

Abstract
Islet amyloid polypeptide (IAPP) is responsible for amyloid deposition in type 2 diabetes and plays an important role in the loss of β-cell mass associated with the disease and in the failure of islet transplants, but the mechanism of islet amyloid formation is not understood. The incorrect processing of proIAPP to produce partially processed forms of the peptide has been proposed to play a role in the initiation of islet amyloid in vivo by promoting interactions with proteoglycans of the extracellular matrix. Insulin is a potent inhibitor of the formation of amyloid by IAPP in vitro in a homogeneous solution; however, its ability to inhibit IAPP in the presence of proteoglycans has not been tested, nor has its effect on the formation of amyloid by proIAPP processing intermediates been examined. Here we show that insulin is a much less effective amyloid inhibitor of both IAPP and proIAPP processing intermediates in vitro in the presence of model glycosaminoglycans, but does inhibit the formation of amyloid by proIAPP processing intermediates in a homogeneous solution. This highlights another mechanism by which sulfated proteoglycans could enhance islet amyloid formation in vivo. Interactions with sulfated proteoglycans can directly promote amyloid formation and can also significantly reduce the effectiveness of natural inhibitors.
Amyloid formation is a characteristic feature of many human diseases, including Alzheimer’s disease, Parkinson’s disease, and type 2 diabetes.1,2 Human islet amyloid polypeptide (IAPP or amylin) is a polypeptide hormone that forms extracellular fibrillar amyloid deposits in the pancreatic islets of Langerhans in type 2 diabetes.3,4 IAPP helps regulate gastric emptying, suppression of food intake, and glucose homeostasis,5−7 but formation of islet amyloid contributes to β-cell dysfunction in type 2 diabetes and is associated with the decrease in β-cell mass associated with the disease.8,9 Islet amyloid also makes important contributions to the failure of islet transplantation.10,11 The polypeptide is produced with insulin in the β-cells as a proform, stored in the insulin secretory granule, and cosecreted with insulin. Protein levels are maintained at an ∼1:100 IAPP:insulin ratio in the secretory granule of healthy β-cells, but the concentration of IAPP in the granule is still much higher than that required to promote rapid amyloid formation in vitro, suggesting that there are factors that inhibit its aggregation in the secretory granule. Interactions with insulin have been proposed to play such a role in vivo and have been shown to inhibit IAPP amyloid formation in vitro.12−14
The mechanisms of islet amyloid formation in type 2 diabetes are still not understood, although impairment of the prohormone processing machinery has been thought to play an important role in the initiation and progression of this process.15−18 IAPP is synthesized as an 89-residue precursor, preproIAPP. Removal of the signal sequence generates the 67-residue prohormone, proIAPP, which is further processed by cleavage at two conserved dibasic sites by the same prohormone convertases that process proinsulin.19 The C-terminal prosequence is removed in either the trans-Golgi network or secretory granule, preferentially by the prohormone convertase PC(1/3). The remaining dibasic residues at the C-terminus are cleaved by carboxypeptidase E (CPE),20 and amidation is conducted by the peptidyl amidating monooxygenase complex (PAM) with a conserved glycine residue acting as the nitrogen donor.21 Cleavage of the prosequence at the N-terminus by convertase PC2 gives the 37-residue mature IAPP.22 Additional posttranslational modifications include the formation of a disulfide between Cys2 and Cys7 (Figure 1).23
Figure 1.
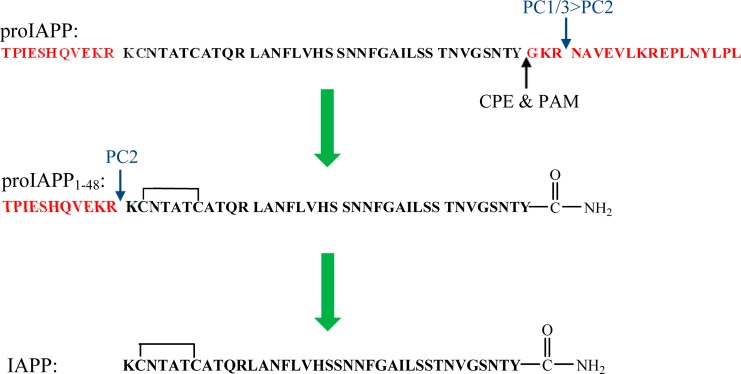
Processing pathway of human proIAPP. The N-terminal and C-terminal flanking regions of proIAPP are colored red. Cleavage of proIAPP occurs at the two dibasic sites denoted with blue arrows. The C-terminal region of proIAPP is removed preferentially by PC(1/3), and the remaining dibasic residues are removed by CPE. Final processing of the C-terminus includes removal of the remaining Gly and amidation of the Tyr by PAM, leading to the processing intermediate proIAPP1–48. The N-terminal region is removed by PC2. There is an intramolecular disulfide bond in proIAPP1–48 and in mature IAPP.
Unprocessed proinsulin and incompletely processed intermediates of proinsulin are present in the early phase of type 2 diabetes,24 and the same is true for IAPP.25 Immunohistochemical studies indicate the presence of the N-terminal prosequence of proIAPP in islet amyloid in vivo, but not the C-terminal region.26,27 This suggests that incomplete processing results in secretion of an intermediate peptide with the N-terminal flanking region of proIAPP, proIAPP1–48, which corresponds to the first 48 residues of proIAPP (Figure 1).
