

Abstract
The rise of drug-resistant Mycobacterium tuberculosis lends urgency to the need for new drugs for the treatment of tuberculosis (TB). The identification of a serine protease, mycosin protease-1 (MycP1), as the crucial agent in hydrolyzing the virulence factor, ESX-secretion-associated protein B (EspB), potentially opens the door to new tuberculosis treatment options. Using the crystal structure of mycobacterial MycP1 in the apo form, we performed an iterative ligand- and structure-based virtual screening (VS) strategy to identify novel, nonpeptide, small-molecule inhibitors against MycP1 protease. Screening of ∼485 000 ligands from databases at the Genomics Research Institute (GRI) at the University of Cincinnati and the National Cancer Institute (NCI) using our VS approach, which integrated a pharmacophore model and consensus molecular shape patterns of active ligands (4D fingerprints), identified 81 putative inhibitors, and in vitro testing subsequently confirmed two of them as active inhibitors. Thereafter, the lead structures of each VS round were used to generate a new 4D fingerprint that enabled virtual rescreening of the chemical libraries. Finally, the iterative process identified a number of diverse scaffolds as lead compounds that were tested and found to have micromolar IC50 values against the MycP1 target. This study validated the efficiency of the SABRE 4D fingerprints as a means of identifying novel lead compounds in each screening round of the databases. Together, these results underscored the value of using a combination of in silico iterative ligand- and structure-based virtual screening of chemical libraries with experimental validation for the identification of promising structural scaffolds, such as the MycP1 inhibitors.
Introduction
Mycobacterium tuberculosis, the agent causing tuberculosis (TB), is responsible for significant worldwide morbidity and mortality, estimated at more than 1.3 million deaths in 2012 according to the World Health Organization.1 This ancient but persistent disease still lacks effective antimicrobial treatment regimens, particularly in cases of multidrug-resistant tuberculosis.2,3 An attractive target for development of new antimicrobials is the recently identified mycobacterial protein-export machinery.4−6M. tuberculosis relies on specialized ESX secretion systems, also called Type VII Secretion (T7S) Systems, to evade the human immune system and promote bacterial survival within host cells,7,8 and it possesses five gene clusters that encode sets of conserved proteins comprising the ESX systems.9 Each of these gene clusters, called ESX-1 through ESX-5, includes essential mycosin proteases, which are named MycP1 through MycP5, respectively.
The mycosins are membrane-bound serine proteases belonging to the subtilisin family of proteases.10,11 The crystal structures of MycP1 and MycP3 revealed that these mycosins are characterized by a relatively deep, broad, substrate-binding groove and by the absence of an autoinhibitory propeptide, making them the first subtilisins that do not undergo post-translational processing.12−14 MycP1 is known to hydrolyze the important virulence factor ESX secretion-associated protein B (EspB).15−17 MycP1 cleaves the unstructured C-terminal part of EspB, possibly activating EspB for phospholipid binding.18 Most importantly, inactivation of MycP1 leads to decreased virulence of M. tuberculosis and increased survival in the mouse model of TB.17 These data, combined with the availability of MycP1 crystallographic structures, make MycP1 an attractive target for development of antimicrobial compounds for the treatment of TB.19,20 The structural characteristics of the MycP1 substrate-binding pocket differ substantially from known subtilisins whose structures were solved with bound inhibitors,21 and the commercially available subtilisin inhibitors and protease inhibitor cocktails displayed little or no inhibition of MycP1 (Supporting Information Table 1).
In vitro High Throughput Screening (HTS) in conjunction with in silico virtual screening represent complementary methods for the identification of MycP1 inhibitors. Although HTS alone has been used successfully to identify new leads in drug discovery, it remains a costly and time-consuming process. Various computational approaches are available at present to complement HTS technologies, including the popular, virtual screening (VS) techniques.22−25 VS consists of approaches that either take the structure of the target protein into account (structure-based screening) or rely solely on structures of known bioactive molecules (ligand-based screening). The ligand- and structure-based VS strategies are not mutually exclusive and are often used in parallel. We and others have reported the successful application of these VS methods for lead structure identification,26−29 and many VS software packages are available,30−32 as summarized by Reddy.33 Although the algorithms for these VS methods exploit different types of structural “fingerprints” and scoring functions, their performance varies significantly depending on the specific targets.34−36
As part of an integrated, reiterative program of virtual in silico lead identification, in vitro screening, and laboratory synthesis, we developed two approaches that enhance the effectiveness of the combined ligand- and structure-based VS and hold promise for the development of new classes of MycP1 inhibitors. Previously, we reported an efficient 3D shape-based similarity algorithm including an effective 3D shape-fitting procedure and a robust scoring function (HWZ score).37 We also improved the VS algorithm using an enhanced molecular shape-density model called Shape Approach Based Routines Enhanced (SABRE),37 and we applied this algorithm to a number of medically relevant proteins.38−42 SABRE is unique in that it takes advantage of the structural features of known ligands to generate a consensus molecular-shape pattern (i.e., a “4D fingerprint”) that filters out unacceptable candidates and identifies desired candidates that fit in the binding pocket. The successful performance of SABRE37,43,44 in ranking screened compounds for the 40 databases of the Directory of Useful Decoys (DUD) prompted us to apply this method to the challenging MycP1 target for which no inhibitors have been thus far reported.
