

Abstract
A newly recognized, primary cause of obesity epidemic is the developmental programming effects of (1) intrauterine growth restricted (IUGR) newborns exposed in utero to undernutrition, and (2) normal or excessive weight newborns exposed to maternal obesity and high fat (HF) diets. The mechanisms contributing to offspring obesity have been extensively studied in animal models with adipose tissue being identified as one of the principal target of programming. IUGR and HF offspring exhibit programmed adipocytes, such that an intrinsic enhanced lipogenesis and adipocyte proliferation contribute to the development of obesity. This is attributed to early induction of adipogenic transcription factor, PPARγ, the activity of which is enhanced under limited or excess nutrient availability. Nonetheless, this occurs via different mechanisms involving PPARγ co-regulators - in IUGR, it is upregulation of co-activators whereas in HF newborns, it is downregulation of co-repressors. Thus, preventive therapeutic interventions will require target-specific modalities dependent upon the primary etiology.
Keywords: adipogenesis, lipogenesis, PPARγ, obesity
Obesity has emerged as a pre-eminent public health problem.1 In the United States, 66% of adults are overweight (BMI 25 to <30 kg/m2) and 33% are obese (BMI≥30 kg/m2), representing a modern health crisis.2 Perhaps of even greater concern is the continued increase in prevalence of obesity among pregnant women, which is associated with high birth weight newborns, and represents a known risk factor for childhood obesity.3;4 To further exacerbate this problem, childhood obesity is known to lead to adult obesity.5 The rapid increase in the prevalence of obesity has been attributed to environmental factors, increasing availability of highly palatable foods and ‘modern’ lifestyles involving less physical work thereby creating a more obesigenic environment. The question arises as to why some individuals are more susceptible to increased adiposity than others. The ‘thrifty phenotype’ or “programming” hypothesis proposes that alteration in nutritional, metabolic and/or hormonal milieu during development modifies obesity susceptibility by perturbing homeostatic regulatory mechanisms.6;7 This concept has been gaining increasing credence supported by strong findings from epidemiologic studies and animal models.(reviewed6–9) Obesity is central to the development of metabolic syndrome which includes a constellation of abnormalities comprised of insulin resistance, elevated triglycerides, hypertension and atherosclerosis.10;11 The underlying factor that links these disorders is dysregulation of adipogenesis and lipid metabolism.11 This review provides an overview of the developmental origins of obesity with specific emphasis on the role of adipose tissue and potential implication for intervention strategy and future studies.
Programmed Obesity
Humans
Epidemiological studies link birth weight to risk of adult obesity and metabolic syndrome. Notably in humans, both low and high birth weights lead to increased risk for childhood and adult obesity, suggesting increased risk of obesity at both ends of the birth weight spectrum.(review12;13) Early epidemiologic studies initially demonstrated that the intrauterine growth restricted (IUGR) newborns or low birth weight infants, particularly those with rapid catch-up growth, have higher risk of adult obesity and metabolic syndrome. The prevalence of metabolic syndrome fell progressively in both men and women, from those who had the lowest to those who had the highest birth weights. Of 64-year-old men whose birth weights were 6.5 pounds or less, 22% had metabolic syndrome. Those with the lowest birth weight were 18 times more likely to have metabolic syndrome as compared to those who were heaviest at birth.14 Most notably, among men born during the Dutch Hunger Winter of 1944–1945, those exposed to famine during the first half of gestation had a markedly increased risk of adult obesity 15. Subsequently, numerous epidemiologic studies from diverse populations have confirmed this relationship.(review12;16)
Obesity in pregnancy has not only adverse effects on maternal health and pregnancy outcome but also on the developing fetus. Specifically, maternal obesity before and during pregnancy, including increased weight gain in pregnancy, is associated with higher birth weight newborns.17;18 The 25–36% increase in maternal BMI over the last decade has translated to an approximately 25% increase in the incidence of high birth weight babies.19 This is of particular importance, as high birth weight newborns show increased adipose tissue mass and an increased risk of obesity and diabetes risk in later life (review20).