Two models have been proposed for how incorrectly processed IAPP might contribute to islet amyloid formation. One hypothesis is that the proIAPP processing intermediate forms intragranular amyloid that causes cell death and results in the release of amyloid that can seed extracellular formation of amyloid by secreted mature IAPP.18 In an alternative model, release of proIAPP1–48 leads to enhanced extracellular amyloid formation by promoting interactions with the glycosaminoglycan (GAG) components of heparan sulfate proteoglycans (HSPGs) of the extracellular matrix.16,28 The HSPG perlecan is found in islet amyloid deposits isolated from patients with type 2 diabetes,29 and HSPGs are associated with nearly all types of amyloid plaques.30−39 The model GAG, heparan sulfate (HS), accelerates the formation of amyloid by both IAPP and proIAPP1–48in vitro.16,40 In addition, the amyloid fibrils formed by proIAPP1–48 in the presence of HS have been shown to seed the formation of amyloid by IAPP in vitro, supporting the hypothesis that proIAPP1–48 may play a role in initiating amyloid formation.40
It is not known whether islet amyloid originates intracellularly or extracellularly, and this is a controversial question. Studies with transgenic animals that overexpress IAPP suggest an intracellular origin, but other studies with islets have shown that amyloid deposition is linked with secretion.41−43 In either case, interactions with insulin could be important for inhibiting amyloid formation in vivo, either in the granule or immediately after release when the local concentration of IAPP and insulin is high. Insulin is known to be an effective inhibitor of the formation of amyloid by IAPP in vitro; however, its effect on the formation of amyloid by proIAPP1–48 has not been investigated, and it is possible that less effective inhibition of aggregation by the pro form could play a role in promoting islet amyloid. In addition, the effects of HSPGs or GAGs on the ability of insulin to inhibit IAPP or ProIAPP1–48 amyloid formation have not been examined. Indeed, there have been very few studies that have examined the effectiveness of IAPP inhibitors in the presence of sulfated proteoglycans or their GAG components. Here we compare the ability of insulin to inhibit the formation of amyloid by mature IAPP and the incompletely processed intermediate proIAPP1–48. We also compare the effect of insulin on the formation of amyloid by the two peptides in the presence of the model GAG, HS.
Materials and Methods
Peptide Synthesis and Purification
Peptides were synthesized on a 0.25 mmol scale with an Applied Biosystems model 433A peptide synthesizer and on a 0.1 mmol scale with a CEM microwave peptide synthesizer utilizing 9-fluoronylmethoxycarbonyl (Fmoc) chemistry. Solvents were ACS-grade. 5-(4′-Fmoc-aminomethyl-3′,5-dimethoxyphenol)valeric acid (PAL-PEG) resin was used to form an amidated C-terminus. Fmoc-protected pseudoproline (oxazolidine) dipeptide derivatives were used as previously described.44,45 Standard Fmoc reaction cycles were used. The first residue attached to the resin, all β-branched residues, the residues directly following a β-branched residue, all pseudoproline dipeptide derivatives, and the residues directly following pseudoproline dipeptide derivatives were double-coupled for the synthesis with the Applied Biosystems model 433A peptide synthesizer. The microwave-assisted synthesis was performed as previously described.45 The peptides were cleaved from the resin through the use of standard trifluoroacetic acid (TFA) methods. Crude peptides were oxidized with dimethyl sulfoxide at room temperature.46 The peptides were purified via reverse-phase high-performance liquid chromatography (RP-HPLC) using a Vydac C18 preparative column. Analytical HPLC was used to check the purity of the peptides before each experiment. The masses of the pure peptides were confirmed by ionization time-of-flight mass spectrometry: wild-type IAPP, 3902.9 (expected) and 3902.7 (observed); proIAPP1–48, 5208.7 (expected) and 5209.4 (observed).
Sample Preparation
Peptide stock solutions were dissolved in 100% hexafluoro-2-proponal (HFIP) at 1.6 mM. Human insulin (recombinant, catalog no. I2643) was purchased from Sigma. Stock solutions of insulin were first prepared by dissolving insulin in 20 mM Tris-HCl buffer (pH 2) and then adding dilute NaOH to adjust the pH to 7.4. Insulin solutions were freshly made before each experiment. High-molecular weight heparan sulfate (10000–14000 Da) was purchased from Sigma. Heparan sulfate stock solutions were prepared by dissolving HS in 20 mM Tris-HCl (pH 7.4) at a concentration of 2.2 mg/mL.
Fluorescence Assays
Amyloid formation was monitored by thioflavin-T binding assays without stirring at 25 °C. Fluorescence measurements were performed using a Beckman Coulter DTX 880 plate reader with a multimode detector with 430 nm excitation and 485 nm emission. For experiments conducted in the presence of 2% HFIP, solutions were prepared by diluting filtered stock solutions of peptides (0.45 μM Acrodisc syringe filter with GHP membrane) in Tris-HCl buffer and a thioflavin-T solution immediately before the measurement. For experiments conducted without HFIP, filtered stock solutions were first lyophilized for 22 h and then the dry peptide was dissolved into Tris-HCl buffer and a thioflavin-T solution immediately before the measurement. The inhibition experiments were performed by first diluting peptide stock solutions (assays with HFIP) or dissolving dry peptides (assays without HFIP) in the buffer, followed by the addition of insulin. HS, when present, was added last. The final conditions were 16 μM peptide and 32 μM thioflavin-T in 20 mM Tris-HCl (pH 7.4). HS, when present, was at a concentration of 1.3 μM.
Circular Dichroism (CD)
Far-UV CD experiments were performed at 25 °C on an Applied Photophysics Chirascan CD spectrophotometer. Aliquots from the kinetic experiments were removed at the end of each experiment, and the spectra were recorded. Spectra were the average of three repeats recorded over a range of 190–260 nm, at 1 nm intervals. A 0.1 cm quartz cuvette was used, and a background spectrum was subtracted from the data.
Transmission Electron Microscopy (TEM)
TEM images were collected at the Life Science Microscopy Center at the State University of New York at Stony Brook. Aliquots (15 μL) of the samples used for the kinetic studies were removed at the end of each experiment, placed on a carbon-coated 300 mesh copper grid for 1 min, and then negatively stained with saturated uranyl acetate for 1 min.
Results and Discussion
Insulin Inhibits the Formation of Amyloid by IAPP and ProIAPP1–48 in a Concentration-Dependent Manner
The ability of insulin to inhibit the formation of amyloid by IAPP and proIAPP1–48 was tested using fluorescence-detected thioflavin-T binding assays. These studies were conducted by diluting solutions of the peptides solubilized in HFIP in buffer. This is a commonly employed approach for biophysical studies of IAPP. Additional experiments were conducted in the absence of HFIP. Thioflavin-T experiences a significant increase in its quantum yield upon binding to amyloid fibrils and provides a convenient probe for monitoring the time course of fibril formation. We first examined the formation of amyloid by different mixtures of IAPP and insulin and observed significant effects of insulin on the formation of amyloid by IAPP at substoichiometric concentrations. This is consistent with prior studies conducted in the presence of low percentages of HFIP.13 The results, shown in Figure 2, confirm that insulin is an effective inhibitor of the formation of amyloid by IAPP. The data are plotted as time normalized by the T50 value of IAPP in the absence of insulin, where T50 is the time required for the amyloid reaction to reach 50% of the final fluorescence intensity. The unnormalized data are included in the Supporting Information. Insulin was still able to inhibit amyloid formation, albeit weakly, when IAPP was in 100-fold excess, as indicated by a 1.7-fold longer T50. The effects of insulin became more significant as the concentration of insulin increased. In a mixture of IAPP and insulin at a 20:1 ratio (IAPP in 20-fold excess), T50 was increased by a factor of 23 under the conditions used in these studies. TEM images and CD spectra confirmed the existence of fibrils with typical amyloid morphology and β-sheet structure at the end of each reaction. This excluded the possibility of false positives from the thioflavin-T binding assays (Figure 2 and Supporting Information). Control experiments conducted with insulin alone confirm that insulin did not form amyloid in the time course of these experiments, as judged by thioflavin-T assays and TEM (Figure 2).
