a number of common cancers, including brain, lung, breast, colon, and prostate, giving credence to the notion that a molecular understanding of EGFR activation will yield clinical benefit. EGFR signaling is generally regarded to be initiated by ligand binding to the extracellular region, which leads to receptor dimerization,4 conformational rearrangements within preformed complexes5−7 and higher-order oligomerization8−10. Subsequent to kinase activation and autophosphorylation, cytoplasmic adaptors are recruited to the EGFR cytoplasmic tail.11−16 Whether these processes are influenced by the size of the EGFR cell-surface clusters is an important question. Here, we address this question with a focus on adaptor binding, which is the first step after receptor activation and connects receptor activation to its intracellular signaling cascades.
Grb2 is a pivotal adaptor first discovered to physically link phosphorylated EGFR to the Ras signaling pathway11,12 through the guanine nucleotide exchange factor Sos.13 Grb2 overexpression has been found in breast cancer cells.14 Since the initial discovery, Grb2 has been linked to a host of other cellular pathways including the actin cytoskeleton and endocytosis.15
Theoretical studies suggest the possibility that EGFR clustering into subcellular domains (or perhaps protein islands) may indeed influence the binding of cytoplasmic adaptors such as Grb2.16 Comparison of randomly dispersed EGFR dimers and clustered EGFR distributions predicted the retention of EGFR–Grb2 complexes in clusters for a longer period than the randomly distributed dimeric EGFR distributions. Consequently, it is important to measure the cluster size of adaptor-bound EGFR and to determine whether there is a relationship between adaptor binding and receptor cluster size.
We have made use of a method developed in our earlier work to determine the relative cluster size of complexed versus uncomplexed molecules on the surface of cells.9 The method, called FRET–FLIM–ICS,17 combines two well-established techniques: (1) lifetime-detected (FLIM) FRET,18 which can be used to measure complex formation, and (2) image correlation spectroscopy (ICS),19−23 which can be used to determine cluster densities and sizes. Sorkin and co-workers previously established that FRET can be used to detect interactions between EGFR and Grb2, but no estimates of cluster sizes were made in that study.32
To ascertain the relative importance of dimers versus higher-order oligomers, one requires a model system wherein higher-order oligomerization has been established and characterized. Such a system is provided by murine BaF/3 cells stably transfected with EGFR coupled at the C-terminus to an enhanced GFP tag (EGFR–eGFP). In prior work with this system, we found that EGFR–eGFP is predominantly dimeric in the absence of ligand (with less than 10% of EGFR–eGFP being monomeric) and forms phosphorylated tetramers upon exposure to ligand.8,24,25 The results reported here were obtained using two related BaF/3 cell lines: the cell line used in prior work,8 which expresses EGFR–eGFP at physiological levels (i.e., 50 000 to 70 000 copies per cell), and a new cell line expressing both EGFR–eGFP and Grb2–mRFP.
Our report is organized as follows. First, we present FLIM data for EGFR–eGFP BaF/3 cells and EGFR–eGFP/Grb2–mRFP BaF/3 cells with different combinations of EGF stimulation. Using the cell-phasor approach to lifetime microscopy,26−29 we detected EGF-dependent FRET between EGFR–eGFP and Grb2–mRFP. Second, we present the results of FRET–FLIM–ICS analysis of individual EGFR–eGFP/Grb2–mRFP BaF/3 cells. The results indicate that Grb2–mRFP associated with EGF-EGFR–eGFP complexes that are more than 4-fold brighter than EGF-EGFR–eGFP complexes not associated with Grb2–mRFP. The brightness ratio, as well as Grb2–mRFP association with EGFR–eGFP, correlates positively with EGFR–eGFP cluster density. Third, we present a rule-based model, which we have used to interpret our experimental data. The model is an extension of our earlier model25 and now includes EGFR–Grb2 binding in addition to the processes considered in the original model (i.e., ligand–receptor binding, self-interactions capable of mediating receptor oligomerization, and receptor autophosphorylation). In the extended model, receptors are considered to be bivalent, with sites of self-interaction in the ectodomain and the cytoplasmic domain. The model allows for the formation of extended polymer-like chains (up to tetramers) as well as the formation of a cyclic receptor tetramer. Oligomers larger than dimers emerge through a combination of ectodomain–ectodomain and kinase–kinase interactions. Significantly, in agreement with our new experimental observations, the model predicts that Grb2 associates predominantly with EGFR tetramers. We conclude that ligand-induced EGFR tetramers can play an important role in sequestering Grb2, Grb2-associated proteins, and possibly other proteins that directly interact with phosphorylated EGFR.
Experimental Procedures
Construction of Grb2–mRFP Plasmid
Human Grb2 (growth factor receptor-bound protein-2) transcript variant 1, as 10 μg of transfection-ready DNA, was purchased from OriGene (catalog no. SC111933) in the vector pCMV6-XL5. Grb2 was amplified from the Origene clone using the Invitrogen Pfx kit according to manufacturer’s instructions with the following primers: forward 5′ GGA TAC GTA GGG TGG CAT TGT GTG TCC CAG (incorporating a SnaB1 site) and reverse 5′ TGA GAC GTT CCG GTT CAC GGG GGT GAC ATA. The PCR product was purified and cloned into pPCRScript Amp, and resulting clones were sequenced. A correct forward-orientation clone was then cloned into pMonoRed using HindIII and SacII. Positive clones were identified by analytical restriction enzyme digest and then confirmed with sequencing. Grb2-pMonoRed was further subcloned into pBABE, a puromycin vector. The fragment was amplified by PCR using the same forward primer as was used previously and the reverse primer 5′ TGA GTC GAC TTA GGC GCC GGT GGA GTG GCG. A band of the correct size was excised from an agarose gel and purified used a QIAGEN gel extraction kit per the manufacturer’s instructions. Both pBABE and the PCR fragment were digested with Sal1 and SnaB1, ligated, and transformed. The resulting Grb2–RFP–pBABE clones were confirmed by analytical digests and sequencing.
Cells and Reagents
The murine hemopoietic cell line BaF/3 expressing C-terminally tagged EGFR–eGFP constructs has been described previously.8 BaF/3 cells expressing EGFR–eGFP and mRFP–Grb2 were produced by cotransfection of EGFR–eGFP and mRFP–Grb2 plasmids. Murine EGF was purified from mouse submaxillary glands as described previously.30
Live-Cell Microscopy
Suitable clones of BaF/3 cells (transfected with EGFR–eGFP or cotransfected with EGFR–eGFP and mRFP–Grb2) were selected using flow cytometry as described previously.8 Cells from each clone were collected by centrifugation (5 mL culture, 1400 rpm, 4 min, 4 °C), serum starved for 3 h at 37 °C in serum-free medium, and then resuspended in PBS containing 0.25% BSA and 10 μM phenyl arsine oxide (to block receptor internalization7−9). Half of the cell suspension was treated with EGF (final concentration: 16 nM) and half with an equivalent volume of buffer. After 20 min, the cells were aliquoted onto a coverslip of an inverted chamber (ambient conditions, ca. 23 °C) and imaged with a frequency-domain lifetime-imaging microscope (100× NA1.2 oil objective, 470 nm LED, FITC filter block, Nikon TE2000U microscope; Nikon Inc., Japan) coupled to a LIFA lifetime attachment (Lambert Instruments, The Netherlands). Lifetime images were corrected for instrument response (pixel-dependent instrument phase and modulation) with a solution of rhodamine 6G in distilled water (lifetime: 4.1 ns).31 BaF/3 cells (nontransfected) were also measured to determine the lifetime characteristics of cell background fluorescence.