We now report the successful application of this ligand- and structure-based virtual screening approach in the discovery of inhibitors that target the active site of the MycP1 protease. Four compounds were identified by SABRE that showed inhibitory activity against MycP1 with IC50 values in the micromolar range and that provide diverse scaffolds as starting points for the development of small-molecule antagonists of MycP1. This study further validated the SABRE 4D fingerprint algorithm as a means of identifying potential inhibitors and providing departure points for laboratory synthesis.
Methods
To identify potential inhibitors of MycP1 protease, we employed VS, according to the flowchart in Figure 1, using a merged compound library from GRI and the NCI downloaded from the ZINC database.45 The structure-based VS (docking) study utilized the recently reported X-ray structure of M. thermoresistibile MycP1 protease (PDB ID: 4HVL).12 We generated multiple conformations of each ligand in the database using OMEGA (OpenEye Scientific Software).46−48 Atom typing, energy calculations, and geometry optimization in OMEGA were performed using the Merck Molecular Force Field (MMFF). The maximum allowed number of conformations per compound was 200, and the energy window, which was the value used to discard high-energy conformations, was set at 10 kcal/mol.
Figure 1.

Flowchart of the VS process.
Ligand Shape-Based Virtual Screening
The ligand 3D-shape-based similarity method of SABRE was used as the first filter in the VS strategy.37,43,44 Briefly, the shape-based VS method takes into account both the structural diversity of each ligand and the HWZ scoring function. Thus, the superimposition of molecules A and B is scored according to the shape-density overlap VAB between the query (molecule A) with shape-density VA and testing structure (B) with shape-density VB. Subsequently, the ligands are ranked according to a uniform scoring function, denoted by Hamza–Wei–Zhan (HWZ) score for convenience.37
The coefficients ak, bk, and ck have been recalibrated from the previous version.
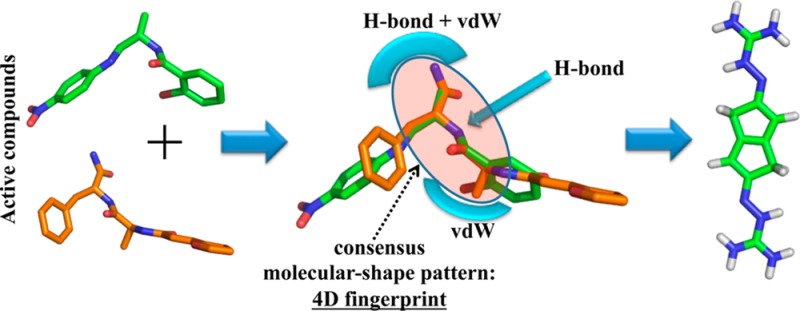
The VS approach includes two stages: the calculation of the optimal coefficients of the shape-density function using a set of known lead compounds and the utilization of these optimal coefficients during the automated ligand/structure shape-based screening of candidate structures. The active structures are ranked according to how well they complement the shape of the binding site and by the degree of differences in molecular shape between the active ligand and inactive (decoy) molecules. SABRE builds a consensus molecular-shape pattern (i.e., a 4D fingerprint) defined by the coefficients [c1, c2, ....,cn] using a set of active ligands, in which the maximum diversity of pharmacophores is taken into account during the screening of the compound library.43,44
Unlike other ligand based shape-overlapping methods,36,49,50 our approach efficiently detects the key pharmacophore groups of the active ligands responsible for binding to the target. The main improvement in our method lies with consideration of “virtual” similar inactive structures (decoys) during the consensus molecular shape pattern detection process. After similarity scoring, the selected structures are further ranked according to the shape complementarity of the receptor-binding site.
In the SABRE algorithm, the shape-density model is enhanced and defined as a linear combination of weighted atomic Gaussian functions.37 Thus, the molecular shape-density is the sum of all individual weighted pharmacophore densities, and the molecular volume can be rewritten as
where Vkpharm is the volume of the pharmacophore k.
The optimal coefficients Ck are determined by iteratively adjusting the coefficients using the set of known active ligands {Ai} and “virtual” decoy structures {Bi} (virtual decoys are chemically possible compounds that are not necessarily synthetically feasible) until they satisfy these two criteria: for A ∈ {Ai}
The algorithm builds an efficient consensus molecular-shape pattern in which the optimal coefficients {Ck} define the 4D fingerprint of the entire set of active ligands and take into account the structural similarity and chemical features of inactive structures (decoys).37,44 Therefore, the “4D fingerprint” encodes the (3D) shapes of the known active ligand structures in their multi conformational states (1D).