Figure 2.
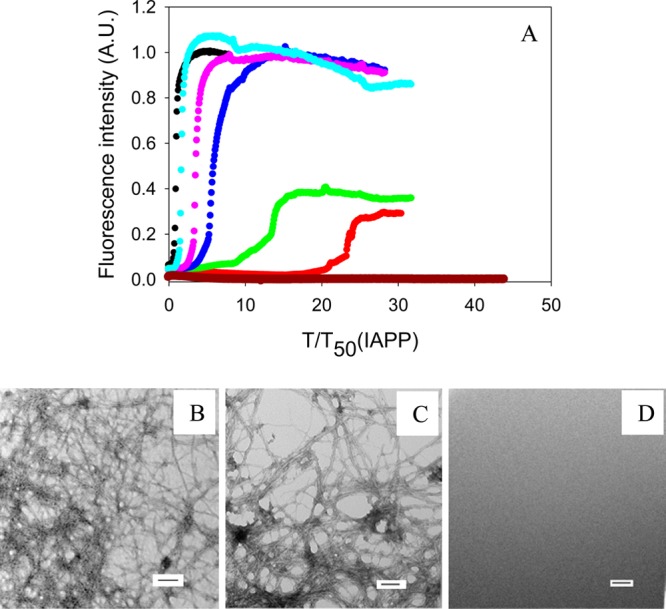
Inhibition of the formation of IAPP amyloid by insulin. (A) The results of thioflavin-T binding assays are displayed. The data are plotted as time normalized by the T50 value of IAPP in the absence of insulin: black, IAPP; red, IAPP and insulin in a 20:1 ratio; green, IAPP and insulin in a 40:1 ratio; blue, IAPP and insulin in a 60:1 ratio; pink, IAPP and insulin in an 80:1 ratio; cyan, IAPP and insulin in a 100:1 ratio; brown, insulin alone at 0.8 μM. (B) TEM image of IAPP. (C) TEM image of a 20:1 mixture of IAPP and insulin. IAPP is in 20-fold excess. (D) TEM image of insulin alone at 0.8 μM. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The IAPP concentration was 16 μM.
The effect of insulin on the formation of amyloid by proIAPP1–48 was also studied using this approach. An increase in T50 was observed for all proIAPP1–48:insulin ratios tested, and the inhibitory effect of insulin was dose-dependent. However, insulin was less effective at inhibiting the formation of amyloid by the processing intermediate proIAPP1–48 than by mature IAPP under these conditions (Figure 3). For example, when the IAPP:insulin or proIAPP1–48:insulin ratio was 20:1, the T50 of the formation of amyloid by proIAPP1–48 was increased 8.3-fold, while the T50 of the formation of amyloid by IAPP was increased by a factor of 23. TEM images and CD spectra confirmed the existence of typical fibril structure at the end of each reaction (Figure 3 and Supporting Information).
Figure 3.
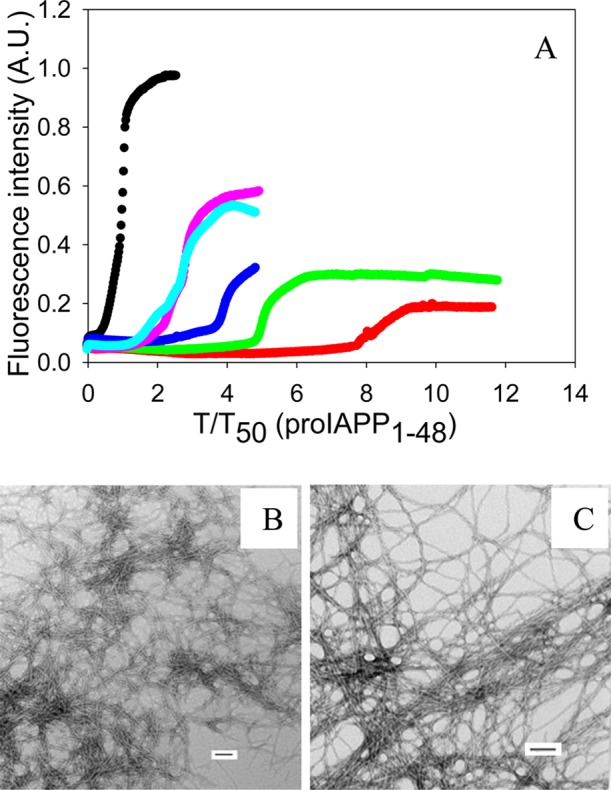
Inhibition of the formation of proIAPP1–48 amyloid by insulin. (A) The results of thioflavin-T binding assays are displayed. The data are plotted as time normalized by the T50 value of proIAPP1–48 in the absence of insulin: black, proIAPP1–48; red, proIAPP1–48 and insulin in a 20:1 ratio; green, proIAPP1–48 and insulin in a 40:1 ratio; blue, proIAPP1–48 and insulin in a 60:1 ratio; pink, proIAPP1–48 and insulin in an 80:1 ratio; cyan, proIAPP1–48 and insulin in a 100:1 ratio. (B) TEM image of proIAPP1–48. (C) TEM image of a 20:1 mixture of proIAPP1–48 and insulin. proIAPP1–48 is in 20-fold excess. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The proIAPP1–48 concentration was 16 μM.