Data Analysis
FLIM
An intensity threshold was applied to isolate the fluorescence from the BaF/3 cells. The fluorescence lifetime measurements were represented in two different ways. First, to get an indication of trends, the average phase lifetime and average modulation lifetime were determined for each BaF/3 cell along with the average values for a number of cells. The second approach utilized the AB plot28 (also referred to as phasor27 or polar plot29) to display the lifetime experiments graphically. This plot represents an experiment by a point in 2D space defined by x = m cos φ and y = m sin φ (where φ is the phase and m is the modulation of the fluorescence signal). This graphical approach has the advantage that the type of fluorescence decay (simple, complex, or excited-state reaction/solvent relaxation) and the complexity of the system trajectory (binary or more complex) can be deduced visually without further analysis. The phasor of single-exponential-decaying fluorophores lies on a semicircle described by m = cos φ that intersects with points (0,0), (0.5,0.5) and (1,0). Phasors from heterogeneous fluorescence decays lie in the region within the semicircle and follow the inequality m < cos φ. The linear combination of two phasors is described by a linear trajectory in AB space. This enables the distinction between optical mixing of two species and FRET to be made.
FRET–FLIM–ICS with Two Species
The FRET–FLIM–ICS procedure for two species was outlined in two previous publications.9,17 In essence, three images (phase, modulation, and intensity) are converted into two images: one that represents the spatial intensity distribution from the FRET species and the other the image of the non-FRET species.
For a given phasor, r(x,y), the fractional fluorescence from the FRET states, fFRET, is given by
| 1 |
where r(x,y)NFRET is the (constant) phasor for the non-FRET state and r(x,y)FRET is the (constant) phasor for the FRET state. These phasor values are fixed and determined using global analysis procedures as outlined.27,28
The fluorescence of the FRET species, IFRET, is a function of the total intensity, ITOT, and the fractional fluorescence resulting from the FRET species, fFRET, according to eq 1,
| 2 |
A similar relationship pertains to the non-FRET species, INON-FRET
| 3 |
Applying eqs 1–3 to each pixel enables fluorescence images of FRET and non-FRET states to be produced. Image correlation spectroscopy techniques are then applied to the fluorescence images representing the FRET and non-FRET species.
The density of clusters (number of FRET clusters per beam area) containing molecules undergoing FRET is given by the reciprocal of the amplitude of the spatial autocorrelation function (g(0))
| 4 |
The cluster density of molecules not undergoing FRET is given by
| 5 |
If the expression level of receptors is known, then the brightness or oligomeric state of the FRET and non-FRET clusters, BFRET and BNON-FRET, can be determined
 |
6 |
A more robust measure, which does not require knowledge of expression level, is the brightness ratio or BR, which is a function of the measured cluster densities (CDFRET and CDNFRET), fractional FRET fluorescence (fFRET), and lifetimes of the FRET and non-FRET states (τFRET and τNFRET)
| 7 |
Equation 8 can be written in more compact form using concentration fraction ratios
| 8 |
where fraction/(1 – fraction) is equal to τNFRETfFRET/τFRET(1 – fFRET).
The brightness ratio is a particularly useful index if there is a correlation between the occurrence of FRET and oligomerization or dissociation. BR = 1 implies no correlation between FRET and oligomeric state, BR > 1 implies oligomerization is linked to FRET, and BR < 1 implies that the lower-order oligomers are associated with FRET.
FRET–FLIM–ICS with Three Species
The total phasor for FRET, non-FRET, and background species is given by the equation
| 9 |
The symbols have been defined above (see eq 1) and r(x,y)BK represents the phasor for the background fluorescence.
Subtracting the background phasor from both sides we have
| 10 |
Similar to the two-species case, the phasor values are fixed, and NFRET and BK can be determined from cells containing donor-only and untransfected cells, respectively. The FRET phasor can be determined using global analysis methods using the approaches previously described.27,28 By inversion of eq 10 (using the cosine and sine components of the phasor), the fractional fluorescence contributions from FRET and NFRET can be extracted. Using the values of fFRET and fNFRET, the procedures outlined for the two-species case can then be followed.
Geometric Transformation of Phasor Approach
If the components of the non-FRET, background, and FRET phasor values are known, then eq 10 can be solved exactly to yield the fraction of three species (i.e., two equations and two unknowns). An alternative procedure based on the linear properties of the phasor representation itself can be utilized that essentially subtracts the contributions from the background and non-FRET states without explicit knowledge of the FRET values. The steps required to achieve this are (i) to add a constant phase to all pixels in the phase image Δ = (π/2 – arctan((m sin φNFRET – m sin φBK)/(m cos φNFRET – m cos φBK)), creating a new m cos(φi + Δ) image, and then (ii) to subtract a constant value (m cos(φb + Δ)) from all pixels in the m cos(φi + Δ) image. In essence, this rotates and translates the phasors such that the line connecting the background and non-FRET phasors is parallel to the y axis and intersects the origin, making (m cos φ)NFRET and (m cos φ)BK both zero. The transformed M cos φ image then takes the form
| 11 |
For a constant FRET phasor value ((m cos(φ + Δ)FRET), the transformed M cos φ is proportional to fFRET, which is the fraction FRET species in the pixel. Because of this proportionality, knowledge of the actual FRET phasor value is not required for the subsequent ICS analysis.
Model
A rule-based model reported earlier25 was extended to include one new rule for Grb2 interaction with phosphorylated EGFR (pEGFR) (Figures 1 and 2). This rule is associated with two rate constants and corresponding mass-action rate laws for Grb2–pEGFR association and dissociation. The rate constants were set at values consistent with an equilibrium dissociation constant (0.713 μM) and a dissociation rate constant (0.31/s) reported in the literature.33 In addition to these parameters, a copy number for Grb2 was also introduced to the model. This parameter was adjusted to account for the high percentage of EGFR–eGFP bound to Grb2–mRFP observed in our experiments. The model was formulated using BNGL; it was simulated using BioNetGen.34 A model specification that can be processed by BioNetGen is provided as a plain-text file in the Supporting Information. The file includes parameter estimates, annotation, and simulation instructions.
Figure 1.

Proteins (EGF, EGFR, and Grb2), EGFR component states, and protein–protein interfaces considered in our computational model. The EGFR ectodomain is taken to be free or bound to EGF. A cytoplasmic domain of EGFR, comprising the juxtamembrane region (JM) and kinase domain, is taken to be locked (i.e., unavailable for interaction) or freed (i.e., available for interaction). The C-terminal tail of EGFR is taken to contain, as a simplification, a single docking site for Grb2, which can be unphosphorylated (Y) and inactive or phosphorylated (pY) and active.
Figure 2.

Illustration of the rules for interactions in our computational, rule-based model. The model, which captures the mass-action chemical kinetics of the indicated interactions, consists of 16 rules, which are either reversible (and associated with two rate constants) or unidirectional (and associated with a single rate constant). Each rule represents an interaction. The glyphs used here to represent proteins and protein components are the same as those presented in Figure 1. Here, in illustrating a rule, we use a question mark (?) to indicate a missing protein component or component state that is not depicted explicitly; the missing component or state is taken to have zero influence on the interaction represented by the rule. Similarly, representation of an EGFR ectodomain by a dotted triangle is meant to indicate that the ectodomain may or may not be present in a complex, without influence on the interaction of concern. The model is the same as that presented in our earlier report25 except that a rule for Grb2 binding to phosphorylated EGFR has been added. This rule is illustrated in the lower left box.
The model has several notable features. It includes a cyclic EGFR tetramer and a ligand-triggered conformation change that frees the EGFR kinase domain to participate in kinase–kinase interactions, which are necessary for kinase activity. A 3D structural model with atomic resolution recently constructed by C.-S. Tung35 suggests that the EGFR tetramer considered in the model can feasibly form. Moreover, the model is consistent with observed negative cooperativity in EGF binding to intact EGFR as well as positive linkage between EGF dose and the size of EGF-induced EGFR clusters.