Thus, epidemiologic studies confirm that the relationship between human birth weight and adult obesity, hypertension, and/or insulin resistance is a “U-shaped curve”.21–24 Perhaps most importantly, the relation of fetal growth to offspring obesity and metabolic syndrome is a continuum, rather than a threshold response. There may well be an optimal newborn weight (potentially specific to an individual mother) at which the programming of obesity potential is minimized. However, within ranges of lower or higher birth weights in comparison to mean values, studies indicate a gradation of propensity to programming sequelae. Thus, alterations from “optimal” in utero growth, be it from limited or excess nutrition, increase the relative risk of adult metabolic syndrome.
Animal Models
Animal models of IUGR, using a variety of methods, such as maternal nutrient restriction, placental uterine ligation or glucocorticoid exposure, among others, have effectively demonstrated increased adult adiposity.8;25–27 Similar to human studies, the propensity to obesity is particularly evident in IUGR newborns exhibiting postnatal catch-up growth.28;29 We have established a rat model of maternal undernutrition which results in IUGR pups with decreased plasma leptin levels. When provided normal nursing, the IUGR newborns demonstrate significantly increased food intake, with rapid catch-up growth resulting in adult metabolic syndrome, including obesity, increased percent body fat, lipid abnormalities and insulin resistance.29;30
More recent animal models of maternal over-nutrition, representing the Western diet, have typically used high-fat (HF) diet during pregnancy and lactation.31;32 However, rat studies on maternal HF feeding have reported variable effect on birth weight, ranging from low to normal to high33–35 This varying effect on birth weight may be attributed to other confounding maternal abnormalities, such as diabetes, and to the specific composition of HF diets. Nonetheless, the large majority of studies confirm that perturbations of fetal growth and development lead to adult obesity.29;33;36 Our rat model on maternal obesity and HF diet prior to and during pregnancy results in normal birth weight newborns. But when nursed by HF dams, these HF offspring also demonstrate compensatory, accelerated (catch-up) growth and early onset of obesity with lipid abnormalities, evident by 3 weeks of age.37
Thus, an apparent paradox of increased adiposity at both ends of the birth weight spectrum exists (i.e., higher BMI with higher birth weight and increased central obesity with lower birth weight). It may be that similar underlying mechanism(s) predispose both newborns (IUGR/maternal obesity) to adult obesity. This may include lasting changes in central nervous system appetite control, proportions of fat and lean body mass, adiposity structure and function, and adipokine secretion and regulation. Thus, elucidating the relationship between early life experiences and later body proportions is critical in preventing obesity, particularly because it can have life-long and perhaps multigenerational impact.
Mechanisms of Programmed Obesity
Animal studies have provided important insights into the underlying mechanisms of programmed obesity. Obesity occurs as a result of dysregulated energy balance wherein increased food intake, reduced energy expenditure, altered adipocyte phenotype, and efficient metabolism may all or partly contribute. Appetite and satiety function develops in utero in precocial species in order to prepare for newborn life. In the rat and humans, although neurons which regulate appetite and satiety becomes detectable in the fetal hypothalamus early in gestation, the functional neuronal pathways form during the second week of postnatal life in the rat and likely during the third trimester in humans.38–40 Notably, the obesity (ob) gene product leptin, which is synthesized primarily by adipose tissue and placenta, is a critical neurotrophic factor during development.41 In the fetus/newborn, leptin promotes the development of satiety pathways. In contrast, in the adult, leptin acts as a satiety factor.42
Studies have demonstrated that appetite dysregulation contributes to obese phenotype in IUGR newborns.43;44 Our studies on IUGR specifically indicate dysfunction at several aspects of the satiety pathway, as evidenced by reduced satiety and cellular signaling responses to leptin.45 Most recently, our studies have demonstrated a hypothalamic upregulation of the nutrient sensor SIRT1, a factor which epigenetically regulates gene transcription of factors critical to neural development.46 Importantly, we have demonstrated that neuronal stem cells from IUGR fetuses/newborns demonstrate reduced growth and impaired differentiation to neurons and glial cells.47 Thus, impaired neuronal development (and ultimately reduced satiety pathways) may be a consequence of a reduction in neural stem cell growth potential and reduced leptin-mediated neurotrophic stimulation during periods of axonal development.