Insulin Is a Significantly Less Effective Inhibitor in the Presence of Heparan Sulfate
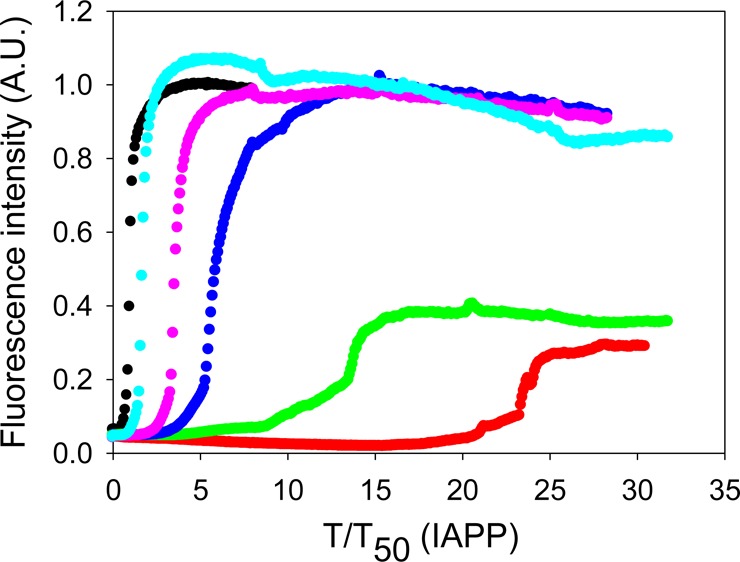
We next conducted inhibition experiments in the presence of the model glycosaminoglycan, HS. As expected, the formation of amyloid by IAPP was greatly enhanced in the presence of HS, consistent with prior reports.16,40 IAPP formed amyloid immediately after the reaction was initiated in the presence of HS, and no lag time was observed (Figure 4A). TEM images confirmed the presence of amyloid fibrils, and the fibrils formed in the presence of HS had a morphology very similar to the morphology of those formed in the absence of HS (Figure 4). CD confirmed that β-sheet structure was formed (Supporting Information).
Figure 4.

Effects of insulin on the formation of amyloid by IAPP in the presence of HS. (A) The results of thioflavin-T binding assays are displayed. The data are plotted as time normalized by the T50 value of IAPP in the presence of HS, but in the absence of insulin: black, IAPP in the presence of HS; red, IAPP and insulin in a 20:1 ratio in the presence of HS; green, IAPP and insulin in a 5:1 ratio in the presence of HS; blue, IAPP and insulin in a 1:1 ratio in the presence of HS; pink, IAPP and insulin in a 1:5 ratio in the presence of HS; brown, a mixture of 80 μM insulin and 1.3 μM HS. (B) Expansion of the first five time units of panel A. The same color coding is used. (C) TEM image of IAPP in the presence of HS. (D) TEM image of a 1:5 mixture of IAPP and insulin in the presence of HS. Insulin is in 5-fold excess. (E) TEM image of the insulin/HS mixture. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The IAPP concentration was 16 μM. The HS concentration was 1.3 μM.
Insulin was a much less effective inhibitor when HS was present, although there were modest effects on the time required to complete amyloid formation, and the effects of insulin were dose-dependent. However, even at a 1:5 IAPP:insulin ratio, IAPP formed amyloid without an apparent lag phase in the presence of HS (Figure 4A). A rapid initial rise in fluorescence intensity was observed followed by a more gradual rise to the final value. Although no significant lag phase was observed, even with relatively high concentrations of insulin (1:1 and 1:5 IAPP:insulin ratios), amyloid formation required several hours to reach completion after the initial rapid increase in the thioflavin-T fluorescence intensity. TEM images revealed the presence of fibrils at the end of each experiment (Figure 4 and Supporting Information). Similar behavior has been observed in experiments using other inhibitors in the presence of HS and has been proposed to be due to the rapid formation of a GAG–peptide intermediate.47 CD spectra of mixtures with a low concentration of insulin showed typical β-sheet structure, while those from samples with a higher concentration of insulin (1:1 and 1:5 IAPP:insulin ratios) exhibited a broad peak centered at 219 nm, likely due to contributions from the insulin α-helical structure (Supporting Information). Interestingly, in the presence of HS, the CD signal of either IAPP or mixtures of IAPP and insulin appears to be more intense than in the absence of HS (Supporting Information). This may be caused by the fact that HS has a solubilizing effect on the aggregates formed so they are less prone to bind to the walls of the microplates or segregate to the air–water interface or pellet. The exact reason for the differencies in the intensity of the CD spectra is not clear, but the shape of the spectrum reports on the secondary structure of the soluble aggregates.
Insulin is also much less effective at inhibiting the formation of amyloid by proIAPP1–48 in the presence of HS (Figure 5). Insulin barely showed any inhibitory effect at 20:1 and 5:1 proIAPP1–48:insulin ratios in the presence of HS. Increasing the amount of insulin to a ratio of 1:1 and 1:5 slightly increased the time required to complete amyloid formation, but no lag time was observed. TEM images and CD spectra confirmed the results of thioflavin-T fluorescence and showed that amyloid was formed (Supporting Information).
Figure 5.

Effects of insulin on the formation of amyloid by proIAPP1–48 in the presence of HS. (A) The results of thioflavin-T binding assays are displayed. The data are plotted as time normalized by the T50 value of proIAPP1–48 in the presence of HS, but in the absence of insulin: black, proIAPP1–48 in the presence of HS; red, proIAPP1–48 and insulin in a 20:1 ratio in the presence of HS; green, proIAPP1–48 and insulin in a 5:1 ratio in the presence of HS; blue, proIAPP1–48 and insulin in a 1:1 ratio in the presence of HS; pink, proIAPP1–48 and insulin in a 1:5 ratio in the presence of HS. (B) Expansion of the first five time units of panel A. The same color coding is used. (C) TEM image of proIAPP1–48 in the presence of HS. (D) TEM image of a 1:5 mixture of proIAPP1–48 and insulin in the presence of HS. Insulin is in 5-fold excess. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The proIAPP1–48 concentration was 16 μM. The HS concentration was 1.3 μM.
Insulin is able to aggregate; thus, to exclude the possibility that the results discussed above were caused by interactions of insulin with HS, we monitored the thioflavin-T fluorescence of a mixture of insulin and HS. No increase in fluorescence was observed during the time course of the study (Figure 4A), and TEM showed that no fibrils were formed (Figure 4E). The CD spectrum revealed helical structure that is similar to that observed from insulin samples that did not contain HS (Supporting Information).