Structure Shape-Based Virtual Screening
The “docking option” available within the SABRE program37,42,44 placed the filtered conformations of each ligand into the binding cavity of the MycP1 X-ray structure12 (PDB ID: 4HVL). During this shape-fitting process, the specific conformation was placed into a grid box encompassing all active-site atoms within the receptor-binding site. The volume of the receptor-binding pocket was calculated using a smooth Gaussian function.44 The docking strategy exhaustively explored all possible positions of each ligand in the binding site and generally focused upon two parameters: shape-fitting and ligand-pose optimization, where ligand-pose is defined as a specific ligand–receptor conformation. The ligand-pose ensemble was then filtered to reject arrangements that did not have sufficient complementarity with the active site of the protein and was next filtered to reject those arrangements lacking significant van der Waals contacts with nearby residues. For MycP1 protease, the ligands were filtered based on van der Waals contacts with the Ser202, Ala236, S334, and Thr333 side chains. Finally, the screened ligands were ranked using the HWZ scoring function.37
Molecular Modeling of the Binding of the Octapeptide AVKAASLG with MycP1
We utilized reported procedures for the docking and solvated molecular dynamics (MD) simulations procedures of the MycP1–peptide complex.27,42 The docking and MD simulations procedures are described in the Supporting Information.
In Vitro Assay of MycP1mth Inhibitors
Recombinant M. thermoresistibile MycP1 was expressed and purified as reported previously.12 In quenched fluorescent peptide assays, MycP1 (1 μM) was used to digest 20 μM of a fluorescent peptide substrate, AbzAVKAASLGK(Dnp)OH (GenScript Inc.). Potential MycP1 inhibitors were dissolved in DMSO at 30 mM final concentrations and subsequently diluted with a buffer (50 mM HEPES pH 7.5, 100 mM NaCl) to the desired concentrations. Inhibitors were screened in duplicate in 96-well format at 150 μM concentrations by adding compounds to MycP1 immediately prior to the addition of the fluorescent peptide. Hydrolysis of the fluorescent peptide was measured by following the increase in fluorescence in a SpectraMaxGemini XPS plate reader (Molecular Devices, LLC) with the following settings: excitation = 319 nm, emission = 419 nm. The initial rate for each inhibitor was calculated using the Softmax Pro software (Molecular Devices, LLC). Inhibitors with high intrinsic fluorescence at time zero were eliminated from further consideration. Rates of the remaining compounds were compared to controls (DMSO-buffer blank) in order to estimate the percent inhibition of individual compounds.
The same in vitro assay was utilized to measure IC50 of compounds that displayed at least 50% inhibition at 150 μM concentrations. Inhibitors were added at 0, 5, 10, 50, 100, 200, 350, and 500 μM concentrations, and DMSO was added such that the final DMSO concentration in each well was the same (2% v/v). Initial rates of fluorescent peptide hydrolysis were measured in triplicate and converted to percentages of baseline activity. Dose–response curves were calculated using the sigmoidal dose–response option in GraphPad Prism version 4.00 (GraphPad Software, Inc.). IC50 values were calculated using GraphPad plotted rates of MycP1 activity as a function of log([inhibitor]) and fitted to a sigmoidal curve constrained to 100% activity (top) and 0% activity (bottom) (Supporting Information Figure 1).
Results and Discussion
The MycP1 protease recognized the cleavage site sequence ...–AVKAA358|SLG–... within an unstructured portion of EspB.12,13 Consequently, the MycP1 protease readily processed a fluorescently labeled peptide analogue, AbzAVKAASLGK(Dnp)OH, that had a Km of 60 μM.12 Molecular docking of the peptide revealed relatively few, low-energy MycP1–peptide complexes. The MycP1–peptide complex that oriented the expected peptide residues in positions P1 and P2, which lie near the catalytic Ser334 side-chain, had the largest number of low-energy binding conformations or poses. The docked peptide was further refined by performing MD simulations in which the stability of the MD-simulated MycP1–peptide complex was described by the time dependence of the root-mean-square deviation (RMSD) values of the peptide backbone (Supporting Information Figure 2). The refined MycP1–peptide complex displayed a peptide backbone bound to the catalytic active site of MycP1 through hydrogen bonds with the backbone of the protein (Figure 2). These hydrogen bonds included contacts between (H)AVKAASLGK(OH) with Thr333, Ser202, Glu203, and Ala331. In addition, we observed that the Lys side chain of the peptide was stabilized through H-bonding and long-range, electrostatic interactions with the Gln242 and Asp243 side-chains of the MycP1 binding pocket.
Figure 2.
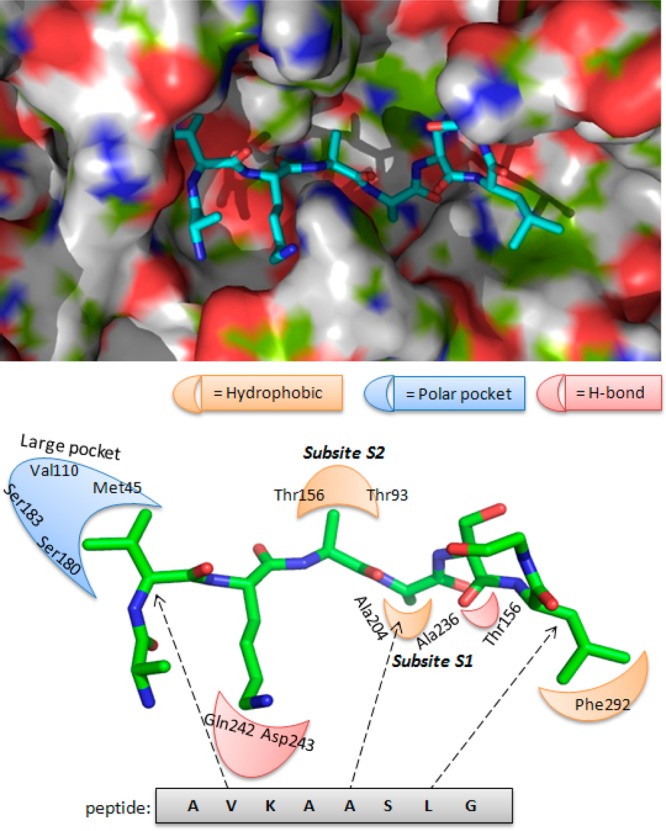
(Top) Preferred binding conformation of the peptide substrate in the MycP1 active site. (Bottom) Pharmacophoric model describing key interactions of the peptide with the MycP1 residues.