Results
FRET–FLIM Studies of the Interaction of EGFR–eGFP with Grb2–mRFP
Previously, Sorkin et al.32 used FRET microscopy to image the association of EGFR–CFP with Grb2–YFP in porcine aortic cells using filter-based fluorescence imaging. FRET is particularly sensitive to such interactions because of the highly nonlinear dependence of FRET rate, R, on distance, D, between donor and acceptor labels (R is proportional to D–6 on the length scale of 1–10 nm). To quantitatively measure the interaction between EGFR–eGFP and Grb2–mRFP in BaF/3 cells, we utilized lifetime-detected FRET–FLIM microscopy, which enables a robust evaluation of FRET efficiency based on the quenching of donor lifetime in the presence of acceptor.
Table 1 summarizes the time-resolved fluorescence parameters obtained using frequency-domain FLIM from measurements on several sets of cells. Figure 3 portrays the time-resolved experiments in terms of a polar plot, which is also called an AB or phasor plot.26−29
Table 1. Summary of FLIM Parameters for EGFR–GFP in Living BaF/3 Cells.
| ligand | adaptor | τphasea | τmodb | m cos(φ)c | m sin(φ)c | Nd | τe | Ef |
|---|---|---|---|---|---|---|---|---|
| no | no | 2.82 | 2.81 | 0.67 | 0.47 | 48 | 2.82 | n.a. |
| yes | no | 2.82 | 2.76 | 0.67 | 0.48 | 23 | 2.85 | n.a. |
| no | yes | 2.67 | 3.26 | 0.64 | 0.43 | 60 | 2.76 | 0.03 |
| yes | yes | 2.46 | 3.20 | 0.78 | 0.67 | 57 | 2.50 | 0.11 |
| background | 2.30 | 5.50 | 0.51 | 0.29 | 80 | n.a | n.a. |
Lifetime calculated from the phase of the fluorescence at 40 MHz (±0.011 ns).
Lifetime calculated from the modulation of the fluorescence at 40 MHz (±0.015 ns).
Components of the cell population FLIM phasor: m represents the modulation and φ represents the phase.
Number of cells.
Apparent lifetime calculated by subtraction of background phasor.
Apparent FRET efficiency calculated as E = 1 – (apparent lifetime(donor + adaptor)/2.8 ns).
Figure 3.
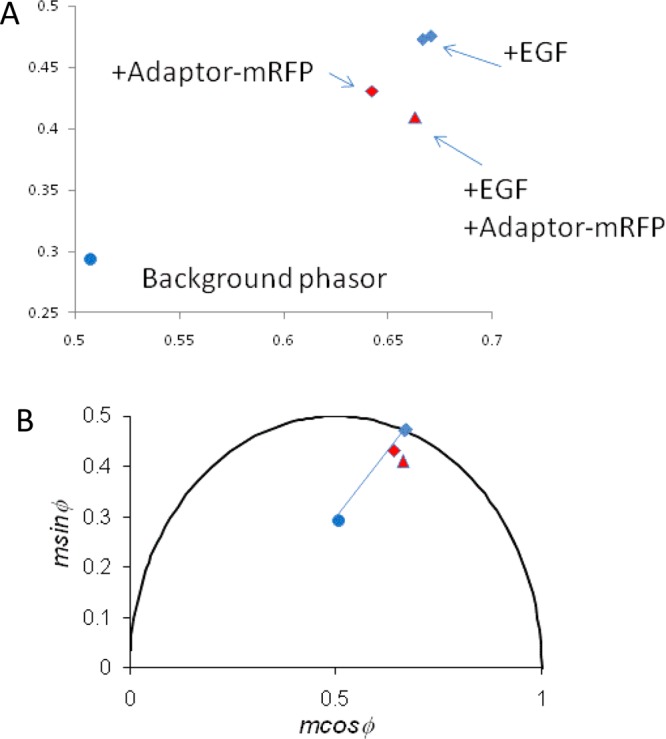
FLIM data of living BaF/3 cell populations represented on a phasor diagram. (A) Phasor diagram over a limited data range. (B) Phasor diagram on an expanded scale. Individual data points represent the cell-phasor components [x = m cos(φ); y = m sin(φ)] averaged from >20 cells. Data points correspond to BaF/3 cells transfected with EGFR–eGFP alone (blue diamond), EGFR–eGFP + EGF (second blue diamond), EGFR–eGFP/Grb2–mRFP (red-filled diamond), EGFR–eGFP/Grb2–mRFP + EGF (red-filled triangle), and untransfected control cells (blue filled circle).
In the absence of EGF or Grb2–mRFP, the emission from BaF/3 cells expressing EGFR–eGFP was characterized by a phase lifetime of 2.82 ± 0.01 ns and a modulation lifetime of 2.81 ± 0.02 ns. Importantly, addition of EGF did not appreciably affect phase or modulation of EGFR–eGFP fluorescence (Table 1).
BaF/3 cells cotransfected with EGFR–eGFP and Grb2–mRFP displayed perturbations to time-resolved fluorescence that was reflected in the lifetime parameters (Table 1). The EGFR–eGFP phase lifetime decreased from 2.82 ± 0.01 (in the absence of Grb2–mRFP) to 2.76 ± 0.02 ns (in the presence of Grb2–mRFP), and the modulation lifetime increased. In the presence of both EGF and Grb2–mRFP, the phase lifetime of EGFR–eGFP decreased further to 2.46 ± 0.01 ns, whereas the modulation lifetime remained at the value in the presence of Grb2–mRFP.
Insight into the physical mechanisms responsible for the observed changes is gained by inspection of the phasor plot (Figure 3) together with the phasor components (m cos(φ), m sin(φ)) of the time-resolved emission from the cells and background. The position of the phasor corresponding to BaF/3 cells containing EGFR–eGFP is close to, but not on, the universal circle, which indicates non-exponential behavior from the eGFP fluorophore of EGFR–eGFP (Figure 3, blue diamond, and Table 1).27−29 As expected, EGF treatment does not significantly change the phasor position of EGFR–eGFP (second blue diamond and Table 1). However, in the cells containing both EGFR–eGFP and Grb2–mRFP, the phasor lies further inside the semicircle (Figure 3A, red diamond). This change in phasor position can be explained by fractional emission because of the cell’s background fluorescence (i.e., by background mixing only). Background mixing only (i.e., no FRET) is apparent because the background phasor (Figure 3B, blue circle), the EGFR–eGFP phasor (Figure 3B, blue diamond), and the EGFR–eGFP/Grb2–mRFP phasor (Figure 3B, red diamond) are almost collinear (Figure 3). In contrast, EGF treatment of EGFR–eGFP/Grb2–mRFP cells moves the phasor of EGFR–eGFP/Grb2–mRFP in a clockwise direction such that it is no longer collinear with the background and EGFR–eGFP cell phasors (red triangles). These results provide qualitative evidence for an interaction between EGFR–eGFP and Grb2–mRFP after EGF treatment.
Using the approach of Caiolfa et al.,36 we can determine whether FRET is significant by removing the background contribution. By drawing a line connecting the background phasor to the observed phasor, an apparent lifetime (i.e., lifetime in the absence of background) can be obtained from the intersection point of the line with the semicircle. The apparent lifetimes calculated in this manner are listed in Table 1. This analysis yielded an EGFR–eGFP lifetime of 2.76 ± 0.03 ns in EGFR–eGFP/Grb2–mRFP cells compared with 2.82 ± 0.03 ns in cells lacking Grb2–mRFP. A Student’s t test revealed that the difference between 2.81 and 2.76 ns was not statistically significant at the 95% confidence interval (p > 0.1, standard error about the mean 0.03 ns, sample size = 50). Using the same background subtraction procedure, a lifetime of 2.5 ± 0.03 ns was obtained for EGFR–eGFP in the presence of EGF and Grb2–mRFP. A Student’s t test revealed that the difference in apparent lifetimes (2.85 vs 2.50 ns) was highly significant (p < 0.0001, standard error about the mean 0.03 ns, sample size = 50). These results provide quantitative evidence for an EGF-dependent interaction between EGFR–eGFP and Grb2–mRFP.