The Observed Effects Are Not Due to the Presence of Organic Cosolvents
The results outlined above were collected using assays with 2% HFIP by volume as a cosolvent. Even this low percentage of organic solvent accelerates the kinetics of the formation of amyloid by IAPP significantly.48,49 Initial kinetic studies of the effects of insulin on IAPP amyloid formation made use of HFIP as a cosolvent,13 and subsequent studies showed that similar relative effects were obtained in the absence of HFIP in many cases.50 We compared the ability of insulin to inhibit the formation of amyloid by both IAPP and proIAPP1–48 in the absence and presence of HS using a different assay that avoided the use of HFIP to exclude the possibility that our results were a consequence of the conditions used. Control experiments conducted with insulin in the absence of HFIP or IAPP showed that it did not form amyloid under these conditions during the time course of the experiments (Supporting Information). The results showed that in the absence of HFIP, at a 1:20 ratio (IAPP is in 20-fold excess), insulin increased the T50 of the formation of amyloid by IAPP by a factor of 3.3 in a homogeneous solution (Figure 6A). For comparison, insulin at the same ratio increased the T50 of the formation of amyloid by IAPP by a factor of 23 in the presence of HFIP. The reason for the proportionally larger effect in HFIP is not clear but may reflect the acquisition of structure in HFIP that facilitates interactions of IAPP with insulin. Peptide mapping studies have suggested that the region corresponding to residues 7–19 of IAPP interacts with insulin. This segment of IAPP has been proposed to form a transient helical structure during amyloid formation,51−55 and even low levels of HFIP can promote helical structure.
Figure 6.

Inhibition by insulin of the formation of amyloid by IAPP in the presence and absence of HS in buffer without HFIP. (A) Inhibition in the absence of HS monitored by thioflavin-T assays: black, IAPP; red, IAPP and insulin in a 20:1 ratio. Data are plotted as time normalized by the T50 value of IAPP in the absence of insulin. (B) Inhibition in the presence of HS monitored by thioflavin-T assays. Data are plotted as time normalized by the T50 value of IAPP in the presence of HS, but in the absence of insulin. The same color coding is used here as in panel A. (C) TEM image of IAPP in the absence of HS. (D) TEM image of a 20:1 mixture of IAPP and insulin in the absence of HS (IAPP was in 20-fold excess). (E) TEM image of IAPP in the presence of HS. (F) TEM image of a 20:1 mixture of IAPP and insulin in the presence of HS (IAPP was in 20-fold excess). Scale bars represent 100 nm. Aliquots were removed at the end of each experiment for TEM analysis. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) without stirring at 25 °C. The IAPP concentration was 16 μM. The HS concentration was 1.3 μM (when present).
When HS was present, the formation of amyloid by IAPP was greatly accelerated, as indicated by the lack of a lag phase. In addition, insulin was a much less effective inhibitor of the formation of amyloid by IAPP in the presence of HS (Figure 6B). At a 20:1 IAPP:insulin ratio, the time required to complete the formation of amyloid by IAPP was increased by a factor of only 1.3 when HS was in the mixture. These results are consistent with the trends observed in the presence of HFIP and indicate that the observations are not an artifact caused by residual HFIP. TEM images were collected at the end of each experiment and confirmed that amyloid was formed (Figure 6).
Very similar results were obtained with proIAPP1–48. In the absence of HFIP, insulin increased the T50 of the formation of amyloid by proIAPP1–48 by 2.7-fold when proIAPP1–48 and insulin were in a 20:1 ratio in a homogeneous solution (Figure 7A). In contrast, at the same proIAPP1–48:insulin ratio, T50 was increased by a factor of 8.3 in the presence of HFIP.
Figure 7.

Inhibition by insulin of the formation of amyloid by proIAPP1–48 in the presence and absence of HS in buffer without HFIP. (A) Inhibition in the absence of HS monitored by thioflavin-T assays. Data are plotted as time normalized by the T50 value of proIAPP1–48 in the absence of insulin: black, proIAPP1–48; red, proIAPP1–48 and insulin in a 20:1 ratio. (B) Inhibition in the presence of HS monitored by thioflavin-T assays. Data are plotted as time normalized by the T50 value of proIAPP1–48 in the presence of HS, but in the absence of insulin. The same color coding is used here as in panel A. (C) TEM image of proIAPP1–48 in the absence of HS. (D) TEM image of a 20:1 mixture of proIAPP1–48 and insulin in the absence of HS. (E) TEM image of proIAPP1–48 in the presence of HS. (F) TEM image of a 20:1 mixture of proIAPP1–48 and insulin in the presence of HS. Scale bars represent 100 nm. Aliquots were removed at the end of each experiment for TEM analysis. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) without stirring at 25 °C. The proIAPP1–48 concentration was 16 μM. The HS concentration was 1.3 μM (when present).
We also tested the ability of insulin to inhibit the formation of amyloid by proIAPP1–48 in the presence of HS, but in the absence of HFIP. Insulin is a much less effective inhibitor of the formation of amyloid by proIAPP1–48 when HS is present. In the presence of HS, proIAPP1–48 immediately aggregated to form amyloid fibrils without any observable lag phase. The time required to complete the formation of amyloid by proIAPP1–48 in the presence of insulin is nearly the same as that without insulin (Figure 7B). TEM images were collected at the end of each experiment and confirmed that amyloid was formed (Figure 7).
Conclusions
The data show that insulin is a potent amyloid inhibitor for both IAPP and the proIAPP1–48 processing intermediate in homogeneous solutions. Figure 8 shows a comparison of the ability of insulin to inhibit the formation of amyloid by IAPP and proIAPP1–48 as judged by the T50 values in the presence of HFIP. Insulin, at relatively high concentrations, is more potent in slowing the formation of amyloid by IAPP than by proIAPP1–48, and the inhibition of the formation of IAPP amyloid by insulin is more sensitive to changes in the insulin level than is the inhibition of the formation of amyloid by proIAPP1–48. For example, a 5-fold change in insulin concentration in the mixture (IAPP:insulin ratio ranging from 100:1 to 20:1) led to a significant increase in the T50 of the formation of amyloid by IAPP, from 1.7- to 23-fold relative to that with IAPP alone. In contrast, increasing the insulin ratio by the same amount had a smaller effect on proIAPP1–48; T50 increased from 2.7- to 8.3-fold relative to that with proIAPP1–48 alone. However, insulin is still an effective inhibitor, arguing that interactions between proIAPP1–48 and insulin that are weaker than those between IAPP and insulin are unlikely to account for islet amyloid formation in vivo.
Figure 8.

Bar graph comparing the T50 values for the formation of amyloid by IAPP and proIAPP1–48 in the presence of 2% HFIP at different ratios of IAPP or proIAPP1–48 to insulin: black bars, mixtures of IAPP and insulin; gray bars, mixtures of proIAPP1–48 and insulin. Values of T50 were derived from the kinetic curves shown in Figures 2 and 3. Data are plotted as T50 normalized by the value of T50 of either IAPP or proIAPP1–48 in the absence of insulin. Experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The IAPP or proIAPP1–48 concentration was 16 μM.