Studies on the effects of maternal HF diets have similarly shown increased offspring food intake associated with increased expression of orexigenic peptides in the offspring,31 prolonged neonatal leptin surge and dysregualted leptin signaling.34;48
Adipose Tissue
In addition to appetite/satiety dysfunction, mechanisms regulating adipose tissue (AT) development and function (lipogenesis) may be a key factor in the development of programmed obesity. Increase in AT mass or adipogenesis occurs primarily during the prenatal and postnatal development, though some adipogenesis continues throughout adulthood.49 The process of adipogenesis requires highly organized and precisely controlled expression of a cascade of transcription factors within the preadipocyte. Further, AT is now acknowledged as a major secretory and endocrine organ involved in multitude functions that ultimately impacts homeostasis and disease states. Lastly, AT differ from many other tissues in that they occur in multiple, dispersed sites around the body, differentially impacting metabolism. We briefly summarize each of these three characteristics of AT.
Adipogenesis and Lipogenesis
The cellular development associated with adipose tissue growth involves both cellular hypertrophy (increase in cell size) and hyperplasia (increase in cell number). Hypertrophy is the result of excess triglyceride accumulation in existing adipocytes due to a positive energy balance (energy intake in excess of energy expenditure). Hyperplasia, also referred to herein as ‘adipogenesis’, results from the recruitment of new adipocytes from precursor cells in AT and involves the proliferation and differentiation of preadipocytes.50–52 Both adipocyte hypertrophy and hyperplasia occur in association with positive energy balance during normal growth and during the development of obesity, with hypertrophy often preceeding hyperplasia in a cyclic manner.53
Adipogenesis regulation involves an association of transcription factors promoting differentiation, both directly, and via functional interplay among each other.54;55 The PPAR and C/EBP family members play a cooperative role in the induction of adipocyte differentiation.56–58 These transcription factors appear to be part of a cascade in which C/EBPβ and C/EBPδ are involved in the induction of PPARγ and C/EBPα59;60 PPARγ activation induces dimerization with retinoid X receptors (RXRα) which leads to adipocyte differentiation. PPARγ2 is the principal regulator of adipogenesis and activated PPARγ2 facilitates lipogenesis by induction of lipogenic transcription factor, sterol regulatory element binding protein 1 (SREBP1).61–63 However, SREBP1 can also activate PPARγ, by both stimulating the production of an endogenous ligand,64 as well as by inducing PPARγ promoter activity via a E-box motif.61;64 These data are suggestive of a feed-forward mechanism, in which PPARγ activates SREBP1 and vice-versa, and which is aimed at promoting adipogenesis and lipogenesis.62 SREBP1 has been shown to induce the expression of extracellular lipolytic enzyme (lipoprotein lipase) and lipogenic enzyme (fatty acid synthase) that in turn, lead to an increase in fatty acid uptake and synthesis, promoting lipid accumulation within the adipocyte.65;66 The release of free fatty acid from adipocytes is facilitated by an intracellular lipolytic enzyme, hormone-sensitive lipase67 (Figure 1).
Figure 1. Sequential Regulation of Adipogenesis and Lipogenesis.

(1) C/EBPδ and C/EBPβ upregulate PPARγ and C/EBPα; (2) PPARγ2 upregulates C/EBPα and vice versa, resulting in adipocyte differentiation; (3) PPARγ2 upregulates lipogenic transcription factor, SREBP1c; (4) SREBP1c upregulates extracellular lipolytic enzyme, lipoprotein lipase facilitating fatty acid uptake by adipocytes; (5) SREBP1c upregulates lipogenic enzyme, fatty acid synthase facilitating lipogenesis within adipocytes; (6) Intracellular lipolytic enzyme, hormone sensitive lipase acts on triglyceride to release fatty acid from adipocyte.
