

Abstract

Red fluorescent proteins (RFPs) are indispensable tools for deep-tissue imaging, fluorescence resonance energy transfer applications, and super-resolution microscopy. Using time-resolved optical spectroscopy this study investigated photoinduced dynamics of three RFPs, KillerRed, mRFP, and DsRed. In all three RFPs, a new transient absorption intermediate was observed, which decays on a microsecond–millisecond time scale. This intermediate is characterized by red-shifted absorption at 1.68–1.72 eV (λmax = 720–740 nm). On the basis of electronic structure calculations, experimental evidence, and published literature, the chemical nature of the intermediate is assigned to an unusual open-shell dianionic chromophore (dianion-radical) formed via photoreduction. A doubly charged state that is not stable in the isolated (gas phase) chromophore is stabilized by the electrostatic field of the protein. Mechanistic implications for photobleaching, blinking, and phototoxicity are discussed.
I. Introduction
The discovery of the green fluorescent protein (GFP) and its ensuing applications as a genetically encoded fluorescent label have significantly advanced our understanding of the complex biochemical processes in living systems.1 Mutations of wtGFP and similar fluorescent and chromoproteins gave rise to a palette of biomarkers covering the entire visible range from blue to far-red.2,3 Red fluorescent proteins (RFPs) are particularly important for in vivo imaging as they enable better penetration depth and signal separation from cellular autofluorescence.4 FPs enable studies of protein–protein interactions, gene expression, protein localization, and intracellular protein targeting.5,6 The unique properties of FPs have also been exploited for development of pH,7 metal,8,9 redox10,11 and hydrogen peroxide sensors,12 and phototoxic agents.13,14 However, FPs as fluorescent markers suffer from irreversible (photobleaching) and reversible (blinking or flickering) loss of fluorescence15,16 that limits their applications. Despite being crucial for engineering of better fluorescent labels, the mechanistic understanding of these processes in FPs is quite rudimentary, in stark contrast to similar phenomena in synthetic dyes.17
Several photobleaching and blinking pathways have been determined using a combination of theoretical and experimental approaches. Photoinduced rapture of the π-conjugated system in IrisFP resulting in a (dark) chromophore with sp3-hybridized Cα was shown to be responsible for blinking.18,19 Similar chromophore structures with partially destroyed π-conjugation have been suggested to explain the X-ray structure of a bleached state of KillerRed.20,21 Among other suggested blinking/flickering pathways are transitions to triplet states, reversible conformational changes, and changes of the chromophore H-bonding network and its protonation state.17
Although photobleaching of organic dyes is often initiated by electron attachment to (or detachment from) electronically excited chromophores,17 it is unclear whether such photoreduction and photo-oxidation processes play a role in FPs. There is only limited evidence of radical involvement in the photobleaching of FPs. It was suggested that formation of radical species precedes photobleaching of IrisFP upon X-ray irradiation.21 Photoinduced electron transfer in FPs leads to the so-called oxidative redding, the photoconversion resulting in the green-to-red shift of the fluorescence.22 However, the mechanism of oxidative redding and even the chemical identity of the red form are still unknown.23,24 Apart from their relevance to photostability, formation of long-living radical species may have implications for the phototoxicity of FPs. Indeed, an EPR spectrum was reported for the KillerRed protein, the most phototoxic FP, although the structure of the long-lived radical is not known.25 KillerRed is strongly phototoxic when irradiated with light of 540–580 nm (2.30–2.14 eV) in the presence of oxygen; the phototoxicity was attributed to the formation of reactive oxygen species (ROS).14,25,26
Figure 1 illustrates major pathways of ROS formation including superoxide radical and singlet oxygen. The key point is that electronic excitation increases both reducing and oxidizing abilities of the chromophore, because 1Chro∗– and 3Chro– can accept an extra electron into the highest occupied molecular orbital (HOMO) or donate an electron from the lowest unoccupied molecular orbital (LUMO), in contrast to the ground-state chromophore (1Chro–) that accepts an electron into the LUMO and donates one from the HOMO. The photo-oxidation/photoreduction leads to a formation of doublet radicals, 2Chro and 2Chro2–, respectively. The latter can then donate an electron to a nearby oxygen molecule forming 2O2–. Superoxide can also be formed by direct oxidation of 1Chro∗– by oxygen. Singlet oxygen can be produced via photosensitization by 3Chro–. Contrary to superoxide formation, 1O2 production must proceed through the triplet state of the chromophore, thus requiring an intersystem crossing step.
Figure 1.
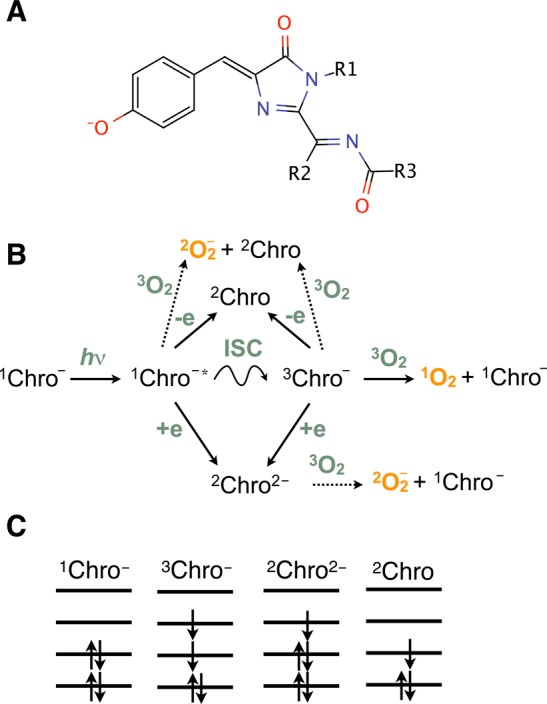
(A) RFP chromophore in the anionic form. The chromophore in KillerRed is linked to the protein via Glu68 (R1) and Ile64 (R3), and R2 is a side chain of Gln forming the chromophore. (B) Photoinduced processes that can lead to the formation of reactive oxygen species (ROS), 1O2 (singlet oxygen), and O2•– (superoxide). Relevant chromophore states are the ground and excited singlet states (1Chro– and 1Chro∗–), a triplet state (3Chro–), and electron-attached and electron-detached doublet states (2Chro2– and 2Chro). (C) Electronic configurations of the ground-state singlet, lowest excited singlet and triplet states, and two doublet radicals derived by electron attachment and detachment from 1Chro–. Solid arrows denote pathways supported by the previous and present studies, whereas dotted lines represent merely speculative (at this moment) pathways of ROS formation.