The effects of insulin on the formation of amyloid by IAPP and proIAPP1–48 in the presence of HS are quantitatively compared in terms of T50 in Figure 9. The ability of insulin to inhibit amyloid formation is greatly weakened in the presence of HS for both peptides. When peptide and insulin are combined at a 20:1 ratio, the T50 for the formation of amyloid by IAPP is increased by a factor of only 1.6, and no obvious increase in T50 is observed for proIAPP1–48. Even at the highest concentrations of insulin (1:1 and 1:5 ratios of IAPP or proIAPP1–48 to insulin), T50 is increased by <3-fold for both peptides in the presence of HS. The formation of amyloid by IAPP is slightly more effectively inhibited than is the formation of amyloid by proIAPP1–48 in the presence of HS. However, insulin is clearly a significantly worse inhibitor of the formation of amyloid by either peptide in the presence of HS.
Figure 9.

Bar graph comparing the T50 values for amyloid formation in buffer with HFIP for mixtures of IAPP with insulin and for mixtures of proIAPP1–48 with insulin in the presence of HS. Values are plotted for different ratios of IAPP or proIAPP1–48 to insulin: black bars, mixtures of IAPP and insulin in the presence of HS; gray bars, mixtures of proIAPP1–48 and insulin in the presence of HS. The data are plotted as T50 normalized by the T50 value of either IAPP or proIAPP1–48 in the presence of HS, but in the absence of insulin. Values of T50 were derived from the kinetic curves in Figures 4 and 5. Experiments were conducted in 20 mM Tris-HCl (pH 7.4) and 2% (v/v) HFIP without stirring at 25 °C. The IAPP or proIAPP1–48 concentration was 16 μM. The HS concentration was 1.3 μM.
Our control experiments show that the results are not due to the presence of HFIP. Insulin is a much less effective inhibitor of amyloid formation in the presence of HS even in the absence of HFIP. The results are summarized in Figure 10. The T50 for the formation of amyloid by IAPP and proIAPP1–48 in the absence of HFIP is increased by 3.3- and 2.7-fold, respectively, at a 20:1 ratio of IAPP or proIAPP1–48 to insulin in a homogeneous solution. However, in the presence of HS, insulin, at the same ratio, increased the time required for the formation of amyloid by IAPP by a factor of only 1.3, while the kinetics of the formation of amyloid by proIAPP1–48 was virtually unaltered. No lag phase was observed for the formation of amyloid by either IAPP or proIAPP1–48 even at a 20:1 ratio of IAPP or proIAPP1–48 to insulin.
Figure 10.

Bar graph comparing the T50 values for the formation of amyloid by mixtures of IAPP with insulin and mixtures of proIAPP1–48 with insulin in the absence of HFIP. (A) HS is absent in the assay. The data are plotted as T50 normalized by the T50 value of IAPP or proIAPP1–48 in the absence of insulin. (B) HS is present in the assay. The data are plotted as T50 normalized by the T50 value of IAPP or proIAPP1–48 in the presence of HS without insulin. The ratio of IAPP or proIAPP1–48 to insulin was 20:1. Both panels: black bars, mixtures of IAPP and insulin; gray bars, mixtures of proIAPP1–48 and insulin. Values of T50 were derived from the kinetic curves in Figures 6 and 7. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4) without stirring at 25 °C. The IAPP or proIAPP1–48 concentration was 16 μM. The HS concentration was 1.3 μM (when present).
HS is not the only factor that can accelerate the formation of amyloid by IAPP. Anionic vesicles also catalyze the formation of amyloid by IAPP. Insulin still inhibits the process in the presence of anionic model membranes;50,56 however, insulin is noticeably less effective.50 Those studies together with this work highlight the importance of considering the role of the heterogeneous environment in amyloid formation. The deleterious effects of HS on insulin’s ability to act as an amyloid inhibitor could be due to stronger interactions of IAPP or proIAPP1–48 with HS than with insulin. Alternatively, HS might bind to insulin and sequester the inhibitor, or both effects could play a role. The data presented here clearly show that HS accelerates the formation of amyloid by proIAPP1–48 and IAPP, and this has been established to occur via direct interactions between the peptides and the GAG.47,57 These observations, along with the fact that HS did not induce the formation of amyloid by insulin under the conditions used in our studies, suggest that interactions between IAPP or proIAPP1–48 and HS outcompete interactions between insulin and IAPP or proIAPP1–48.
Our observations suggest another mechanism by which HS can promote amyloid formation. Proteoglycans can accelerate amyloid formation by direct interactions with proIAPP1–48 or IAPP and can also significantly reduce the inhibitory effects of insulin.
Acknowledgments
We thank Dr. Fanling Meng for helpful discussions.
Glossary
Abbreviations
- IAPP
islet amyloid polypeptide
- CPE
carboxypeptidase E
- PAM
peptidyl amidating monooxygenase complex
- HS
heparan sulfate
- Fmoc
9-fluoronylmethoxycarbonyl
- PAL-PEG
5-(4′-fmoc-aminomethyl-3′,5-dimethoxyphenol)valeric acid
- TFA
trifluoroacetic acid
- RP-HPLC
reverse-phase high-performance liquid chromatography
- HFIP
hexafluoro-2-proponal
- CD
circular dichroism
- TEM
transmission electron microscopy.