Finally, the interactions between the peptide and active site amino acids were analyzed, and pharmacophore features, such as hydrogen bond acceptors and donors, and hydrophobic areas were mapped. Guided by this docking study, we generated a primary topographical interaction model to guide the subsequent VS process (Figure 2). We reasoned that inhibitors that interacted with the MycP1 binding site in a fashion similar to (H)AVKAASLGK(OH) should also compete effectively with EspB in the active site. Because of our prior successes, we utilized the SABRE program for the VS campaign and identified potential inhibitors that occupied the active site of MycP1 (Figure 2) and displayed binding energies comparable or superior to the peptide (H)AVKAASLGK(OH).
In the first stage of the VS protocol, we used the bound peptide structure as a query to search collections of small molecules for “hits” that exhibited 3D shapes similar to the octapeptide (H)AVKAASLGK(OH) in Figure 2. To limit the number of structures, we evaluated the multiconformational states of each potential inhibitor from the GRI/NCI database using the ligand shape-based similarity screening technique in the SABRE program. In recent reports, we described the high efficiency of the SABRE algorithm to identify active ligands at different stages (0.1% to 10%) of the screened DUD databases.43,44
We docked approximately the top 10% of the active ligands into the MycP1 active site using the fast-and-rigid-docking method of the SABRE program. All active ligands that passed the receptor-shape-fitting filter were scored. Visual inspection of the top 1000 structures (top ∼0.3% of the database) served to exclude “false positives” resulting from protein side-chain distortion and/or erroneous ligand binding that overestimated the scoring value of an active ligand with the receptor site. As a consequence, we selected molecular structures that fit into the binding pocket (i.e., had shape complementarity to the octapeptide substrate) and that adopted a conformation similar to the octapeptide backbone in the active site of MycP1.
On the basis of these docking calculations, we tested 81 compounds (15 from NCI and 66 from GRI databases) for their inhibitory activity against the recombinant MycP1. The in vitro assays were performed with a single concentration (150 μM), and these assays disclosed a series of compounds with ∼10% to 60% inhibitory activity against MycP1. Finally, we selected two of the best inhibitors, namely compounds 1 and 2 (2.5% of the hits; Figure 3), which inhibited more than 40% of the MycP1 activity (Table 1). In comparing the binding of compounds 1 and 2 to (H)AVKAASLGK(OH), we noted that the octapeptide exploited the entire MycP1 binding site, including a deep pocket containing subsite S1 into which the side-chain of (H)AVKAA358SLGK(OH) inserted as well as a long cavity where the backbone of (H)AVKAASLGK(OH) fitted. Compounds 1 and 2 were predicted to use the same deep subsite S1 cavity: the amide moiety in compound 1 was crucial to its binding, and the methyl moiety in compound 2 was crucial to its binding in this deep cavity.
Figure 3.
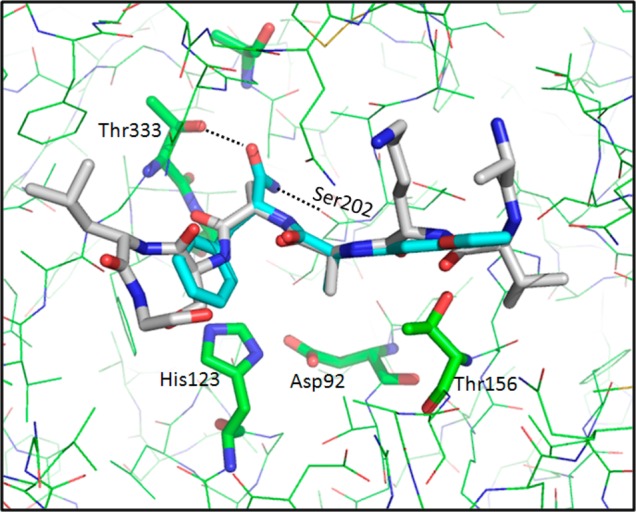
Binding mode of compound 1 (NSC-334943) in the MycP1 active site. For comparison, the docked peptide is represented in gray.