An Endocrine Organ
AT was conventionally characterized as a metabolically inactive tissue, storing fat during calorie abundance and releasing fatty acids in times of energy shortage. As evidence emerged of AT’s ability to secret hormones and cytokines, it established AT as an important endocrine organ playing a crucial role in multiple regulatory and homeostatic functions (Figure 2).68;69 The first discovery of an adipocyte-derived signaling molecule was an appetite regulating hormone, leptin. Since then a number of proteins secreted by adipocytes have been identified. These include adiponectin which is associated with insulin sensitivity, fatty acid oxidation, and reduced hepatic glucose output; resistin and retinol binding protein-4 are associated with adiposity and insulin resistance; metallothionein is a metal-binding and stress-response protein which may have an antioxidant role; and other secretory proteins (angiotensinogen, adipsin, acylation-stimulating protein, retinol-binding protein, plasminogen activator inhibitor-1 and tissue factor) are involved in vascular homeostasis or the complement system. More recent studies demonstrate that adipocyte-derived macrophages are the major source of inflammatory cyokines, such as TNFα and IL-6. Increased TNFα and IL-6 often seen in obesity, contributes to a chronic low-grade inflammatory state that has been linked to the development of insulin resistance and diabetes. These proteins, known as adipokines, have autocrine or paracrine effects, and are central to the dynamic control of energy metabolism, communicating the nutrient status with the specific tissues responsible for regulating both energy intake and expenditure as well as insulin sensitivity. As such, the investigation of the pathophysiological consequences of altered adipocyte protein production due to changes in adiposity is imperative. There is already considerable evidence linking increased production of some adipocyte factors with obesity and metabolic abnormalities.70
Figure 2. Adipose Tissue as an Endocrine Organ.
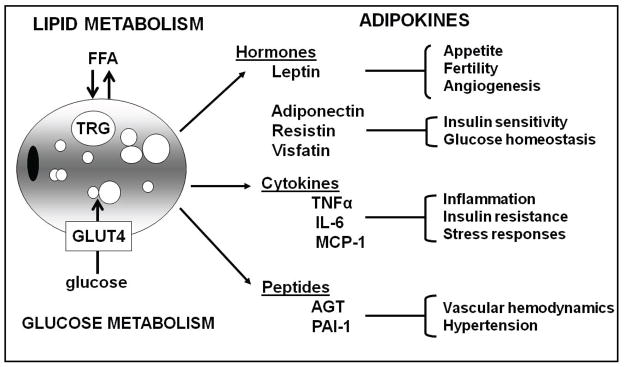
Adipocytes secrete adipokines that regulate energy homeostasis, appetite regulation, insulin sensitivity, inflammatory response, and blood pressure. Imbalanced secretion of some of these adipokines is associated with obesity and metabolic syndrome. Tumor Necrosis Factor alpha (TNF-α), Interleukin 6 (IL-6), Monocyte chemotactic protein-1 (MCP-1), Angiotensinogen (AGT) and Plasminogen Activator Inhibitor (PAI).
Regional Fat Depots
The location of the excess body fat (regional fat distribution) is a major determinant of the degree of excess morbidity and mortality due to obesity.71 At least three components of body fat are associated with obesity-related adverse health outcomes: the total amount of body fat (expressed as a percentage of body weight), the amount of subcutaneous AT or abdominal fat (upper body fat), and the amount of visceral fat located in the abdominal cavity.72 These three components are partly correlated with each other but exhibit a fairly high degree of independence. Each of these components of body fat are gender specific,73 and are associated with varying degrees of metabolic abnormalities and independently predict adverse health outcomes.74;75 White AT is located primarily in two major anatomical AT depots, subcutaneous (inguinal, axillary, interscapular fat depots) and visceral (mesenteric, omental, perirenal, retroperitoneal, epididymal, parametrial).
Subcutaneous AT are made up of smaller, more insulin sensitive adipocytes and act as a sink or powerful ‘buffers,’ avidly absorbing circulating fatty acids and triglycerides in the postprandial period.51;72;76;77 This prevents their diversion to non-adipose tissues, thereby protecting against ectopic fat syndrome and metabolic syndrome.78;79 In contrast, excessive visceral AT amount leads to increased secretion of fatty acids, adipokines and inflammatory molecules that contribute to insulin resistance, glucose intolerance, dyslipidemia, hypertension and coronary artery disease.