The lack of singlet oxygen emission25 suggests that KillerRed is a radical-based type I photosensitizer, although superoxide’s relatively low toxicity is difficult to reconcile with the very strong phototoxicity of KillerRed.
Photoreduction/photo-oxidation of the chromophore does not necessarily involve external redox agents; that is, nearby amino acids may also serve as electron donors/acceptors. For example, photoinduced electron transfer from neighboring Glu to the chromophore is believed to be an initial step in the photoinduced decarboxylation of GFP27−30 and DsRed.31
In sum, the bulk of knowledge of synthetic dyes properties, along with emerging data on FPs, suggests that understanding the dynamics of photoinduced electron transfer events in RFPs as well as lifetimes and chemical identity of the species involved in these processes is of central importance for understanding the photobleaching and phototoxicity of FPs.
Aiming to elucidate photoinduced transformations of FPs, we performed a time-resolved spectroscopic study of RFP dynamics in the micro- to millisecond range bridging the gap between two commonly studied regimes, femto- to microseconds and seconds to minutes. This time scale is typical for processes involving triplet states and radical species in related systems.15−17 Here, we report the first broadband transient absorption (TA) in the microsecond to second time domain of three RFPs, DsRed, mRFP, and KillerRed. All three share an identical anionic chromophore (Figure 1A) and have similar steady-state absorption/emission properties; however, variations in the local environment and the barrel’s structure lead to different photostability, brightness, and phototoxicity. DsRed is a tetrameric RFP from which many other FPs, including mRFP, have been derived. Despite the common chromophore structure, these proteins differ dramatically in their phototoxicity in the order DsRed < mRFP < KillerRed.12,14 This difference is attributed to the chromophore accessibility.20 Interestingly, DsRed shows better photostability than, for example, monomeric RFPs from the mFruit series, which is attributed to the structural weakness of their β-barrels facilitating the diffusion of small species in and out of the protein interior.32,33 To understand their photocycle, we undertook a comparative study of the photoinduced dynamics of these RFPs. Electronic structure calculations facilitate structural assignment of the observed spectral features.
The structure of the paper is as follows. Section II describes experimental and computational details (additional information is provided in the Supporting Information (SI)). The results are discussed in section III. Our concluding remarks are given in section IV.
II. Experimental Setup and Computational Details
A. Experimental Methods
Time-resolved emission and absorption spectra were acquired using the instruments described below. Fluorescence lifetimes were measured using an Edinburgh Instruments time-correlated single photon counting (TCSPC) system. In these measurements, picosecond diode laser (Picoquant) and subnanosecond LED excitation pulses (Edinburgh Instruments) emitting at 467 and 590 nm, respectively, were used as excitation light sources. The detection system consisted of a high-speed MicroChannel Plate PhotoMultiplier Tube (MCP-PMT, Hamamatsu R3809U-50) and TCSPC electronics. The time resolution of the system was 30 ps after deconvolution with an IRF signal.
Subpicosecond excited-state absorption spectra of RFPs were measured using a commercially available pump–probe spectroscopic system (Helios, Ultrafast systems) pumped by the femtosecond laser system consisting of a Ti:sapphire regenerative amplifier (Solstice, Spectra-Physics, 800 nm, 1 kHz) and an optical parametric amplifier (OPA, TOPAS-C, Spectra-Physics, 290–2800 nm, ∼100 fs fwhm). Other experimental conditions were the same as in earlier studies.34
Transient absorption spectroscopy measurements in the microsecond-to-second time domain were performed using a custom-built kinetic setup based on ANDOR and Basler Vision microarray imaging cameras, allowing the broadband TA measurements with 1 μs time resolution after a single pulse excitation. This setup, previously used to study multiheme O2-reducing electron-transfer enzymes,35−37 is located at the University of Helsinki (Helsinki, Finland), where measurements were performed.
Additional details of the experimental setup are presented in the SI.
B. Computational Details
We employed computational protocols developed and validated in our earlier studies of FPs.24,33,38,39 Unit A from the 3GB3 (PDB:ID) X-ray structure20 was used as a model of the KillerRed protein. The protonation states of the protein residues were assigned using PROPKA.40 The only amino acids found in the nonstandard protonation states are GLU68 and GLU218; they were protonated in our model. The protonation states of the amino acids were consistent with the hydrogen-bonding pattern in the X-ray structure (see the SI for detailed discussion). The protein was placed in the 100 × 100 × 100 Å water box. Eight Cl– and 15 Na+ counterions were added to neutralize the protein’s surface charges. Molecular dynamics (MD) simulations were performed to sample the ground-state structure of the protein. All MD simulations were done using the GROMACS 4.6 package.41 The system was first equilibrated during consecutive NVT (T = 300 K) and NPT (P = 1 atm, T = 300 K) 100 ps simulations. Modified Berendsen thermostat42 and Parinello–Rahman barostat43 were employed. Equilibration stage was followed by a 1 ns production run. Periodic boundary conditions were used in all simulations. All atoms were allowed to move during the MD simulation.