Table 1. Experimentally Determined Inhibitory Activity of the 10 Compounds Selected from the Virtual Screening.
| compound | NSC or UC/GRI | % inhibition at 150 μM | IC50 (μM)a | hits tested | % yield of active compounds |
|---|---|---|---|---|---|
| 1 | NSC-334943 | 61% | 135 | 81 | 2.5% |
| 2 | UC-521228 | 42% | ND | ||
| 3 | NSC-334344 | 67% | 146 | 40 | 10% |
| 4 | NSC-657705 | 43% | ND | ||
| 5 | NSC-112182 | 43% | ND | ||
| 6 | NSC-25812 | 41% | ND | ||
| 7 | NSC-176297 | 56% | ND | 25 | 16% |
| 8 | NSC-106893 | 68% | 95 | ||
| 9 | NSC-97914 | 59% | ND | ||
| 10 | NSC-357905 | 73% | 48 |
ND: Not Determined.
We observed that the compounds 1 and 2 were ranked at positions 296 and 31 of the top 1000 selected ligands of the VS and were ranked at positions 25 and 7 in the list of 81 tested structures. These results are encouraging and are in good agreement with our previous studies in which the SABRE program provided high enrichment factors (EF) at EF0.1% (top 100) and EF1% (top 1000) after screening a large database (95 000 ligands).44
To test whether our iterative VS scheme would successfully identify novel inhibitors, the pharmacophore model was used once again to screen the GRI/NCI database. Our approach is distinct from other VS methods in that it uses a “consensus molecular shape pattern” or what we call “4D fingerprints” of active ligands to search out similar structures.43,44 A visual depiction of this consensus molecular shape pattern is shown in Figure 4. We recently reported that searches using this consensus molecular shape pattern constituted an effective procedure for searches for active compounds within data sets of varying size and structural diversity.43,44 Using the consensus molecular shape pattern derived from compounds 1 and 2 avoided the need to screen large databases using several different queries and then to rank the ligands using time-consuming, data-fusion methods that in the past often produced false positives.35
Figure 4.
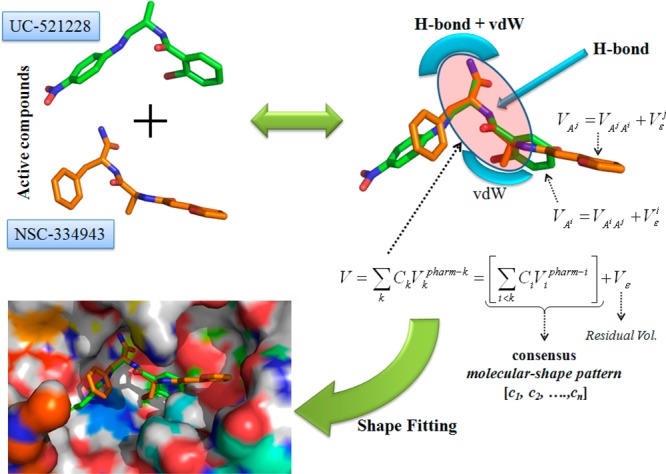
Schematic description of the consensus molecular pattern or 4D fingerprint derived from compounds 1 and 2.
We started the second round of virtual screening of the GRI/NCI database using the structure of compound 1 as a query and the computed 4D fingerprint from compounds 1 and 2. As before, the top 1000 compounds that resulted from the VS process were selected and visually inspected to eliminate those that did not efficiently span the MycP1 binding site. Forty compounds from the NCI database were then obtained and tested experimentally at a dose of 150 μM in the high throughput in vitro inhibition assay described previously. Four compounds, 3 to 6 (Figure 5; 10% of the hits), showed more than 40% inhibitory activity. At this stage, the query structure 1 permitted the identification of a similar scaffold (compound 3) to compound 1 as well as highly diverse structural scaffolds (compounds 4, 5, and 6). The resulting “hits” simultaneously served as a means of validating the docking model (since compounds 1 and 3 were similar) and assisted in the identification of new putative compounds for the next iteration of VS. As displayed in Figure 5 and as expected, the amide moiety of compound 3 bound into the S1 site of the MycP1 cavity and formed two H-bonds with Thr333 and Ser202 residues. In addition, the ligand backbone was stabilized in the binding cavity through H-bonding with Thr156 side-chain.
Figure 5.
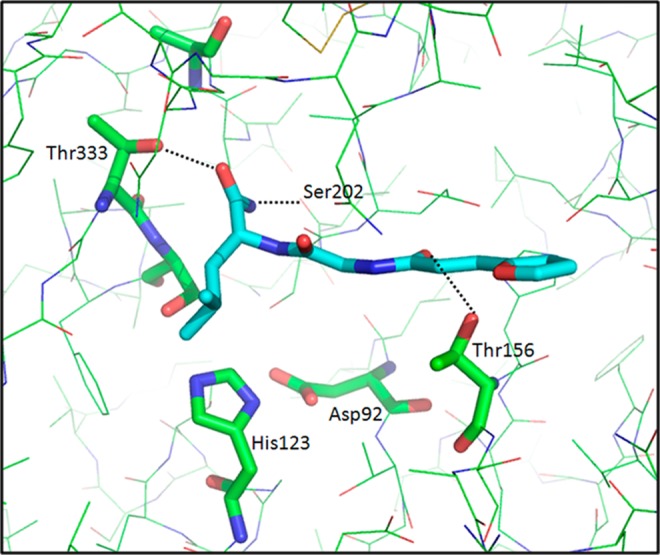
Binding mode of compound 3 (NSC-334344) in the MycP1 active site.