Mechanism of Programmed Adipogenesis
IUGR Offspring
Mechanistic studies on enhanced adipogenesis or alteration in function/response of adipocytes in IUGR offspring are limited. Past studies on maternal protein restriction model have demonstrated smaller adipocytes with increased expression of insulin receptor, hypertrophic adipocytes and upregulation of genes involved in adipocyte differentiation in adult offspring that already exhibit obesity.80–82 Our model of IUGR offspring specifically demonstrates that hyperleptinemia and hypertrophic adipocytes, including increased de novo fatty acid synthesis, all indicative of increased propensity for fat storage, occurs prior to onset of obesity. 29;83;84. Hence, our focus has been to investigate primary mechanism(s) early in life that lead to enhanced adipogenesis in IUGR newborns. We have shown that at 1 day of age, IUGR male offspring have upregulated adipogenic signaling cascade, specifically increased AT expression of PPARγ.83 Further, enzymes influencing adipocyte lipid synthesis, storage and release indicate increased susceptibility to retain fat in IUGR adipocytes. As these changes are evident early in life, it suggests a programmed pathway of increased adipocyte differentiation and lipogenesis which likely promotes the development of obesity and metabolic abnormalities in IUGR offspring.
We next explored the putative mechanism for the paradoxical increased PPARγ activity. PPARγ activity is modulated by select co-repressors and co-activators. The co-repressors include SIRT1 (an NAD+-dependent histone deacetylase85 and chromatin-silencing factor), NCoR (nuclear receptor corepressor), and SMRT (silencing mediator for retinoid and thyroid hormone receptor). The complex SIRT1/PPARγ/NCoR is recruited to specific DNA sequences (PPRE) in the promoter region of PPARγ target genes and inhibits their transcription.86;87 Co-activators include members SRC1 (steroid receptor co-activator 1) and TIF2 (transcriptional intermediary factor 1) which interact directly with PPARγ to initiate its transactivation.88;89 In our study, IUGR newborns at 1 day of age showed significantly increased protein expression of co-repressors (SIRT1, SMRT) and the co-activator SRC1, suggesting a key role of co-activator in modulating PPARγ activity.90;91
These results further raise the question of whether the increased adipogenic potential of IUGR adipocytes is due to an intrinsic cellular change. To address this, we utilized primary adipocyte cell cultures and demonstrated that indeed IUGR adipocytes retain the basal phenotypic characteristic of programmed upregulation of adipogenic PPARγ in cell culture, as evident by significantly increased mRNA and protein expression.92 Thus, the adipocyte itself exhibits “programmed” adipogenesis/lipogenesis, independent upon the IUGR hormonal milieu. Despite this programming, the primary adipocyte cell cultures from 1 day old IUGR were able to respond to a synthetic PPARγ repressor-ligand, BADGE (bisphenol A diglycidyl ether)93 with downregulation of PPARγ expression. Interestingly, primary adipocyte cell cultures from 3 week old IUGR offspring were non-responsive to BADGE.94 Thus, the normal suppressive response to the repressor at 1 day of age suggests that IUGR adipocytes may respond to pharmacologic approaches to prevent obesity, though within a certain limiting period.