A CHARMM27 force field was used for all standard protein residues. Water was described using the TIP3P model. The chromophore parameters were generated as follows. The equilibrium parameters (bond lengths and angles) were taken from the optimized ωB97X-D/6-31+G(d,p) (see ref (44)) KillerRed chromophore structure. NBO45 partial charges computed with ωB97X-D/6-31+G(d,p) were used. Force constants were taken from ref (46) and from the CHARMM27 parameters for the chemically similar moieties. The list of all force field parameters for the KillerRed chromophore is given in the SI.
All excitation/attachment/detachment energies were computed using the geometries taken from the MD snapshots for KillerRed with the chromophore in the closed-shell anionic form. An electrostatic embedding scheme was used. The QM part included the chromophore, as shown in Figure S10 in the SI. The rest of the system was represented by the MM force field point charges. In our previous studies of GFP and mStrawberry, we found that the effects of extending the QM part by including nearby residues are insignificant.24,33 In the present study, we revisited this question and investigated the effect of including Arg94 in the QM part. As discussed in the SI, the shifts in excitation energies caused by including the Arg94 side chain into the QM part were negligible (SI Table S1). The MM charges for the atoms at the QM–MM border were set to zero and only 1–4 interactions were considered. The resulting nonzero total charge on a truncated residue was redistributed equally between the rest of the residue’s atoms.
SOS-CIS(D)47 and TDDFT(B5050LYP) were used in the QM calculations for the anion and dianion excitation energies, respectively. SOS-CIS(D) was demonstrated to provide accurate results for the excitation energies of the GFP chromophore and its analogues.24,33,48,49 However, the benchmark calculations for the dianion radical showed that SOS-CIS(D) based on the UHF reference suffers from spin contamination (⟨S2⟩ ∼ 1.5), whereas ROHF calculations exhibit problematic convergence behavior. We found that for this open-shell dianionic system, the TDDFT description is more reliable (⟨S2⟩ ∼ 0.8–0.9); thus, this method was used for excitation energy calculation. Electron attachment and detachment energies were computed with ωB97X-D (see ref (44)) within the same QM/MM scheme. ωB97X-D was previously shown to provide reliable estimates of ionization energies of organic molecules, for example, DNA bases.50 B5050LYP functional was employed for excitation energy calculations. We note that the computed excitation energies are almost identical for TDDFT(ωB97X-D), TDDFT(B5050LYP), and SOS-CIS(D) with a ROHF-like reference (Table 1). The quantitative agreement between the three different approaches validates the applicability of the chosen TDDFT scheme for calculation of excitation energies of the open-shell dianion. The 6-31G(d,p) basis was used for more expensive excitation energies calculations (50 calculations for the MD trajectory). The cc-pVDZ basis was employed for attachment/detachment energy calculations. In addition, extrapolation to aug-cc-pVTZ was performed using the following protocol. In calculations of fluctuation of the excitation and electron attachment/detachment energies along the MD trajectory, the calculations were performed for the MD trajectory snapshots extracted each 20 ps using the 6-31G(d,p) and cc-pVDZ bases for excitation and attachment/detachment energies, respectively. Each 200 ps the same calculations were performed with the aug-cc-pVTZ basis set. These data were used for extrapolation to the aug-cc-pVTZ basis set.
Table 1. Excitation Energies of the Dianionic Chromophore (2Chro2– → 2Chro2–∗) Computed with Different Methods.
| method | basis | ΔE, eV |
|---|---|---|
| SOS-CIS(D)a | 6-31G(d,p) | 1.68 |
| TDDFT(B5050LYP) | 6-31G(d,p) | 1.71 |
| TDDFT(B5050LYP) | aug-cc-pVTZ | 1.65 |
| TDDFT(ωB97X-D) | 6-31G(d,p) | 1.65 |
UHF reference converged to the solution close to the ROHF one using maximum overlap method;59⟨ S2⟩ = 1.2.
All quantum-chemical calculations were performed using the Q-Chem package.51
III. Results and Discussion
Table 2 summarizes spectral features and lifetimes of the TA components in microsecond-to-second and subpicosecond decays.
Table 2. Spectral Features and Lifetimes of the TA Components for KillerRed, mRFP, and DsReda.
| Killer Red | mRFP | DsRed | |
|---|---|---|---|
| steady-state absorption | λmax = 585 nm (2.12 eV) | λmax = 585 nm (2.12 eV) | λmax = 561 nm (2.21 eV) |
| steady-state emission | λmax = 609 nm (2.04 eV) | λmax = 609 nm (2.04 eV) | λmax = 595 nm (2.08 eV) |
| light-minus-dark spectrum | λbleach = 585 nm (2.12 eV) | λbleach = 585 nm (2.12 eV) | λbleach = 560 nm (2.21 eV) |
| shoulder ∼640 nm | shoulder ∼640 nm | shoulder ∼660 nm | |
| λmax = 735 nm (1.69 eV) | λmax = 722 nm (1.72 eV) | λmax = 745 nm (1.66 eV) | |
| multiexponential decomposition of a | τ2 = 290 μs: | τ2 = 270 μs: | τ2 = 910 μs: |
| laser flash-induced transient absorption | λmax = 731 nm (1.70 eV) | λmax = 722 nm (1.72 eV) | λmax = 740 nm (1.68 eV) |
| decay (2 μs–4 s span) | |||
| subpicosecond transient abs decays | τ = 1.45 ns | τ = 1.8 ns | τ = 3.9 ns |
| fluorescence lifetimes | τ = 1.6 ns | τ = 2.0 ns | τ = 3.6 ns |
All data are taken in PBS buffer, pH 7.5 at 25 °C.