To gain further insight into the interaction, we examined the phasors of individual EGFR–eGFP/Grb2–mRFP BaF/3 cells in the presence of EGF (Figure 4). It is apparent that some cells have phasors that are close to the phasor of the EGFR–eGFP donor. Our intepretation is that this population of cells exhibits no or low FRET. This interpretation is supported by the fact that these cells appear close to the line joining the EGFR–eGFP phasor and the background phasor (Figure 4, blue line). The other populations of cells have phasors that lie on a line connecting background, donor, and a highly quenched fluorescence. Using linear extrapolation28 (Figure 4, red line), we calculate that the lifetime of the FRET state is 0.71 ± 0.03 ns. This FRET state lifetime is physically reasonable and is similar to the 0.75 ns lifetime determined for EGFR–eGFP interacting with Cy3-labeled antiphosphotyrosine antibodies.27 According to this interpretation, the different phasor positions for individual cells can be explained by different proportions of free EGFR–eGFP and EGFR–eGFP/Grb2–mRFP complexes. This information can be used to determine whether there is a link between EGFR cluster size and Grb2–mRFP binding.
Figure 4.
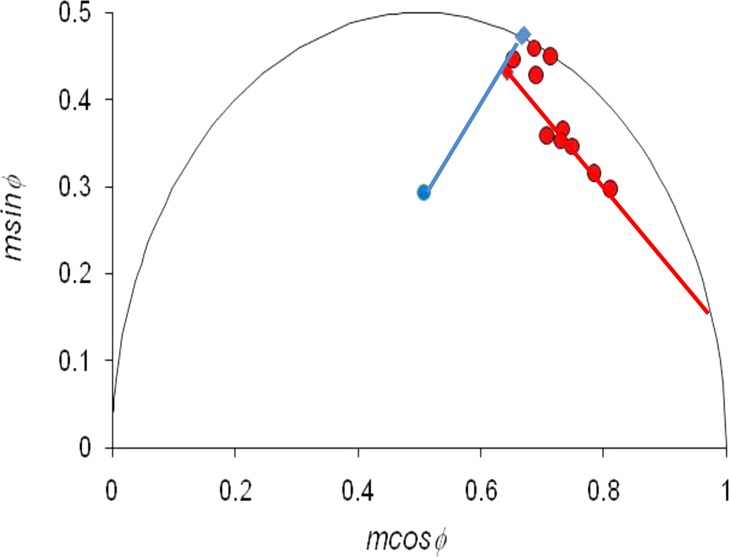
FLIM data of individual living BaF/3 cells represented on a phasor diagram. Data points correspond to BaF/3 cells transfected with EGFR–eGFP alone (blue diamond), EGFR–eGFP + EGF (second blue diamond), EGFR–eGFP/Grb2–mRFP (red-filled diamond), EGFR–eGFP/Grb2–mRFP + EGF (red-filled triangle), and untransfected control cells (blue filled circle). Blue solid line denotes trajectory for mixtures of background and EGFR–eGFP. Red line indicates trajectory for EGFR–eGFP/Grb2–mRFP FRET complex mixing with background and EGFR–eGFP fluorescence.
FRET–FLIM–ICS Measurements Reveal That Grb2 Is Associated with Higher-Order EGFR Clusters
To assess the relative sizes of Grb2–bound versus Grb2-free EGF–EGFR–eGFP oligomers in EGFR–eGFP/Grb2–mRFP BaF/3 cells, we used a FRET–FLIM–ICS approach that was modified to take background fluorescence into account. Figure 5A displays a typical fluorescence image from EGF–EGFR–eGFP/Grb2–mRFP complexes, and Figure 5B displays the corresponding 2D spatial autocorrelation function image. The amplitude of the autocorrelation function is inversely related to the cluster density of an EGF–EGFR–eGFP/Grb2–mRFP complex (eq 4). Analogous images of the EGF–EGFR–eGFP complexes were also obtained, and the cluster densities were calculated (eq 5). A summary of lifetime parameters, fraction of EGFRs undergoing FRET, cluster densities, and brightness ratios obtained from the analysis of several cells is shown in Table 2. The fraction FRET values are positively correlated with the cluster density FRET values (correlation coefficient 0.81) and also positively correlated with the brightness ratio values (correlation coefficient 0.74), implying that EGFR clustering and Grb2 adaptor association are linked. We shall use the data in Table 2 to extract the information on the sizes of EGFR–Grb2 complexes.
Figure 5.
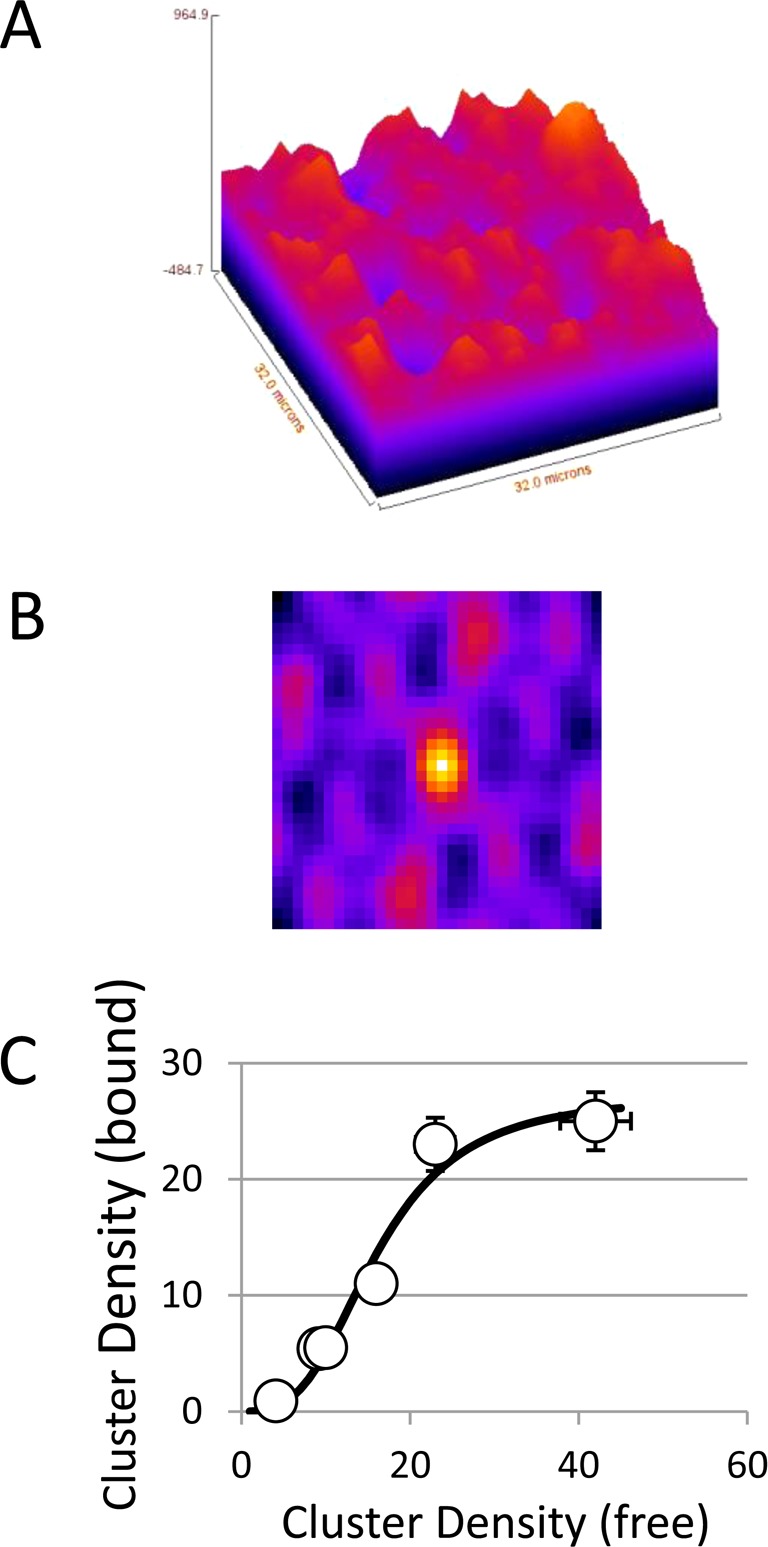
FRET–FLIM–ICS on living BaF/3 cells cotransfected with EGFR–eGFP and Grb2–mRFP. (A) Fluorescence image of EGFR–eGFP/Grb2–mRFP complexes. (B) Spatial autocorrelation image of EGFR–eGFP/Grb2–mRFP complexes. (C) Density of Grb2–mRFP-bound EGFR–EGFP clusters as a function of the density of Grb2–free EGFR–eGFP clusters. The solid line is fit to a Hill function (CDbound = A/(1 + ((Kd/CDfree)(N−1)), with N = 4.1, A = 27, and Kd = 18 clusters).