Maternal High Fat Diet Offspring
The mechanism of programming resulting from in utero overnutrition likely involve a triad of effects: preexisting maternal obesity, high fat diet effects, and varying degrees of glucose intolerance. Thus, human studies of obese mothers with gestational diabetes indicate an increased risk for offspring adiposity.95
Currently, mechanistic studies on programmed adipogenesis due to maternal obesity or high fat diet are lacking. Our studies show that despite the putative nutritional difference, HF offspring demonstrate enhanced adipogenesis, akin to IUGR newborns. Thus both under- and overnutrition programs increased adipogenesis. Although, the underpinning contributory factor is once again attributed to upregulated PPARγ, it is mediated via different mechanisms. In contrast to IUGR newborn, HF newborns demonstrate downregulated co-repressors (SIRT1, NCoR, SMRT) with unchanged co-activator (TIF2), suggesting a key role of PPARγ co-repressors.96
Therapies, Applications and Conclusions
The obesity epidemic represents one of the major public health challenges in the 21st century. Devising effective policy and practice to combat childhood obesity is a high priority for many governments and health professionals. There is irrevocable evidence that departures from optimal growth in utero, whether from limited or excess nutrition, increase the relative risk of adult obesity. This essentially provide the basis for the ‘programming’ hypothesis and present a challenge to elucidate the mechanisms by which gene-nutrient interactions during embryonic or fetal development set the stage for an adult’s susceptibility to multiple metabolic abnormalities. Emerging evidence suggests that alterations in epigenomics may be a key mechanism by which nutritional exposure in utero can influence gene expression, and therefore, phenotype. Indeed, PPARγ is epigenetically regulated,97 and though less characterized, NCoR and SRC1 genes may also be regulated by epigenetic modifications. This may in part explain upregulated PPARγ and increased adipogenesis under both limited and excess nutrient availability, though occurring via divergent mechanisms. In HF offspring, it may be downregulation of co-repressors whereas IUGR offspring demonstrate upregulation of co-activators. In this scenario, therapeutic interventions for the prevention of offspring obesity will require target-specific modalities dependent upon the primary etiology. Alternatively, overall suppression of PPARγ may be achieved through antagonist treatment or administration of a selective repressor-ligand (BADGE), which is known to decrease adipocyte cell size and prevent obesity. Whether the demonstrated in vitro and in vivo effectiveness98 of this approach can be replicated in human remains to be determined.
Acknowledgments
Our work reported was supported by the American Heart Association 0865125F, National Institute of Child Health and Human Development Grant R03HD060241 and National Institute of Diabetes and Digestive and Kidney Grant R56DK081756, UCLA-UCSD DERC (Diabetes and Endocrinology Research Center) to MD, and National Institute of Child Health and Human Development Grant R01HD054751 to MGR.
Contributor Information
Mina Desai, Email: mdesai@obgyn.humc.edu.
Michael G. Ross, Email: mikeross@ucla.edu.
Reference List
- 1.James PT, Leach R, Kalamara E, Shayeghi M. The worldwide obesity epidemic. Obes Res. 2001;9 (Suppl 4):228S–233S. doi: 10.1038/oby.2001.123. [DOI] [PubMed] [Google Scholar]
- 2.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 3.Ehrenberg HM, Mercer BM, Catalano PM. The influence of obesity and diabetes on the prevalence of macrosomia. Am J Obstet Gynecol. 2004;191:964–968. doi: 10.1016/j.ajog.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 4.ACOG Committee Opinion number 315. September 2005 Obesity in pregnancy. Obstet Gynecol. 2005;106:671–675. doi: 10.1097/00006250-200509000-00054. [DOI] [PubMed] [Google Scholar]
- 5.Guo SS, Wu W, Chumlea WC, Roche AF. Predicting overweight and obesity in adulthood from body mass index values in childhood and adolescence. Am J Clin Nutr. 2002;76:653–658. doi: 10.1093/ajcn/76.3.653. [DOI] [PubMed] [Google Scholar]
- 6.Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–1239. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- 7.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 8.Desai M, Hales CN. Role of fetal and infant growth in programming metabolism in later life. Biol Rev Camb Philos Soc. 1997;72:329–348. doi: 10.1017/s0006323196005026. [DOI] [PubMed] [Google Scholar]