Owing to their structural similarity, all three RFPs have similar steady-state spectra. KillerRed and mRFP have identical absorption and emission maxima at 585 nm (2.12 eV) and 609 nm (2.04 eV). DsRed has absorption and emission maxima at 561 nm (2.21 eV) and 595 nm (2.08 eV), respectively, which were close to the previously reported values.14,25,31
A spectroscopic signature of blinking or bleaching behavior is delayed recovery of the ground-state absorption relative to the decay of the bright excited state. To test whether bleaching is significant at our conditions, we measured the transient absorption spectra with subpicosecond time resolution in the 0–3 ns time window, a common time scale for the fluorescent-state lifetime. The subpicosecond transient absorption (TA) spectra of the three RFPs show that the recovery of the ground-state absorption occurs on a longer time scale than the decay of the bright excited state. All three RFPs exhibit TA bands in the 800–1400 nm (1.55–0.89 eV) range and a band at 430 nm (2.88 eV). Analysis of the transient decays measured at different wavelengths yields lifetimes of 1.5, 1.8, and 3.9 ns in KillerRed, mRFP, and DsRed, respectively. All lifetimes were equal to or close to the fluorescence lifetime of the proteins measured using the time-correlated single photon counting technique (see the SI). Therefore, we attribute these transients to the simple decay of the excited singlet-state population. The S1 lifetimes decrease in the order DsRed > mRFP > KillerRed, revealing the increased quenching in this order. This effect can be attributed to the increased permeability of the β-barrels by the external quenchers, variations of the local structure around the chromophore, and/or increase of the ISC rate.
Altogether, the properties of the S0 → S1 and S1 → S0 transitions of the three RFPs are similar. However, we note that the recovery of the ground-state bleach is slower than the decay of the S1 state, indicating other possible deactivation pathways for 1Chro–∗. Therefore, we investigated these pathways using measurements at longer (μs-to-s) time scales. The photophysics of RFPs on this time scale was previously studied using fluorescence correlation spectroscopy and single-molecule spectroscopy techniques.16,52,53 Complex flickering dynamics was observed; it was interpreted in the framework of kinetic models involving several dark and bright states. However, the chemical nature of the states has not been unequivocally established.
Figure 2 shows TA spectra of the three RFPs in the micro- to millisecond range. The main features of the spectra are ground-state bleach and one major red-shifted TA feature separated by isosbestic points. The best exponential fit (global) of the TA data gave three components for all RFPs studied (see the SI for the kinetic fit details). Deconvolution analysis was performed to give the spectrum for each component. The kinetic fits with their residuals at selected wavelengths are shown in Figure S3 in the SI. The major component, τ2, for all three RFPs has a maximum centered close to 740 nm (Table 2) and absorption that spans beyond 1040 nm, where we reach the limit of our detector. The minor features beyond 800 nm could be attributed to the vibronic structure of the same absorption band. Note that ground-state bleach recovery in KillerRed and mRFP is slower than the decay of 730 nm transient, which suggests that the recovery may proceed through additional intermediates or that there are additional pathways for reversible and irreversible bleaching. The detailed understanding of the mechanism of interconversion between the bright state of KillerRed protein and the red-shifted intermediate is the subject of future work and is beyond the scope of the current study. Here, we focus on the assignment of the chemical nature of the intermediate.
Figure 2.

Micro- to millisecond transient absorption of KillerRed (A), mRFP (B), and DsRed (C) at various time points. For experimental details, see the SI.
Notably, all three proteins exhibit a major similar transient feature peaking at 731, 722, and 740 nm for KillerRed, mRFP, and DsRed, respectively. To elucidate the structure of this red-shifted intermediate (RSI) (720–740 nm), QM/MM calculations have been performed for KillerRed protein. A relatively short lifetime of this intermediate (∼μs) suggests that the spectral changes are likely due to changes in the electronic structure, conformation, or protonation state of the chromophore, rather than dramatic changes in the chemical nature (processes such as chromophore maturation and oxidative redding occur on the minutes to hours time scale).
Figure 3 illustrates the energetics of the relevant electronic states of KillerRed protein: the ground state (1Chro–), the first excited state (1Chro–∗), the lowest and excited triplets (3Chro– and 3Chro–∗), electronic states formed by electron detachment (2Chro and 2Chro*) from and electron attachment (2Chro2– and 2Chro2–∗) to the anionic chromophore. The left panel shows the values averaged along molecular dynamics trajectory sampling ground-state geometries of the protein.
Figure 3.

Energy diagram for relevant electronic states of KillerRed protein. (A) Excitation energy of 1Chro– (black), electron attachment energy (blue), excitation energy of 2Chro2– (red), electron detachment energy (orange), and excitation energy of 2Chro (violet). Excitation energies were computed using a QM/MM protocol with the MM part represented by point charges. The values are averaged over 50 snapshots taken from the MD trajectory of the KillerRed protein. (B) Fluctuation of the excitation and electron attachment/detachment energies along the MD trajectory extrapolated to the aug-cc-pVTZ values. The same color scheme as in (A) is used. See section B and the SI for computational details.