Table 2. Fluorescence Parameters Including Lifetime, Average Degree of Adaptor Binding, and Relative Brightness for EGF-Treated Live Cells Expressing Both EGFR–eGFP and Grb2–mRFP.
| experiment | τphasea | τmodb | fractionboundc | CDboundd | CDfreed | relative brightnesse |
|---|---|---|---|---|---|---|
| 1 | 1.41 | 2.20 | 0.86 | 23 | 23 | 5.9 |
| 2 | 1.60 | 2.51 | 0.81 | 11 | 16 | 6.2 |
| 3 | 1.46 | 2.32 | 0.85 | 25 | 42 | 9.3 |
| 4 | 1.84 | 2.73 | 0.72 | 5.5 | 10 | 4.7 |
| 5 | 1.92 | 2.87 | 0.69 | 5.4 | 9.2 | 3.8 |
| 6 | 2.37 | 3.03 | 0.42 | 0.9 | 4.1 | 3.3 |
| average | 0.725 | 11.7 | 17.3 | 5.5 | ||
| SEM | 0.06 | 4 | 5 | 0.9 |
Lifetime calculated from the phase of the fluorescence at 40 MHz (±0.011 ns).
Lifetime calculated from the modulation of the fluorescence at 40 MHz (±0.015 ns).
Fraction of receptors bound to adaptor (i.e., fraction EGFR–eGFP bound to Grb2–mRFP).
Cluster densities or number of aggregates per square micrometer. The label “bound” refers to images containing only EGFR–eGFP bound to Grb2–mRFP. The label “free” refers to images containing only EGFR–eGFP uncomplexed with Grb2–mRFP.
Relative brightness (RB) of EGFR–eGFP/Grb2–mRFP complexes compared to uncomplexed EGFR–eGFP. Calculated from the equation RB = (fraction)g(0)bound/(1 – fraction)g(0)free
One estimate of cluster size comes from measurement of the cell-averaged cluster densities (Table 2 and eq 6). Grb2-bound EGFR clusters were dispersed at an average of 11 clusters/μm2, whereas the Grb2-free EGFR population had a spatial organization characterized by an average cluster density of 17 clusters/μm2. Considering that on average 74% of EGFRs make up the Grb2-bound population and 36% the unbound population, the normalized density for 100% Grb2-bound EGFR clusters would be 16 clusters/μm2 and 100% Grb2-free EGFR clusters would be 63 clusters/μm2. These cluster density estimates agree remarkably well with cluster densities previously determined for EGFR–eGFP in BaF/3 cells,8 namely, a tetramer density of 17 clusters/μm2 and a monomer density of 70 clusters/μm2. Accordingly, the estimated size of clusters containing Grb2-bound EGFR would be about 4.2 ± 1 receptors/cluster and the estimated size of clusters free of Grb2 would be about 1.1 ± 0.3 receptors/cluster.
Examination of cluster densities across different cells is also informative (Table 2). The local EGFR 2D concentration sampled in experiments varies over a wide range (Table 2; CDfree: 4.1–40 clusters/μm2, CDbound: 0.9–25 clusters/μm2), with a positive correlation between increases in CDfree with CDbound. A plot of CDbound as a function of CDfree is depicted in Figure 5C and closely resembles a sigmoidal binding curve. In the context of a simple local equilibrium between EGFR and EGFR–Grb2 complexes of size N, the concentration of EGFR–Grb2 clusters as a function of EGFR is given by a Hill function of the form CDbound = A/(1 + ((Kd/CDfree)(N−1)), where A, Kd, and N are constants. Nonlinear least-squares fit to the data (solid line, Figure 5C) revealed (N – 1) = 3.1 and therefore N = 4.1. This result suggests that in the context of a simple monomer/N-mer equilibrium the cluster size of the Grb2-bound EGFR is approximately 4.
A third method of analysis utilizes a direct calculation of the brightness ratio for each individual cell (eq 8). The brightness ratios obtained in this manner ranged from 3.3 to 9.3, with a cell average of 5.5 ± 1. It is difficult to determine whether the observed range of brightness ratios represents true cell-to-cell variation in the proportions of different oligomeric states or errors in the determination of cluster densities and fraction FRET values. Using the error calculation procedure from Wiseman’s laboratory,37 we estimate that the error in the cluster densities is approximately 10–20% about the mean value. We take an error of 30% in the cluster density to be conservative. The FRET fraction error is estimated to be 0.05. By error propagation analysis of eq 8, the BR has a 95% confidence interval given by BR ± (2 × 0.43)BR. It is clear that N = 2 cannot explain the spread of BR values because it covers the range of BR = [0.3,3.7]. In order to match to the experimentally determined range BR = [3.3,9.3], the error in the BR would have to be 150% of N = 2, which is unrealistic. However, models with oligomers of higher order than dimers can account for the data, with a BR of 4 to 5 accounting for the majority of observed BR values.
To summarize, using the combined data from the cells examined, we obtained an estimated average cluster size of approximately 4 ± 1 receptors/cluster for the adaptor-bound receptor pool. We next compare these data with our model for receptor aggregation and adaptor binding.
Theoretical Model of Receptor Oligomerization and Adaptor Binding
We recently presented a model for ligand binding, higher-order receptor oligomerization, and receptor phosphorylation that reproduced our biophysical and biochemical experiments with EGFR–eGFP in BaF/3 cells.25 Key ingredients of the model are (i) monomer–dimer equilibrium in the absence of ligand, (ii) retention of negative cooperativity in the ligand-binding step using features and parameters of the model of Pike and Macdonald,38 (iii) inclusion of a ligand-induced conformational transition leading to liberation of the kinase domain in the ligand-bound receptor (making it available for interaction with a neighboring kinase domain, also in the liberated state), (iv) receptor phosphorylation in oligomers of size 2 or larger, (v) ectodomain and kinase domain interactions between receptors leading to linear polymer-like aggregates up to tetramers (i.e., dimers, trimers, and tetramers) as well as cyclic tetramers, and (vi) phosphorylation and dephosphorylation reactions.