Supporting Information Available
TEM images and CD spectra of mixtures of IAPP and insulin at different ratios of IAPP to insulin in the absence and presence of HS, TEM images and CD spectra of mixtures of proIAPP1–48 and insulin at different ratios of proIAPP1–48 to insulin in the absence and presence of HS, CD spectrum of insulin in the absence of HS, CD spectrum of a mixture of insulin and HS, plots of unnormalized kinetic data, and thioflavin-T curves and TEM images of insulin control in the presence and absence of HS without HFIP. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grant GM078114 to D.P.R.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sipe J. D. (1994) Amyloidosis. Crit. Rev. Clin. Lab. Sci. 31, 325–354. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M.; Zurdo J.; MacPhee C. E.; Dobson C. M. (2003) Protein folding and misfolding: A paradigm of self-assembly and regulation in complex biological systems. Philos. Trans. R. Soc., A 361, 1205–1222. [DOI] [PubMed] [Google Scholar]
- Cooper G. J.; Willis A. C.; Clark A.; Turner R. C.; Sim R. B.; Reid K. B. (1987) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U.S.A. 84, 8628–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P.; Wernstedt C.; O’Brien T. D.; Hayden D. W.; Johnson K. H. (1987) Islet amyloid in type 2 human diabetes mellitus and adult diabetic cats contains a novel putative polypeptide hormone. Am. J. Pathol. 127, 414–417. [PMC free article] [PubMed] [Google Scholar]
- Hebda J. A.; Miranker A. D. (2009) The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: Insights from type II diabetes. Annu. Rev. Biophys. 38, 125–152. [DOI] [PubMed] [Google Scholar]
- Rushing P. A.; Hagan M. M.; Seeley R. J.; Lutz T. A.; D’Alessio D. A.; Air E. L.; Woods S. C. (2001) Inhibition of central Amylin signaling increases food intake and body adiposity in rats. Endocrinology 142, 5035–5038. [DOI] [PubMed] [Google Scholar]
- Clementi G.; Caruso A.; Cutuli V. M. C.; deBernardis E.; Prato A.; AmicoRoxas M. (1996) Amylin given by central or peripheral routes decreases gastric emptying and intestinal transit in the rat. Experientia 52, 677–679. [DOI] [PubMed] [Google Scholar]
- Clark A.; Wells C. A.; Buley I. D.; Cruickshank J. K.; Vanhegan R. I.; Matthews D. R.; Cooper G. J.; Holman R. R.; Turner R. C. (1988) Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. Clin. Pract. 9, 151–159. [PubMed] [Google Scholar]
- Kahn S. E.; Andrikopoulos S.; Verchere C. B. (1999) Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48, 241–253. [DOI] [PubMed] [Google Scholar]
- Andersson A.; Bohman S.; Borg L. A.; Paulsson J. F.; Schultz S. W.; Westermark G. T.; Westermark P. (2008) Amyloid deposition in transplanted human pancreatic islets: A conceivable cause of their long-term failure. Exp. Diabetes Res. 2008, 562985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K. J.; Abedini A.; Marek P.; Klimek A. M.; Butterworth S.; Driscoll M.; Baker R.; Nilsson M. R.; Warnock G. L.; Oberholzer J.; Bertera S.; Trucco M.; Korbutt G. S.; Fraser P. E.; Raleigh D. P.; Verchere C. B. (2010) Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts. Proc. Natl. Acad. Sci. U.S.A. 107, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaikaran E.; Nilsson M. R.; Clark A. (2004) Pancreatic β-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation. Biochem. J. 377, 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J. L.; Miranker A. D. (2004) The mechanism of insulin action on islet amyloid polypeptide fiber formation. J. Mol. Biol. 335, 221–231. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Li Z. C.; Westermark G. T.; Leckstrom A.; Steiner D. F. (1996) Effects of β cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett. 379, 203–206. [DOI] [PubMed] [Google Scholar]
- Paulsson J. F.; Westermark G. T. (2005) Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes 54, 2117–2125. [DOI] [PubMed] [Google Scholar]
- Abedini A.; Tracz S. M.; Cho J. H.; Raleigh D. P. (2006) Characterization of the heparin binding site in the N-terminus of human pro-islet amyloid polypeptide: Implications for amyloid formation. Biochemistry 45, 9228–9237. [DOI] [PubMed] [Google Scholar]
- Marzban L.; Rhodes C. J.; Steiner D. F.; Haataja L.; Halban P. A.; Verchere C. B. (2006) Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 55, 2192–2201. [DOI] [PubMed] [Google Scholar]
- Paulsson J. F.; Andersson A.; Westermark P.; Westermark G. T. (2006) Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in β cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 49, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Sanke T.; Bell G. I.; Sample C.; Rubenstein A. H.; Steiner D. F. (1988) An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J. Biol. Chem. 263, 17243–17246. [PubMed] [Google Scholar]
- Marzban L.; Trigo-Gonzales G.; Zhu X. R.; Rhodes C. J.; Halban P. A.; Steiner D. F.; Verchere C. B. (2004) Role of β-cell prohormone convertase (PC) 1/3 in processing of pro-islet amyloid polypeptide. Diabetes 53, 141–148. [DOI] [PubMed] [Google Scholar]
- Marzban L.; Soukhatcheva G.; Verchere C. B. (2005) Role of carboxypeptidase E in processing of pro-islet amyloid polypeptide in β-cells. Endocrinology 146, 1808–1817. [DOI] [PubMed] [Google Scholar]
- Badman M. K.; Shennan K. I. J.; Jermany J. L.; Docherty K.; Clark A. (1996) Processing of pro-islet amyloid polypeptide (proIAPP) by the prohormone convertase PC2. FEBS Lett. 378, 227–231. [DOI] [PubMed] [Google Scholar]
- Roberts A. N.; Leighton B.; Todd J. A.; Cockburn D.; Schofield P. N.; Sutton R.; Holt S.; Boyd Y.; Day A. J.; Foot E. A.; et al. (1989) Molecular and functional characterization of amylin, a peptide associated with type 2 diabetes mellitus. Proc. Natl. Acad. Sci. U.S.A. 86, 9662–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward W. K.; Lacava E. C.; Paquette T. L.; Beard J. C.; Wallum B. J.; Porte D. (1987) Disproportionate elevation of immunoreactive proinsulin in type-2 (non-insulin-dependent) diabetes-mellitus and in experimental insulin resistance. Diabetologia 30, 698–702. [DOI] [PubMed] [Google Scholar]
- Zheng X. Y.; Ren W.; Zhang S. H.; Liu J. J.; Li S. F.; Li J. C.; Yang P.; He J.; Su S. C.; Li P. (2010) Serum levels of proamylin and amylin in normal subjects and patients with impaired glucose regulation and type 2 diabetes mellitus. Acta Diabetol. 47, 265–270. [DOI] [PubMed] [Google Scholar]