The lowest excited state of the neutral radical (photooxidation product) lies ∼0.5 eV above the RSI absorption maximum and is a dark state (fL < 0.01). The only bright transitions that are energetically close to the observed intermediate absorption (1.70 eV or 731 nm) are 3Chro– → 3Chro–∗ (1.3 eV or 954 nm) and 2Chro2– → 2Chro2–∗ (1.7 eV or 729 nm). Because the upper bound for triplet lifetime in KillerRed is 40 μs,25 which is 1 order of magnitude shorter than the transient centered at 731 nm, we rule out the triplet state as a possible candidate for the RSI. Thus, the only viable candidate responsible for the transient intermediate with λmax = 731 nm is an electron-attached dianionic state of the chromophore (2Chro2–). Such a doubly charged state is unstable in the gas phase; however, it is stabilized by the interactions with the nearby charged amino acid residues and is ∼1.7 eV below the ground anionic state of the protein-bound chromophore. We attribute this stabilization to interaction with the positively charged Arg94 residue forming an H-bond with the chromophore. Indeed, calculations with Arg excluded from the MM system show no stabilization of the dianion − electron attachment becomes energetically unfavorable (+1.0 eV).
Although electron attachment to the anionic chromophore in KillerRed is 1.7 eV exothermic (computed value) even for the ground-state chromophore, there should be a reducing agent providing an electron with matching or greater oxidation potential. However, amino acids have much higher ionization energies,54 and it is unlikely that the environmental effects can make electron transfer from nearby amino acids to the ground-state chromophore energetically favorable (otherwise, 1Chro– would undergo spontaneous reduction). Thus, we posit that the intermediate can be formed only via an excited state of the chromophore. The preliminary TA data taken with nanosecond resolution demonstrate that the RSI is formed within the 0.5 ns pulsewidth of the excitation laser.55 This indeed supports the direct formation of this intermediate from the Chro singlet excited states. Direct formation of the dianion from the lowest singlet excited state of the chromophore will be accompanied by a ∼4 eV energy gain. We note that the ionization energies of the amino acids are still higher; for example, ionization energy of 7.3 ± 0.2 eV was reported for tryptophan on the basis of vacuum–ultraviolet single-photon ionization mass spectrometry experiments.56 However, the local environment may significantly alter the ionization/attachment energies as well as the resulting oxidation/reduction potentials of the amino acids, similarly to the observed effect of the nearby residues on the chromophore’s energetics.
To interrogate the identity of the main transient, we investigated the effect of external oxidative/reducing agents on the observed TA for all three RFPs. First, the TA was measured for samples in ambient, aerobic (oxygen-saturated), and fully anaerobic conditions. The lifetime of the main TA component decreased significantly in oxygen-saturated conditions for KillerRed and mRFP (Table 3), whereas in DsRed it remained mostly unchanged (SI Figure S5). The effects of several oxidant and reductants (cytochrome c, β-mercaptoethanol, NAD+, potassium ferricyanide, flavin mononucleotide) were also tested on the kinetics of the RSI feature in KillerRed, mRFP, and DsRed. In all three cases, the kinetics was unaffected. These oxidants and reductants are likely too large or hydrophilic and, therefore, are unable to access the chromophore buried inside the protein barrels. Thus, we tested a smaller molecule, H2O2, that can act as either a relatively strong oxidant or a weak reductant. The results are shown in Figure 4. Remarkably, H2O2 had a profound effect on the lifetime of the transient in KillerRed, decreasing the lifetime to 95 μs in the presence of 5 μM H2O2. The effect was much weaker in mRFP and DsRed, for which the lifetimes decreased to 147 and 810 μs, respectively. We attribute this phenomenon to the unique structural feature of KillerRed’s β-barrel interior, a long water-filled channel.20 It has been hypothesized that this channel facilitates the escape of ROS from the protein, thereby contributing to KillerRed’s phototoxicity.20,21 In the control experiments, we observe that the same levels of H2O2 have little to no effect on the ground-state absorption spectrum in all three proteins (SI). Thus, the transient is efficiently quenched by H2O2, which is consistent with the proposed strongly reducing dianionic state. The dianionic radical chromophore (2Chro2–) can then donate an electron to O2 to generate superoxide, which is likely the main mechanism of phototoxicity in KillerRed.
Table 3. Lifetimes (in μs) of the Major TA Component for KillerRed and mRFP under Ambient, Aerobic (Oxygen-Saturated), and Anaerobic Conditions and in the Presence of H2O2 (See the SI for Details).
| protein | τ2, ambient | τ2-O2 | τ2-Ar | τ2-H2O2 |
|---|---|---|---|---|
| KillerRed | 290 | 162 | 418 | 95 |
| mRFP | 270 | 231 | 295 | 147 |
| DsRed | 910 | 910 | ∼920 | 810 |
Figure 4.
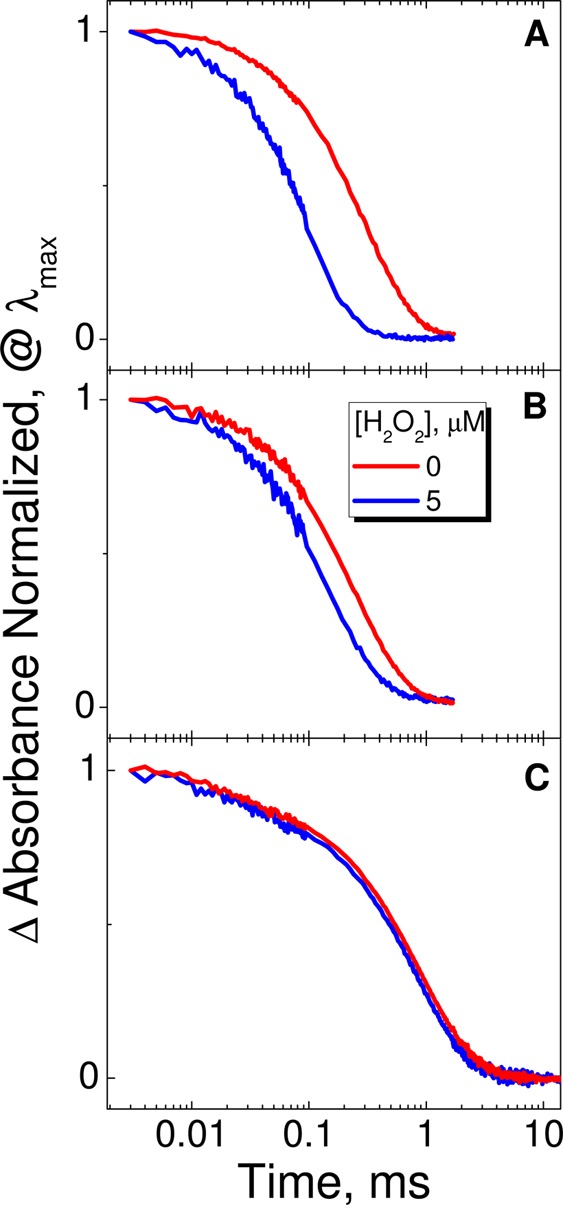
Kinetics of transient absorption signal at 731 nm (1.70 eV) for KillerRed (top), at 722 nm (1.72 eV) for mRFP (middle), and at 745 nm (1.66 eV) for DsRed in the absence and presence H2O2. For experimental details, see the SI.