To relate the present experimental data to models of adaptor binding, we augmented our previous model to include a Grb2-binding step characterized by parameters determined via surface plasmon resonance.33 In the model, we allowed Grb2 to bind a phosphorylated receptor regardless of its aggregation state. This aspect of the model is consistent with the conventional view that adaptors bind exposed phosphotyrosine residues on receptors and our experiments showing that there is only measurable FRET between EGFR and Grb2 after EGF stimulation. We calculated the predicted cluster size distribution of EGFR and EGFR–Grb2 complexes. Figure 6A shows that the dominant Grb2-bound cluster is the EGFR tetramer at all concentrations of EGF. Figure 6B displays the proportion of each cluster bound to Grb2 from a simulation with 10 nM EGF and parameters fixed from our previous publication. Tetramers are the dominant species bound to Grb2, with nearly 95% of tetramers containing at least one bound Grb2 (Figure 6B). The trimers and dimers make up only a small fraction of the total population (<1% of total population); however, the majority of both the trimeric and dimeric pools do not contain any bound Grb2 (Figure 6B). The second largest species in terms of population is the EGFR monomer, which contributes to 28% of the clusters. Ninety-eight percent of the EGFR monomer is not bound to Grb2 (Figure 6B). These results agree well with the cluster size estimates from experiments. The average value of 4 ± 1 EGFR’s per EGFR–Grb2 complex from the FRET–FLIM–ICS experiments is in excellent agreement with the model prediction that the EGFR tetramer is the dominant Grb2-bound form, whereas the dominant Grb2-free form is the monomer, which also agrees well with the cluster size estimate of 1 ± 0.3 EGFR’s from the cell-averaged data.
Figure 6.
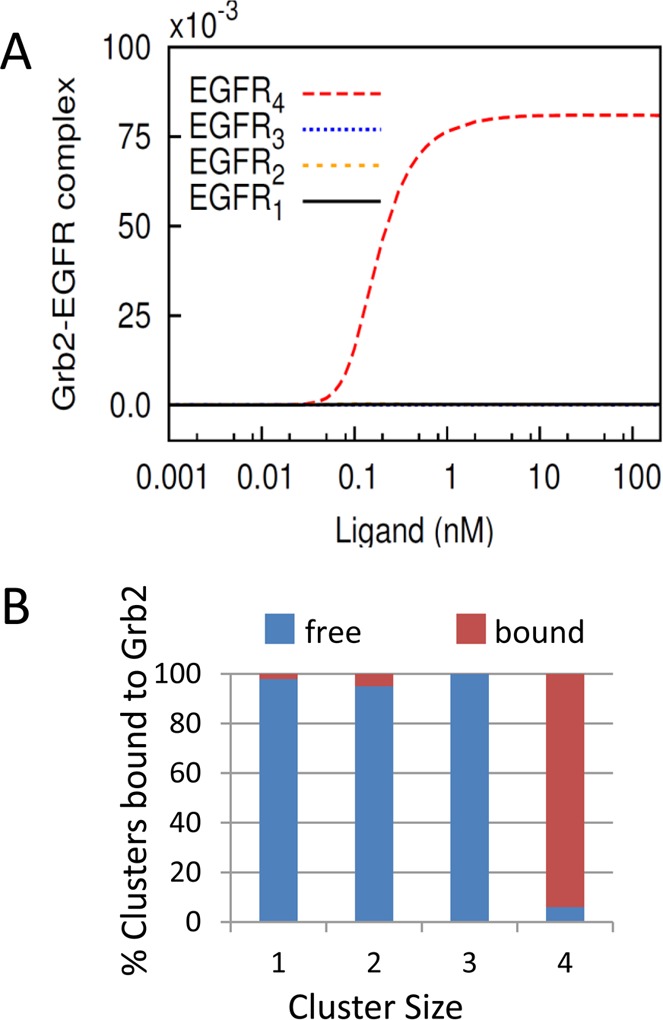
(A) Plot of simulation results depicting the cluster distribution of Grb2-bound EGFR as a function of EGF concentration. Note that at all concentrations of EGF the EGFR tetramer is the predominant form associated with Grb2. The curves corresponding to dimer and trimer are indistinguishable from monomer because the total number of these oligomeric forms bound to Grb2 is almost negligible. (B) Cluster size distribution of EGFR bound to Grb2 and unbound (free) to Grb2 from simulation with 10 nM EGF.
Discussion
Alternative Interpretations
The model above assigns the receptor clustering and adaptor binding to an oligomeric complex formed entirely from extracellular and kinase-activated intracellular domains of the EGFR. However, it is important to consider the following alternative explanations of the observed behavior:
Coated Pits
It is known that after activation EGFR is transported to coated-pit regions of the cell membrane, where Grb2 is also colocalized. Therefore, a trivial explanation might be that the EGFR–Grb2 clusters we observed are multiple copies of activated EGFR dimers that have assembled in coated pits. For example, Nagy et al. reported pentamers of EGFR that were associated with coated pits.39 However, there is evidence that EGFR clusters can also form outside coated pits. First, the measured average cluster density of the EGFR–Grb2 complexes (Table 2, CDbound = 11 clusters/μm2) is an order of magnitude larger than typical coated pit densities (CDcoated-pits = 0.5 clusters/μm2).40 Second, high-resolution imaging has established that EGFR nanoclusters appear outside coated-pit regions of the cell membrane.41 Therefore, EGFR clustering cannot be solely attributed to EGFR accumulation in coated pits.
Subcellular Domains
Another possible explanation is that activated EGFR dimers are corralled into subcellular domains mediated via membrane rafts or possibly interactions with the cytoskeleton. If we assume that after EGFR dimer activation domain entry is a random partition process, then the domain occupancy distribution will be a Poissonian aggregate distribution and the equilibrium distribution between domain and nondomain sites will be given by a simple linear partition function. However, the CDfree versus CDbound plot (Figure 5C) does not fit as well to a linear model as it does to a sigmoidal Hill function; the residual sum of squares is approximately 4.6-fold greater for the linear model than for the Hill function. We suggest that although subcellular domains or lipid platforms may increase local concentrations to enhance oligomerization41 our data clearly does not support a simple domain accumulation model of activated dimers in the absence of higher-order oligomerization.
Adaptor-Mediated Receptor Cross-Linking
It has been reported that Sos and Grb2 can form a trimeric complex and this trimeric Grb2–Sos–Grb2 complex can cross-link phosphorylated transmembrane proteins.42 Such a mechanism might also account for the observation that Grb2-bound EGFR is in a higher-order oligomeric complex relative to Grb2-unbound EGFR and for the positive correlation between Grb2–mRFP binding and EGFR–eGFP clustering. The activated EGFR dimer has at least four phosphorylation sites (two per receptor monomer) that recognize Grb2, allowing for the possibility of multivalent (receptor dimer)–bivalent (adaptor complex) interactions. Our model does not include this adaptor-mediated cross-linking mode of interaction. Determination of the role of adaptor-mediated receptor cross-linking versus receptor-mediated oligomerization requires further investigation with cells that contain defined concentrations of Sos, Grb2, and EGFR.
Phenyl Arsine Oxide-Mediated Phosphorylation and Clustering
Our experiments were conducted in the presence of phenyl arsine oxide to block receptor internalization and to ensure that we are measuring cell-surface activation and clustering processes. The possibility remains that the phenyl arise oxide may influence receptor phosphorylation and clustering by keeping receptor activity/phosphorylation at an artificially high level. We argue that these effects are modest on the basis of experimental and theoretical grounds. First, as discussed in detail in our previous paper on receptor clustering and receptor phosphorylation,25 we see negligible effects of phenyl arise oxide on EGF-dose-dependent receptor cluster size and phosphorylation in our BaF/3 cell system at the concentrations of phenyl arsine oxide employed. Second, a sensitivity analysis, performed in our earlier modeling study,25 indicates that receptor tetramer formation is insensitive to changes in the values of the model parameters that govern receptor phosphorylation and dephosphorylation.
Receptor clustering was recognized more than 30 years ago as being important in the activation and biological functioning of the EGFR.43 Notably, Schlessinger’s laboratory revealed that the biological effects of EGF on cells could be mimicked using bivalent or polyvalent antibodies against the receptor but not using monovalent antibody fragments.43,44 Results from biochemical and structural studies in noncellular environments have produced refined models leading to the conclusion that an asymmetric kinase dimer is required for initial kinase activation (see refs (1, 7, and 45) for recent reviews). These studies are so elegant that the role of the higher-order oligomers or clusters, as distinct from the dimers, has been largely overlooked, although oligomerization has received some notable attention recently.7−9,20,46−52 In other cell-surface receptor systems, receptor oligomerization or clustering, as distinct from the initial activation event in dimers, is seriously considered as a biological control mechanism.53−58 In this study, we examined the possibility that the first step in the assembly of signaling complexes at the cell membrane, the binding of adaptor to activated receptor, might be influenced by receptor clustering. We did this by measuring the relationship between Grb2 binding and EGFR cluster size.