In the next stage of this process, the SABRE program utilized the best structure identified in the previous step, namely compound 3, as the query, and the 4D fingerprint generated from compounds 1 to 6 for the screening of the GRI/NCI database. From the final top 1000 structures, 25 compounds were assayed for their inhibitory activity against MycP1 protease. As shown in Table 1, four compounds 7–10 (Figure 6; 16% of the hits) displayed inhibitory activity of more than 50% at the initial 150 μM concentration. Although the second-query compound 3 displayed structural similarity to the first-query compound 1, it permitted the identification of a more diverse subset of compounds than those identified by compound 1. Thus, the computed 4D fingerprint in the second VS round increased the structural diversity of the scaffolds over those seen in the first VS round (Table 1). The predicted binding conformations of compounds 8 and 10 and the key interactions with MycP1 are shown in Figure 6. The ligands make van der Waals contacts with the active site and span the S1 subsite, thereby blocking access to the catalytic Ser334–His123–Asp92 triad. Specifically, compound 8 formed hydrogen bonds with the side-chain hydroxyl group of Ser334 and Thr333 in the S1 subsite while the methoxyphenyl moiety of the structure formed hydrogen bonds with the hydroxyl group of the Thr156 side chain and long-range electrostatic interactions with the carboxylate group of the Glu203 residue. Compound 10 interacted mainly via the hydrogen bonds between the primary amino group of the ligand and the Thr333, Ser202, and Thr156 side-chains of MycP1 as well as with long-range electrostatic interactions with the Glu203 side-chain. These interactions are accompanied by van der Waals interactions between the scaffold of the ligand and the Thr156 side-chain. It is striking that compound 10 displays low scaffold similarity to the reference compounds (query) while maintaining a comparable binding mode with highest affinity to MycP1. A superposition of the putative complex structure of compounds (1, 3, 8, and 10) in the MycP1 active site is displayed in Supporting Information Figure 4. Structural scaffolds of MycP1 inhibitors identified during the VS campaign are shown in Figure 7.
Figure 6.
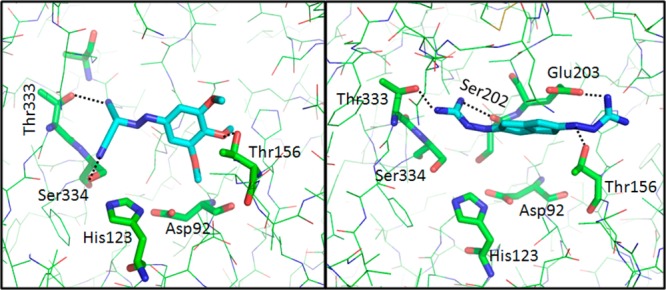
Binding mode of compounds 8 (NSC-106893) and 10 (NSC-357905) in the MycP1 active site.
Figure 7.

Structural scaffolds of MycP1 inhibitors identified during the VS campaign.
To highlight the structural diversity of the lead compounds, a 2D heat map of pairwise MACCS fingerprints among the 10 compounds was calculated using the babel program (Figure 8).51 The 2D heat map shows that most of the compounds have a pairwise similarity ranging from 0.2 to 0.6 (red and green colors), which indicates high structural diversity between the 10 lead compounds. It is interesting to note that the pairwise similarities decrease going from compound 1 to 10 (the pairwise values are reported in Supporting Information Table 2) suggesting the evolution of increasing structural diversity in each VS round.
Figure 8.
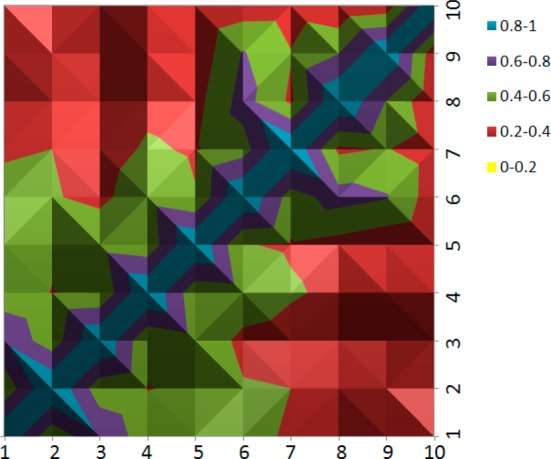
2D heat map of pairwise MACCS fingerprints among the 10 lead compounds.
In summary, the application of the consensus molecular shape pattern-based virtual screening approach, using the SABRE program, identified diverse structural lead compounds for inhibition of MycP1 protease. This study validated the SABRE 4D fingerprint algorithm as a means of enriching compound libraries for active compounds during the VS campaign. The ligand- and structure-based virtual screening approach allowed the utilization of relatively small compound libraries while still discovering a number of interesting lead compounds. These small libraries coupled with the high throughput nature of the in vitro screening assay allowed quick and efficient discovery of novel active scaffolds. These initial scaffolds should prove useful for the development of additional, more potent MycP1 antagonists than those discussed here in a drug discovery process that is now underway. Furthermore, the screening approach described here can be applied for the identification of novel inhibitors to many other proteins of known structure.