Among the three cytotoxic proteins studied, the phototoxicity and radical production increase in the order DsRed < mRFP < KillerRed.12,14 Thus, there is a direct correlation between phototoxicity and our quenching experiments in which the main TA component (RSI) is quenched with O2 and H2O2. For example, the transient in the most phototoxic fluorescent protein, KillerRed, shows the greatest quenching with H2O2 and O2, whereas the lifetime of the transient in DsRed remains mostly unaffected. This observation suggests using weak reducing/oxidative pairs to control the phototoxicity and photostability of FPs, as successfully done for organic dyes in solution.57 It is quite interesting that an agent as small as O2 is unable to penetrate the β-barrel of DsRed; however, it is not that surprising because previous researchers had to use guanadinium–HCl to add flexibility to a GFP mutant to detect singlet oxygen production.58 Recent studies of RFPs from the mFruit family attribute their poor photostability to the structural weakness of their β-barrel that facilitates oxygen diffusion.32,33
IV. Conclusions
We report an observation of a novel strongly red-shifted (λmax 720–740 nm) photoinduced intermediate that is common for KillerRed, mRFP, and DsRed and decays on the microsecond time scale. On the basis of the electronic structure calculations we interpret the signal as absorption by the dianionic open-shell state of the chromophore formed by photoreduction. The RSI is effectively quenched by oxygen and hydrogen peroxide in KillerRed, which features a large water channel facilitating access to the chromophore, whereas its lifetimes in DsRed and mRFP are not strongly affected. The results raise the question of whether photoreduction is common for all FPs and if the corresponding electron-attached states are common gateway states for photobleaching and blinking. In such a case, understanding the protein environment effects on the formation and lifetime of the radical species is crucial for development of more photostable FPs, of phototoxic agents for photodynamic therapy, and of more stable FPs with respect to photoinduced decarboxylation. Further studies including time-resolved EPR, IR, and Raman spectroscopies are needed for a detailed characterization of the structure and, especially, of the formation mechanism of these species, which possibly play a crucial role in the photochemistry of photoactive proteins.
Acknowledgments
This work was supported by the U.S. National Science Foundation (CHE-1213047, L.M.T. and K.M.S.; and CHE-1264018, A.I.K.) and the U.S. National Institutes of Health (R21EB009976-01, A.S.B.). D.A.B and M.V. were supported by grants from Biocentrum Helsinki, the Sigrid Jusélius Foundation, and the Academy of Finland. We thank Konstantin Lukyanov and Delmar Larsen for stimulating discussions. We are grateful to the members of Joseph Perry’s group for help with fs TA measurements.
Supporting Information Available
Computational details, experimental setup, and additional results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Status
▽ Deceased.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tsien R. Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [DOI] [PubMed] [Google Scholar]
- Verkhusha V. V.; Lukyanov K. A. The molecular properties and applications of anthozoa fluorescent proteins and chromoproteins. Nat. Biotechnol. 2004, 22, 289–296. [DOI] [PubMed] [Google Scholar]
- Subach F. V.; Verkhusha V. V. Chromophore transformations in red fluorescent proteins. Chem. Rev. 2012, 112, 4308–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J.; Schnitzer M. J. An infrared fluorescent protein for deeper imaging. Nat. Biotechnol. 2011, 29, 715716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans B. N. G.; Adams S. R.; Ellisman M. H.; Tsien R. Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217. [DOI] [PubMed] [Google Scholar]
- Chalfie M.; Tu Y.; Euskirchen G.; Ward W. W.; Prasher D. C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [DOI] [PubMed] [Google Scholar]
- Kneen M.; Farinas J.; Li Y.; Verkman A. S. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 1998, 74, 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souslova E. A.; Belousov V. V.; Lock J. G.; Strömblad S.; Kasparov S.; Bolshakov A. P.; Pinelis V. G.; Labas Y. A.; Lukyanov S.; Mayr L. M.; et al. Single fluorescent protein-based Ca2+ sensors with increased dynamic range. BMC Biotechnol. 2007, 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barondeau D. P.; Kassmann C. J.; Tainer J. A.; Getzoff E. D. Structural chemistry of a green fluorescent protein zn biosensor. J. Am. Chem. Soc. 2002, 124, 3522–3524. [DOI] [PubMed] [Google Scholar]
- Meyer A. J.; Dick T. P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [DOI] [PubMed] [Google Scholar]
- Lukyanov K. A.; Belousov V. V. Genetically encoded fluorescent redox sensors. Biochim. Biophys. Acta 2014, 1840, 745–756. [DOI] [PubMed] [Google Scholar]
- Belousov V. V.; Fradkov A. F.; Lukyanov K. A.; Staroverov D. B.; Shakhbazov K. S.; Terskikh A. V.; Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [DOI] [PubMed] [Google Scholar]