- Westermark G. T.; Steiner D. F.; Gebre-Medhin S.; Engstrom U.; Westermark P. (2000) Pro islet amyloid polypeptide (ProIAPP) immunoreactivity in the islets of langerhans. Upsala J. Med. Sci. 105, 97–106. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Engstrom U.; Westermark G. T.; Johnson K. H.; Permerth J.; Betsholtz C. (1989) Islet amyloid polypeptide (Iapp) and pro-Iapp immunoreactivity in human islets of Langerhans. Diabetes Res. Clin. Pract. 7, 219–226. [DOI] [PubMed] [Google Scholar]
- Park K.; Verchere C. B. (2001) Identification of a heparin binding domain in the N-terminal cleavage site of pro-islet amyloid polypeptide: Implications for islet amyloid formation. J. Biol. Chem. 276, 16611–16616. [DOI] [PubMed] [Google Scholar]
- Young I. D.; Ailles L.; Narindrasorasak S.; Tan R.; Kisilevsky R. (1992) Localization of the basement membrane heparan sulfate proteoglycan in islet amyloid deposits in type II diabetes mellitus. Arch. Pathol. Lab. Med. 116, 951–954. [PubMed] [Google Scholar]
- Ancsin J. B. (2003) Amyloidogenesis: Historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 10, 67–79. [DOI] [PubMed] [Google Scholar]
- Watson D. J.; Lander A. D.; Selkoe D. J. (1997) Heparin-binding properties of the amyloidogenic peptides Aβ and amylin: Dependence on aggregation state and inhibition by Congo red. J. Biol. Chem. 272, 31617–31624. [DOI] [PubMed] [Google Scholar]
- Snow A. D.; Wight T. N. (1989) Proteoglycans in the pathogenesis of Alzheimers-disease and other amyloidoses. Neurobiol. Aging 10, 481–497. [DOI] [PubMed] [Google Scholar]
- Castillo G. M.; Ngo C.; Cummings J.; Wight T. N.; Snow A. D. (1997) Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer’s disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J. Neurochem. 69, 2452–2465. [DOI] [PubMed] [Google Scholar]
- Inoue S. (2001) Basement membrane and β amyloid fibrillogenesis in Alzheimer’s disease. Int. Rev. Cytol. 210, 121–161. [DOI] [PubMed] [Google Scholar]
- Castillo G. M.; Cummings J. A.; Yang W. H.; Judge M. E.; Sheardown M. J.; Rimvall K.; Hansen J. B.; Snow A. D. (1998) Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 47, 612–620. [DOI] [PubMed] [Google Scholar]
- Potter-Perigo S.; Hull R. L.; Tsoi C.; Braun K. R.; Andrikopoulos S.; Teague J.; Verchere C. B.; Kahn S. E.; Wight T. N. (2003) Proteoglycans synthesized and secreted by pancreatic islet β-cells bind amylin. Arch. Biochem. Biophys. 413, 182–190. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Yamaguchi I.; Hasegawa K.; Tsutsumi S.; Goto Y.; Gejyo F.; Naiki H. (2004) Glycosaminoglycans enhance the trifluoroethanol-induced extension of β(2)-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 15, 126–133. [DOI] [PubMed] [Google Scholar]
- Suk J. Y.; Zhang F. M.; Balch W. E.; Linhardt R. J.; Kelly J. W. (2006) Heparin accelerates gelsolin amyloidogenesis. Biochemistry 45, 2234–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejía L. M.; Ramirez-Alvarado M. (2013) Systemic Amyloidoses. Annu. Rev. Biochem. 82, 745–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Abedini A.; Song B.; Raleigh D. P. (2007) Amyloid formation by pro-islet amyloid polypeptide processing intermediates: Examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry 46, 12091–12099. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Andersson A.; Westermark G. T. (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826. [DOI] [PubMed] [Google Scholar]
- Gurlo T.; Ryazantsev S.; Huang C. J.; Yeh M. W.; Reber H. A.; Hines O. J.; O’Brien T. D.; Glabe C. G.; Butler P. C. (2010) Evidence for proteotoxicity in β cells in type 2 diabetes toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 176, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Mourney K.; Hull R. L.; Zraika S.; Udayasankar J.; Subramanian S. L.; Kahn S. E. (2011) Exendin-4 increases islet amyloid deposition but offsets the resultant β cell toxicity in human islet amyloid polypeptide transgenic mouse islets. Diabetologia 54, 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Raleigh D. P. (2005) Incorporation of pseudoproline derivatives allows the facile synthesis of human IAPP, a highly amyloidogenic and aggregation-prone polypeptide. Org. Lett. 7, 693–696. [DOI] [PubMed] [Google Scholar]
- Marek P.; Woys A. M.; Sutton K.; Zanni M. T.; Raleigh D. P. (2010) Efficient microwave-assisted synthesis of human islet amyloid polypeptide designed to facilitate the specific incorporation of labeled amino acids. Org. Lett. 12, 4848–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Singh G.; Raleigh D. P. (2006) Recovery and purification of highly aggregation-prone disulfide-containing peptides: Application to islet amyloid polypeptide. Anal. Biochem. 351, 181–186. [DOI] [PubMed] [Google Scholar]
- Wang H.; Cao P.; Raleigh D. P. (2013) Amyloid formation in heterogeneous environments: Islet amyloid polypeptide glycosaminoglycan interactions. J. Mol. Biol. 425, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Raleigh D. P. (2005) The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry 44, 16284–16291. [DOI] [PubMed] [Google Scholar]
- Padrick S. B.; Miranker A. D. (2002) Islet amyloid: Phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry 41, 4694–4703. [DOI] [PubMed] [Google Scholar]
- Knight J. D.; Williamson J. A.; Miranker A. D. (2008) Interaction of membrane-bound islet amyloid polypeptide with soluble and crystalline insulin. Protein Sci. 17, 1850–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilead S.; Wolfenson H.; Gazit E. (2006) Molecular mapping of the recognition interface between the islet amyloid polypeptide and insulin. Angew. Chem., Int. Ed. 45, 6476–6480. [DOI] [PubMed] [Google Scholar]
- Wiltzius J. J. W.; Sievers S. A.; Sawaya M. R.; Eisenberg D. (2009) Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 18, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Raleigh D. P. (2009) A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng., Des. Sel. 22, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Raleigh D. P. (2009) A role for helical intermediates in amyloid formation by natively unfolded polypeptides?. Phys. Biol. 6, 015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. A.; Miranker A. D. (2007) Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 16, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin D.; Yan L. M.; Kapurniotu A.; Winter R. (2010) Suppression of IAPP fibrillation at anionic lipid membranes via IAPP-derived amyloid inhibitors and insulin. Biophys. Chem. 150, 73–79. [DOI] [PubMed] [Google Scholar]
- Jha S.; Patil S. M.; Gibson J.; Nelson C. E.; Alder N. N.; Alexandrescu A. T. (2011) Mechanism of amylin fibrillization enhancement by heparin. J. Biol. Chem. 286, 22894–22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.