Acknowledgments
The authors thank Professors Jürgen Bosch at the Johns Hopkins University and Jon Thorson at the University of Kentucky for valuable discussions. We acknowledge the Drug Synthesis and Chemistry Branch of National Cancer Institute for supplying most of the compounds used in the virtual screening and testing. We thank the University of Kentucky Information Technology department and Center for Computational Sciences for computing time on the Lipscomb High Performance Computing Cluster. We acknowledge the University of Kentucky Organic Synthesis Core that is partially supported by grant P20GM103486 from the National Institute of General Medical Sciences (to D.S.W.). This study was supported by NIH/NIGMS grant P20GM103486 (to K.V.K.), and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NIGMS.
Supporting Information Available
Computational procedures for the MD simulations and additional Figures and Tables are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- WorldHealthOrganization. WHO Global tuberculosis report, 2013. http://www.who.int/tb/publications/global_report/en/.
- Mdluli K.; Spigelman M. Novel targets for tuberculosis drug discovery. Curr. Opin. Pharmacol. 2006, 6, 459–467. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Arcus V. L.; Lott J. S.; Johnston J. M.; Baker E. N. The potential impact of structural genomics on tuberculosis drug discovery. Drug Discovery Today 2006, 11, 28–34. [DOI] [PubMed] [Google Scholar]
- Zhao Q. J.; Xie J. P. Mycobacterium tuberculosis proteases and implications for new antibiotics against tuberculosis. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 347–361. [DOI] [PubMed] [Google Scholar]
- Bottai D.; Serafini A.; Cascioferro A.; Brosch R.; Manganelli R. Targeting type VII/ESX secretion systems for development of novel antimycobacterial drugs. Curr. Pharm. Des. 2013, 10.2174/1381612819666131118170717. [DOI] [PubMed] [Google Scholar]
- Simeone R.; Bottai D.; Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 2009, 12, 4–10. [DOI] [PubMed] [Google Scholar]
- Stoop E. J.; Bitter W.; van der Sar A. M. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol. 2012, 20, 477–484. [DOI] [PubMed] [Google Scholar]
- Houben E. N.; Korotkov K. V. Bitter, W. Take five — Type VII secretion systems of Mycobacteria. Biochim. Biophys. Acta 2013, 10.1016/j.bbamcr.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Brown G. D.; Dave J. A.; Gey van Pittius N. C.; Stevens L.; Ehlers M. R.; Beyers A. D. The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene 2000, 254, 147–155. [DOI] [PubMed] [Google Scholar]
- Dave J. A.; Gey van Pittius N. C.; Beyers A. D.; Ehlers M. R.; Brown G. D. Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol. 2002, 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. M.; Evans T. J.; Chen J.; Zhu H.; Houben E. N. G.; Bitter W.; Korotkov K. V. Understanding specificity of the mycosin proteases in ESX/type VII secretion by structural and functional analysis. J. Struct. Biol. 2013, 184, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonson M.; Huesgen P. F.; Wasney G. A.; Watanabe N.; Gruninger R. J.; Prehna G.; Overall C. M.; Strynadka N. C. Structure of the mycosin-1 protease from the Mycobacterial ESX-1 protein type VII secretion system. J. Biol. Chem. 2013, 288, 17782–17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.; Liu Q.; He Y.; Wang C.; Wu F.; Tian C.; Zang J. The putative propeptide of MycP1 in mycobacterial type VII secretion system does not inhibit protease activity but improves protein stability. Protein Cell 2013, 4, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B.; Chon J. S.; MacGurn J. A.; Carlsson F.; Cheng T. L.; Cox J. S.; Brown E. J. A Mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007, 3, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Laine O.; Masciocchi M.; Manoranjan J.; Smith J.; Du S. J.; Edwards N.; Zhu X.; Fenselau C.; Gao L. Y. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol. Microbiol. 2007, 66, 787–800. [DOI] [PubMed] [Google Scholar]
- Ohol Y. M.; Goetz D. H.; Chan K.; Shiloh M. U.; Craik C. S.; Cox J. S. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 2010, 7, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. M.; Zhang M.; Rybniker J.; Boy-Rottger S.; Dhar N.; Pojer F.; Cole S. T. Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA-independent virulence. Mol. Microbiol. 2013, 89, 1154–1166. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Pojer F.; Blasco B.; Cole S. T. Towards anti-virulence drugs targeting ESX-1 mediated pathogenesis of Mycobacterium tuberculosis. Drug Discovery Today: Dis. Mech. 2010, 7, e25–e31. [Google Scholar]
- Roberts D. M.; Personne Y.; Ollinger J.; Parish T. Proteases in Mycobacterium tuberculosis pathogenesis: potential as drug targets. Future Microbiol. 2013, 8, 621–631. [DOI] [PubMed] [Google Scholar]
- McPhalen C. A.; James M. N. Structural comparison of two serine proteinase-protein inhibitor complexes: eglin-c-subtilisin Carlsberg and CI-2-subtilisin Novo. Biochemistry 1988, 27, 6582–6598. [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. [DOI] [PubMed] [Google Scholar]