- McLean M. A.; Rajfur Z.; Chen Z.; Humphrey D.; Yang B.; Sligar S. G.; Jacobson K. Mechanism of chromophore assisted laser inactivation employing fluorescent proteins. Anal. Chem. 2009, 81, 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulina M. E.; Chudakov D. M.; Britanova O. V.; Yanushevich Y. G.; Shkrob M. A.; Lukyanov S.; Lukyanov K. A. A genetically encoded photosensitizer. Nat. Biotechnol. 2006, 24, 95–99. [DOI] [PubMed] [Google Scholar]
- Dean K. M.; Lubbeck J. L.; Binder J. K.; Schwall L. R.; Jimenez R.; Palmer A. E. Analysis of red-fluorescent proteins provides insight into dark-state conversion and photodegradation. Biophys. J. 2011, 101, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S.; Ivanchenko S.; Röcker C.; Wiedenmann J.; Nienhaus G. U. Photodynamics of red fluorescent proteins studied by fluorescence correlation spectroscopy. Biophys. J. 2004, 86, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T.; Tinnefeld P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution and imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam V.; Carpentier P.; Violot S.; Lelimousin M.; Darnault C.; Nienhaus G. U.; Bougeois D. Structural basis of X-ray induced transient photobleaching in a photactivable green fluorescent protein. J. Am. Chem. Soc. 2009, 131, 18063–18065. [DOI] [PubMed] [Google Scholar]
- Roy A.; Field M. J.; Adam V.; Bourgeois D. The nature of transient dark states in photoactivatable fluorescent protein. J. Am. Chem. Soc. 2011, 133, 18586–18589. [DOI] [PubMed] [Google Scholar]
- Pletnev S.; Gurskaya N. G.; Pletneva N. V.; Lukyanov K. A.; Chudakov D. M.; Martynov V. I.; Popov V. O.; Kovalchuk M. V.; Wlodawer A.; Dauter Z.; et al. Structural basis for phototoxicity of the genetically encoded photosensitizer KillerRed. J. Biol. Chem. 2009, 284, 32028–32039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier P.; Violot S.; Blanchoin L.; Bourgeois D. Structural basis for the phototoxicity of the fluorescent protein killerred. FEBS Lett. 2009, 583, 2839–2842. [DOI] [PubMed] [Google Scholar]
- Bogdanov A. M.; Mishin A. S.; Yampolsky I. V.; Belousov V. V.; Chudakov D. M.; Subach F. V.; Verkhusha V. V.; Lukyanov S.; Lukyanov K. A. Green fluorescent proteins are light-induced electron donors. Nat. Chem. Biol. 2009, 5, 459–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epifanovsky E.; Polyakov I.; Grigorenko B. L.; Nemukhin A. V.; Krylov A. I. The effect of oxidation on the electronic structure of the green fluorescent protein chromophore. J. Chem. Phys. 2010, 132, 115104. [DOI] [PubMed] [Google Scholar]
- Bravaya K.; Khrenova M. G.; Grigorenko B. L.; Nemukhin A. V.; Krylov A. I. The effect of protein environment on electronically excited and ionized states of the green fluorescent protein chromophore. J. Phys. Chem. B 2011, 8, 8296–8303. [DOI] [PubMed] [Google Scholar]
- Vegh R. B.; Solntsev K. M.; Kuimova M. K.; Cho S.; Liang Y.; Loo B. L. W.; Tolbert L. M.; Bommarius A. S. Reactive oxygen species in photochemistry of the red fluorescent protein “Killer Red”. Chem. Commun. 2011, 47, 4887–4889. [DOI] [PubMed] [Google Scholar]
- Bulina M. E.; Lukyanov K. A.; Britanova O. V.; Onichtchouk D.; Lukyanov S.; Chudakov D. M. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat. Protoc. 2006, 1, 947–953. [DOI] [PubMed] [Google Scholar]
- Grigorenko B. L.; Nemukhin A. V.; Morozov D. I.; Polyakov I.; Bravaya K. B.; Krylov A. I. Towards molecular-level characterization of photo-induced decarboxylation of the green fluorescent protein: Accessibility of the charge-transfer states. J. Chem. Theory Comput. 2012, 8, 1912–1920. [DOI] [PubMed] [Google Scholar]
- Ding L.; Chung L. W.; Morokuma K. Reaction mechanism of photoinduced decarboxylation of the photoactivatable green fluorescent protein: an ONIOM(QM:MM) study. J. Phys. Chem. B 2013, 117, 1075–1084. [DOI] [PubMed] [Google Scholar]
- van Thor J. J.; Gensch T.; Hellingwerf K. J.; Johnson L. N. Phototransformation of green fluoresecent protein with UV and visible light leads to decarboxilation of glutamate 222. Nat. Struct. Biol. 2002, 9, 37–41. [DOI] [PubMed] [Google Scholar]
- Bell A. F.; Stoner-Ma D.; Wachter R. M.; Tonge P. J. Light-driven decarboxylation of wild-type green fluorescent protein. J. Am. Chem. Soc. 2003, 125, 6919–6926. [DOI] [PubMed] [Google Scholar]
- Habuchi S.; Cotlet M.; Gensch T.; Bednarz T.; Haber-Pohlmeier S.; Rozenski J.; Dirix G.; Michiels J.; Vanderleyden J.; Heberle J.; et al. Evidence for the isomerization and decarboxylation in the photoconversion of the red fluorescent protein dsred. J. Am. Chem. Soc. 2005, 127, 8977–8984. [DOI] [PubMed] [Google Scholar]
- Chapagain P. P.; Regmi C.; Castillo W. Fluorescent protein barrel fluctuations and oxygen diffusion pathways in mCherry. J. Chem. Phys. 2011, 135, 235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent A. D.; Mironov V. A.; Chapagain P. P.; Nemukhin A. V.; Krylov A. I. Exploring structural and optical properties of fluorescent proteins by squeezing: Modeling high-pressure effects on the mStrawberry and mCherry red fluorescent proteins. J. Phys. Chem. B 2012, 116, 12426–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales J. M.; Cozzuol M.; Screen T. E. O.; Anderson H. L.; Perry J. W. Metalloporphyrin polymer with temporally agile, broadband nonlinear absorption for optical limiting in the near infrared. Opt. Exp. 2009, 17, 18478–18488. [DOI] [PubMed] [Google Scholar]