The following lines of evidence point to the enhanced propensity of higher-order oligomers of EGFR to bind the adaptor Grb2. First, the average brightness of EGFR–eGFP complexed with Grb2–mRFP is more than two times greater than uncomplexed EGFR–eGFP. Because the smallest possible aggregation state of the EGFR–eGFP is a monomer, this implies that higher-order EGFR oligomers bind Grb2–mRFP. Second, there is a positive correlation between fraction of EGFR’s bound to Grb2, average cluster size, and overall receptor density. Third, the distribution and proportion of Grb2-bound EGFR oligomers revealed by theoretical modeling indicated a predominance of the EGFR tetramer as the major Grb2-bound form.
What is the significance of adaptor binding to receptor clusters? Theoretical simulations from the Wilson/Edwards laboratories16 revealed that Grb2 association with EGFR is longer-lived if EGFR is clustered nonrandomly. Experimental support for this concept also comes from single-molecule studies that indicate a positive correlation between clustering of phosphotyrosine binding sites and increases in dwell time of SH2-containing proteins near the plasma membrane surface.56
To conclude, EGFR higher-order oligomers bind Grb2 and therefore should be considered, along with the classical EGFR dimer, in models of EGFR signaling. Moreover, the positive correlation between clustering of EGFR and EGFR–Grb2 interaction is consonant with the concept that the nonrandom spatial organization of receptor dimers, in our case, tetramers, can concentrate signaling complexes in space and time.
Glossary
Abbreviations Used
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Grb2
growth factor receptor-bound protein-2
- eGFP
enhanced green fluorescent protein
- mRFP
monomeric red fluorescent protein
- ICS
image correlation spectroscopy
- FRET
Förster resonance energy transfer
- FLIM
fluorescence-lifetime imaging microscopy
- PBS
phosphate-buffered saline solution
Supporting Information Available
Text file used for specification of the theoretical model using BioNetGen34 that includes parameter estimates, annotation, and simulation instructions. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was partially supported by Australian NHMRC Project Grants (nos. 280918 and 433624), NIH/NIGMS grant P50 GM085273, and DOE contract DE-AC52-06NA25396.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lemmon M. A.; Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen R. N.; Walker F.; Pouliot N.; Garrett T. P.; Ward C. W.; Burgess A. W. (2003) Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 284, 31–53. [DOI] [PubMed] [Google Scholar]
- Ekstrand A. J.; Sugawa N.; James C. D.; Collins V. P. (1992) Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. U.S.A. 89, 4309–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadella T. W. Jr.; Jovin T. M. (1995) Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 129, 1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fernandez M.; Clarke D. T.; Tobin M. J.; Jones S. V.; Jones G. R. (2002) Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 82, 2415–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan C. J.; Roberts S. K.; Rolfe D. J.; Clarke D. T.; Loeffler H. H.; Kastner J.; Winn M. D.; Parker P. J.; Martin-Fernandez M. L. (2011) Human epidermal growth factor receptor (EGFR) aligned on the plasma membrane adopts key features of Drosophila EGFR asymmetry. Mol. Cell. Biol. 31, 2241–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F.; Rothacker J.; Henderson C.; Nice E. C.; Catimel B.; Zhang H. H.; Scott A. M.; Bailey M. F.; Orchard S. G.; Adams T. E.; Liu Z.; Garrett T. P.; Clayton A. H.; Burgess A. W. (2012) Ligand binding induces a conformational change in epidermal growth factor receptor dimers. Growth Factors 30, 394–409. [DOI] [PubMed] [Google Scholar]
- Clayton A. H.; Walker F.; Orchard S. G.; Henderson C.; Fuchs D.; Rothacker J.; Nice E. C.; Burgess A. W. (2005) Ligand-induced dimer–tetramer transition during the activation of the cell surface epidermal growth factor receptor–A multidimensional microscopy analysis. J. Biol. Chem. 280, 30392–30399. [DOI] [PubMed] [Google Scholar]
- Clayton A. H.; Orchard S. G.; Nice E. C.; Posner R. G.; Burgess A. W. (2008) Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors 26, 316–324. [DOI] [PubMed] [Google Scholar]
- Webb S. E.; Roberts S. K.; Needham S. R.; Tynan C. J.; Rolfe D. J.; Winn M. D.; Clarke D. T.; Barraclough R.; Martin-Fernandez M. L. (2008) Single-molecule imaging and fluorescence lifetime imaging microscopy show different structures for high- and low-affinity epidermal growth factor receptors in A431 cells. Biophys. J. 94, 803–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein E. J.; Daly R. J.; Batzer A. G.; Li W.; Margolis B.; Lammers R.; Ullrich A.; Skolnik E. Y.; Bar-Sagi D.; Schlessinger J. (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock M.; McGlade J.; Mbamalu G.; Pelicci G.; Daly R.; Li W.; Batzer A.; Thomas S.; Brugge J.; Pelicci P. G.; et al. (1992) Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 360, 689–692. [DOI] [PubMed] [Google Scholar]
- Li N.; Batzer A.; Daly R.; Yajnik V.; Skolnik E.; Chardin P.; Bar-Sagi D.; Margolis B.; Schlessinger J. (1993) Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 363, 85–88. [DOI] [PubMed] [Google Scholar]
- Daly R. J.; Binder M. D.; Sutherland R. L. (1994) Overexpression of the Grb2 gene in human breast cancer cell lines. Oncogene 9, 2723–2727. [PubMed] [Google Scholar]
- Bisson N.; James D. A.; Ivosev G.; Tate S. A.; Bonner R.; Taylor L.; Pawson T. (2011) Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658. [DOI] [PubMed] [Google Scholar]
- Hsieh M. Y.; Yang S.; Raymond-Stinz M. A.; Edwards J. S.; Wilson B. S. (2010) Spatio-temporal modeling of signaling protein recruitment to EGFR. BMC Syst. Biol. 4, 57-1–57-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A. H.; Tavarnesi M. L.; Johns T. G. (2007) Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry 46, 4589–4597. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman E. A.; Jovin T. M. (2006) Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 10, 409–416. [DOI] [PubMed] [Google Scholar]
- St-Pierre P. R.; Petersen N. O. (1990) Relative ligand binding to small or large aggregates measured by scanning correlation spectroscopy. Biophys. J. 58, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre P. R.; Petersen N. O. (1992) Average density and size of microclusters of epidermal growth factor receptors on A431 cells. Biochemistry 31, 2459–2463. [DOI] [PubMed] [Google Scholar]
- Wiseman P. W.; Petersen N. O. (1999) Image correlation spectroscopy. II. Optimization for ultrasensitive detection of preexisting platelet-derived growth factor-beta receptor oligomers on intact cells. Biophys. J. 76, 963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen N. O.; Brown C.; Kaminski A.; Rocheleau J.; Srivastava M.; Wiseman P. W. (1998) Analysis of membrane protein cluster densities and sizes in situ by image correlation spectroscopy. Faraday Discuss. 289–305 and discussion on pp 331–243. [DOI] [PubMed] [Google Scholar]
- Kolin D. L.; Wiseman P. W. (2007) Advances in image correlation spectroscopy: Measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 49, 141–164. [DOI] [PubMed] [Google Scholar]
- Kozer N.; Kelly M. P.; Orchard S.; Burgess A. W.; Scott A. M.; Clayton A. H. (2011) Differential and synergistic effects of epidermal growth factor receptor antibodies on unliganded ErbB dimers and oligomers. Biochemistry 50, 3581–3590. [DOI] [PubMed] [Google Scholar]