- Rester U. From virtuality to reality — Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective. Curr. Opin. Drug Discovery Dev. 2008, 11, 559–568. [PubMed] [Google Scholar]
- Kolb P.; Ferreira R. S.; Irwin J. J.; Shoichet B. K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 20, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Irwin J. J. Docking screens: right for the right reasons?. Curr. Top. Med. Chem. 2009, 9, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks W. H.; McCloskey D. E.; Daniel K. G.; Ealick S. E.; Secrist J. A. 3rd; Waud W. R.; Pegg A. E.; Guida W. C. In silico chemical library screening and experimental validation of a novel 9-aminoacridine based lead-inhibitor of human S-adenosylmethionine decarboxylase. J. Chem. Inf. Model. 2007, 47, 1897–1905. [DOI] [PubMed] [Google Scholar]
- Hamza A.; Zhao X.; Tong M.; Tai H. H.; Zhan C. G. Novel human mPGES-1 inhibitors identified through structure-based virtual screening. Bioorg. Med. Chem. 2011, 19, 6077–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; AbdulHameed M. D.; Hamza A.; Zhan C. G. New inhibitor of 3-phosphoinositide dependent protein kinase-1 identified from virtual screening. Bioorg. Med. Chem. Lett. 2012, 22, 1629–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorna V.; Theisen E. R.; Stephens B.; Warner S. L.; Bearss D. J.; Vankayalapati H.; Sharma S. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 2013, 56, 9496–9508. [DOI] [PubMed] [Google Scholar]
- Taylor R. D.; Jewsbury P. J.; Essex J. W. A review of protein-small molecule docking methods. J. Comput.-Aided Mol. Des. 2002, 16, 151–166. [DOI] [PubMed] [Google Scholar]
- Coupez B.; Lewis R. A. Docking and scoring — theoretically easy, practically impossible?. Curr. Med. Chem. 2006, 13, 2995–3003. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Shoichet B. K.; Peishoff C. E. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 2006, 49, 5851–5855. [DOI] [PubMed] [Google Scholar]
- Reddy A. S.; Pati S. P.; Kumar P. P.; Pradeep H. N.; Sastry G. N. Virtual screening in drug discovery — a computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [DOI] [PubMed] [Google Scholar]
- Vainio M. J.; Puranen J. S.; Johnson M. S. ShaEP: molecular overlay based on shape and electrostatic potential. J. Chem. Inf. Model. 2009, 49, 492–502. [DOI] [PubMed] [Google Scholar]
- Svensson F.; Karlen A.; Skold C. Virtual screening data fusion using both structure- and ligand-based methods. J. Chem. Inf. Model. 2012, 52, 225–232. [DOI] [PubMed] [Google Scholar]
- Yan X.; Li J.; Liu Z.; Zheng M.; Ge H.; Xu J. Enhancing molecular shape comparison by weighted Gaussian functions. J. Chem. Inf. Model. 2013, 53, 1967–1978. [DOI] [PubMed] [Google Scholar]
- Hamza A.; Wei N. N.; Zhan C. G. Ligand-based virtual screening approach using a new scoring function. J. Chem. Inf. Model. 2012, 52, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Hou Y.; Hamza A.; Pain C.; Zhan C. G.; Bugni T. S.; Thorson J. S. Probing the regiospecificity of enzyme-catalyzed steroid glycosylation. Org. Lett. 2012, 14, 5424–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambouli N.; Wei N. N.; Jlizi A.; Aissa S.; Abdelmalek R.; Kilani B.; Slim A.; Tiouiri B. A.; Dridi M.; Hamza A.; Ben Ammar Elgaied A. Structural insight into a novel human CCR5-V130I variant associated with resistance to HIV-1 infection. J. Biomol. Struct. Dyn. 2013, 10.1080/07391102.2013.819297. [DOI] [PubMed] [Google Scholar]
- Stambouli N.; Dridi M.; Wei N. N.; Jlizi A.; Bouraoui A.; Elgaaied A. B. Structural insight into the binding complex: beta-arrestin/CCR5 complex. J. Biomol. Struct. Dyn. 2014, 32, 866–875. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Sviripa V.; Chen X.; Shi J.; Yu T.; Hamza A.; Ward N. D.; Kril L. M.; Vander Kooi C. W.; Zhan C. G.; Evers B. M.; Watt D. S.; Liu C. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 2013, 8, 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Hamza A.; Zhan C. G.; Thorson J. S. Assessing the regioselectivity of OleD-catalyzed glycosylation with a diverse set of acceptors. J. Nat. Prod. 2013, 76, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza A.; Wei N. N.; Hao C.; Xiu Z. L.; Zhan C. G. A novel and efficient ligand-based virtual screening approach using the HWZ scoring function and an enhanced shape-density model. J. Biomol. Struct. Dyn. 2013, 31, 1236–1250. [DOI] [PubMed] [Google Scholar]
- Wei N. N.; Hamza A. SABRE: ligand/structure-based virtual screening approach using consensus molecular-shape pattern recognition. J. Chem. Inf. Model. 2014, 54, 338–346. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC — a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMEGA, version 2.5.1; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com. [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
- Mavridis L.; Hudson B. D.; Ritchie D. W. Toward high throughput 3D virtual screening using spherical harmonic surface representations. J. Chem. Inf. Model. 2007, 47, 1787–1796. [DOI] [PubMed] [Google Scholar]
- Dror O.; Schneidman-Duhovny D.; Inbar Y.; Nussinov R.; Wolfson H. J. Novel approach for efficient pharmacophore-based virtual screening: method and applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.