- Belevich I.; Gorbikova E.; Belevich N. P.; Rauhamäki V.; Wikström M.; Verkhovsky M. I. Initiation of the proton pump of cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18469–18474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov V. B.; Belevich I.; Bloch D. A.; Mogi T.; Verkhovsky M. I. Glutamate 107 in subunit i of cytochrome bd from Escherichia coli is part of a transmembrane intraprotein pathway conducting protons from the cytoplasm to the heme b(595)/heme d active site. Biochemistry 2008, 47, 7907–7914. [DOI] [PubMed] [Google Scholar]
- Belevich I.; Bloch D. A.; Belevich N.; Wikström M.; Verkhovsky M. I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 2685–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravaya K. B.; Subach O.; Korovina N.; Verkhusha V. V.; Krylov A. I. An insight into the common mechanism of the chromophore formation in the red fluorescent proteins: The elusive blue intermediate revealed. J. Am. Chem. Soc. 2012, 134, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravaya K.; Grigorenko B. L.; Nemukhin A. V.; Krylov A. I. Quantum chemistry behind bioimaging: Insights from ab initio studies of fluorescent proteins and their chromophores. Acc. Chem. Res. 2012, 45, 265–275. [DOI] [PubMed] [Google Scholar]
- PROPKA, http://propka.ki.ku.dk.
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435447. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- Nosé S.; Klein M. L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 10551076. [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion interactions. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Weinhold F.. NBO 4.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 1996. [Google Scholar]
- Reuter N.; Lin H.; Thiel W. Green fluorescent proteins: Empirical force field for the neutral and deprotonated forms of the chromophore. Molecular dynamics simulations of the wild type and S65T mutant. J. Phys. Chem. B 2002, 106, 6310–6321. [Google Scholar]
- Rhee Y. M.; Head-Gordon M. Scaled second order perturbation corrections to configuration interaction singles: efficient and reliable excitation energy methods. J. Phys. Chem. A 2007, 111, 5314–5326. [DOI] [PubMed] [Google Scholar]
- Grigorenko B. L.; Nemukhin A. V.; Polyakov I.; Krylov A. I. Triple-decker motif for red-shifted fluorescent protein mutants. J. Phys. Chem. Lett. 2013, 4, 11815–11822. [DOI] [PubMed] [Google Scholar]
- Grigorenko B. L.; Nemukhin A. V.; Polyakov I.; Morozov D.; Krylov A. I. First-principle characterization of the energy landscape and optical spectra of the green fluorescent protein along A-I-B proton transfer route. J. Am. Chem. Soc. 2013, 135, 11541–11549. [DOI] [PubMed] [Google Scholar]
- Bravaya K. B.; Epifanovsky E.; Krylov A. I. Four bases score a run: Ab initio calculations quantify a cooperative effect of h-bonding and pi-stacking on ionization energy of adenine in the AATT tetramer. J. Phys. Chem. Lett. 2012, 3, 2726–2732. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Fusti-Molnar L.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [DOI] [PubMed] [Google Scholar]
- Malvezzi-Campeggi F.; Jahnz M.; Heinze K. G.; Dittrich P.; Schwille P. Light-induced flickering of dsred provides evidence for distinct and interconvertible fluorescent states. Biophys. J. 2001, 81, 1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix J.; Flors C.; Dedecker P.; Hofkens J.; Engelbroghs Y. Dark states of monomeric red fluorescent proteins studies by fluorescence correlation and single molecule spectroscopy. Biophys. J. 2008, 94, 4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close D. M. Calculated vertical ionization energies of the common r-amino acids in the gas phase and in solution. J. Phys. Chem. A 2009, 115, 2900–2912. [DOI] [PubMed] [Google Scholar]
- Larsen D. Private communication.
- Wilson K. R.; Jimenez-Cruz M.; Nicolas C.; Belau L.; Leone S. R.; Ahmed M. Thermal vaporization of biological nanoparticles: fragment-free vacuum untraviolet photoionization mass spectra of tryptophan, phenylalanine-glycine-glycine, and β-carotene. J. Phys. Chem. A 2006, 110, 2106–2113. [DOI] [PubMed] [Google Scholar]
- Vogelsang J.; Kasper R.; Steinhauer C.; Person B.; Heilemann M.; Sauer M.; Tinnefeld P. A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew. Chem. Int. Ed. 2008, 47, 5456–5469. [DOI] [PubMed] [Google Scholar]
- Greenbaum L.; Lavie C.; Rothmann R.; Malik Z. Green fluorescent protein photobleaching: a model for protein damage by endogenous and exogenous singlet oxygen. Biol. Chem. 2000, 381, 12511258. [DOI] [PubMed] [Google Scholar]
- Besley N. A.; Gilbert A. T. B.; Gill P. M. W. Self-consistent-field calculations of core excited states. J. Chem. Phys. 2009, 130, 124308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.