- Kozer N.; Barua D.; Orchard S.; Nice E. C.; Burgess A. W.; Hlavacek W. S.; Clayton A. H. (2013) Exploring higher-order EGFR oligomerisation and phosphorylation–a combined experimental and theoretical approach. Mol. BioSyst. 9, 1849–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari C.; Cinquin A.; Cinquin O.; Digman M. A.; Donovan P. J.; Gratton E. (2011) Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc. Natl. Acad. Sci. U.S.A. 108, 13582–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A.; Caiolfa V. R.; Zamai M.; Gratton E. (2008) The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 94, L14–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A. H.; Hanley Q. S.; Verveer P. J. (2004) Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J. Microsc. 213, 1–5. [DOI] [PubMed] [Google Scholar]
- Redford G. I.; Clegg R. M. (2005) Polar plot representation for frequency-domain analysis of fluorescence lifetimes. J. Fluoresc. 15, 805–815. [DOI] [PubMed] [Google Scholar]
- Burgess A. W.; Lloyd C. J.; Nice E. C. (1983) Murine epidermal growth factor: Heterogeneity on high resolution ion-exchange chromatography. EMBO J. 2, 2065–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley Q. S.; Subramaniam V.; Arndt-Jovin D. J.; Jovin T. M. (2001) Fluorescence lifetime imaging: Multi-point calibration, minimum resolvable differences, and artifact suppression. Cytometry 43, 248–260. [DOI] [PubMed] [Google Scholar]
- Sorkin A.; McClure M.; Huang F.; Carter R. (2000) Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr. Biol. 10, 1395–1398. [DOI] [PubMed] [Google Scholar]
- Chook Y. M.; Gish G. D.; Kay C. M.; Pai E. F.; Pawson T. (1997) The Grb2–mSos1 complex binds phosphopeptides with higher affinity than Grb2. J. Biol. Chem. 271, 30472–30478. [DOI] [PubMed] [Google Scholar]
- Faeder J. R.; Blinov M. L.; Hlavacek W. S. (2009) Rule-based modeling of biochemical systems with BioNetGen. Methods Mol. Biol. 500, 113–167. [DOI] [PubMed] [Google Scholar]
- Chylek L. A.; Harris L. A.; Tung C. S.; Faeder J. R.; Lopez C. F.; Hlavacek W. S. (2014) Rule-based modeling: A computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip. Rev.: Syst. Biol. Med. 6, 13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiolfa V. R.; Zamai M.; Malengo G.; Andolfo A.; Madsen C. D.; Sutin J.; Digman M. A.; Gratton E.; Blasi F.; Sidenius N. (2007) Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J. Cell Biol. 179, 1067–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino S.; Comeau J. W.; Kolin D. L.; Wiseman P. W. (2005) Accuracy and dynamic range of spatial image correlation and cross-correlation spectroscopy. Biophys. J. 89, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald J. L.; Pike L. J. (2008) Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U.S.A. 105, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P.; Claus J.; Jovin T. M.; Arndt-Jovin D. J. (2010) Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. U.S.A. 107, 16524–16529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil A.; Gaidarov I.; Kobylarz K.; Lampson M. A.; Keen J. H.; McGraw T. E. (1999) Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. U.S.A. 96, 6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti N.; Liang H.; Xu Y.; Zhang Y.; Yonekubo Y.; Inder K.; Du G.; Parton R. G.; Hancock J. F.; Plowman S. J. (2010) Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Mol. Cell. Biol. 30, 3795–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtman J. C.; Yamaguchi H.; Barda-Saad M.; Braiman A.; Bowden B.; Appella E.; Schuck P.; Samelson L. E. (2006) Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 13, 798–805. [DOI] [PubMed] [Google Scholar]
- Schechter Y.; Hernaez L.; Schlessinger J.; Cuatrecasas P. (1979) Local aggregation of hormone-receptor complexes is required for activation by epidermal growth factor. Nature 278, 835–838. [DOI] [PubMed] [Google Scholar]
- Schreiber A. B.; Libermann T. A.; Lax I.; Yarden Y.; Schlessinger J. (1983) Biological role of epidermal growth factor-receptor clustering. Investigation with monoclonal anti-receptor antibodies. J. Biol. Chem. 258, 846–853. [PubMed] [Google Scholar]
- Burgess A. W.; Cho H. S.; Eigenbrot C.; Ferguson K. M.; Garrett T. P.; Leahy D. J.; Lemmon M. A.; Sliwkowski M. X.; Ward C. W.; Yokoyama S. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552. [DOI] [PubMed] [Google Scholar]
- Nagy P.; Jenei A.; Kirsch A. K.; Szollosi J.; Damjanovich S.; Jovin T. M. (1999) Activation-dependent clustering of the erbB2 receptor tyrosine kinase detected by scanning near-field optical microscopy. J. Cell Sci. 112, 1733–1741. [DOI] [PubMed] [Google Scholar]
- Hofman E. G.; Bader A. N.; Voortman J.; van den Heuvel D. J.; Sigismund S.; Verkleij A. J.; Gerritsen H. C.; van Bergen en Henegouwen P. M. (2010) Ligand-induced EGF receptor oligomerization is kinase-dependent and enhances internalization. J. Biol. Chem. 285, 39481–39489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarian S.; Li Y.; Elson E. L.; Pike L. J. (2007) Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 93, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. H.; Chernis G. A.; Hoang V. Q.; Landgraf R. (2003) Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc. Natl. Acad. Sci. U.S.A. 100, 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E.; Baron R.; Landgraf R. (2008) Higher-order association states of cellular ERBB3 probed with photo-cross-linkable aptamers. Biochemistry 47, 11992–12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kani K.; Warren C. M.; Kaddis C. S.; Loo J. A.; Landgraf R. (2005) Oligomers of ERBB3 have two distinct interfaces that differ in their sensitivity to disruption by heregulin. J. Biol. Chem. 280, 8238–8247. [DOI] [PubMed] [Google Scholar]
- Kani K.; Park E.; Landgraf R. (2005) The extracellular domains of ErbB3 retain high ligand binding affinity at endosome pH and in the locked conformation. Biochemistry 44, 15842–15857. [DOI] [PubMed] [Google Scholar]
- Fanzo J. C.; Lynch M. P.; Phee H.; Hyer M.; Cremesti A.; Grassme H.; Norris J. S.; Coggeshall K. M.; Rueda B. R.; Pernis A. B.; Kolesnick R.; Gulbins E. (2003) CD95 rapidly clusters in cells of diverse origins. Cancer Biol. Ther. 2, 392–395. [DOI] [PubMed] [Google Scholar]
- Boldog T.; Grimme S.; Li M.; Sligar S. G.; Hazelbauer G. L. (2006) Nanodiscs separate chemoreceptor oligomeric states and reveal their signaling properties. Proc. Natl. Acad. Sci. U.S.A. 103, 11509–11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaji M.; Jin Z. X.; Yamaoka S.; Amakawa R.; Fukuhara S.; Sato S. B.; Kobayashi T.; Domae N.; Mimori T.; Bloom E. T.; Okazaki T.; Umehara H. (2005) Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J. Exp. Med. 202, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh D.; Ogiue-Ikeda M.; Jadwin J. A.; Machida K.; Mayer B. J.; Yu J. (2012) Fast rebinding increases dwell time of Src homology 2 (SH2)-containing proteins near the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 109, 14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch M. F.; Davy D. A.; Willard F. S.; Berven L. A. (2000) Insulin induces epidermal growth factor (EGF) receptor clustering and potentiates EGF-stimulated DNA synthesis in swiss 3T3 cells: A mechanism for costimulation in mitogenic synergy. Immunol. Cell Biol. 78, 408–414. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Park E.; Kani K.; Landgraf R. (2012) Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 13237–13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.