- 9.Ross MG, Desai M. Gestational programming: population survival effects of drought and famine during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2005;288:R25–R33. doi: 10.1152/ajpregu.00418.2004. [DOI] [PubMed] [Google Scholar]
- 10.Flier JS. The adipocyte: storage depot or node on the energy information superhighway? Cell. 1995;80:15–18. doi: 10.1016/0092-8674(95)90445-x. [DOI] [PubMed] [Google Scholar]
- 11.Reilly MP, Rader DJ. The metabolic syndrome: more than the sum of its parts? Circul. 2003;108:1546–1551. doi: 10.1161/01.CIR.0000088846.10655.E0. [DOI] [PubMed] [Google Scholar]
- 12.Gluckman PD, Hanson MA, Morton SM, Pinal CS. Life-long echoes--a critical analysis of the developmental origins of adult disease model. Biol Neonate. 2005;87:127–139. doi: 10.1159/000082311. [DOI] [PubMed] [Google Scholar]
- 13.Harding JE. The nutritional basis of the fetal origins of adult disease. Int J Epidemiol. 2001;30:15–23. doi: 10.1093/ije/30.1.15. [DOI] [PubMed] [Google Scholar]
- 14.Barker DJ, Hales CN, Fall CH, et al. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 15.Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 16.Simmons R. Perinatal programming of obesity. Semin Perinatol. 2008;32:371–374. doi: 10.1053/j.semperi.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 18.Oken E, Rifas-Shiman SL, Field AE, Frazier AL, Gillman MW. Maternal gestational weight gain and offspring weight in adolescence. Obstet Gynecol. 2008;112:999–1006. doi: 10.1097/AOG.0b013e31818a5d50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Surkan PJ, Hsieh CC, Johansson AL, Dickman PW, Cnattingius S. Reasons for increasing trends in large for gestational age births. Obstet Gynecol. 2004;104:720–726. doi: 10.1097/01.AOG.0000141442.59573.cd. [DOI] [PubMed] [Google Scholar]
- 20.Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res. 2008;36:73–84. doi: 10.1159/000115355. [DOI] [PubMed] [Google Scholar]
- 21.Pettitt DJ, Jovanovic L. Birth weight as a predictor of type 2 diabetes mellitus: the U-shaped curve. Curr Diab Rep. 2001;1:78–81. doi: 10.1007/s11892-001-0014-x. [DOI] [PubMed] [Google Scholar]
- 22.Ong KK. Size at birth, postnatal growth and risk of obesity. Horm Res. 2006;65 (Suppl 3):65–69. doi: 10.1159/000091508. [DOI] [PubMed] [Google Scholar]
- 23.Pettitt DJ, Jovanovic L. Low birth weight as a risk factor for gestational diabetes, diabetes, and impaired glucose tolerance during pregnancy. Diabetes Care. 2007;30 (Suppl 2):S147–S149. doi: 10.2337/dc07-s207. [DOI] [PubMed] [Google Scholar]
- 24.Launer LJ, Hofman A, Grobbee DE. Relation between birth weight and blood pressure: longitudinal study of infants and children. BMJ. 1993;307:1451–1454. doi: 10.1136/bmj.307.6917.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 26.Hales CN, Desai M, Ozanne SE. The Thrifty Phenotype hypothesis: how does it look after 5 years? Diabet Med. 1997;14:189–195. doi: 10.1002/(SICI)1096-9136(199703)14:3<189::AID-DIA325>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Seckl JR, Meaney MJ. Glucocorticoid programming. Ann N Y Acad Sci. 2004;1032:63–84. doi: 10.1196/annals.1314.006. [DOI] [PubMed] [Google Scholar]
- 28.Bol VV, Delattre AI, Reusens B, Raes M, Remacle C. Forced catch-up growth after fetal protein restriction alters the adipose tissue gene expression program leading to obesity in adult mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R291–R299. doi: 10.1152/ajpregu.90497.2008. [DOI] [PubMed] [Google Scholar]
- 29.Desai M, Gayle D, Babu J, Ross MG. Programmed obesity in intrauterine growth-restricted newborns: modulation by newborn nutrition. Am J Physiol Regul Integr Comp Physiol. 2005;288:R91–R96. doi: 10.1152/ajpregu.00340.2004. [DOI] [PubMed] [Google Scholar]
- 30.Desai M, Gayle D, Babu J, Ross MG. Permanent reduction in heart and kidney organ growth in offspring of undernourished rat dams. Am J Obstet Gynecol. 2005;193:1224–1232. doi: 10.1016/j.ajog.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 31.Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci. 2008;28:12107–12119. doi: 10.1523/JNEUROSCI.2642-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009 doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- 33.Howie GJ, Sloboda DM, Kamal T, Vickers MH. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol. 2009;587:905–915. doi: 10.1113/jphysiol.2008.163477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferezou-Viala J, Roy AF, Serougne C, et al. Long-term consequences of maternal high-fat feeding on hypothalamic leptin sensitivity and diet-induced obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1056–R1062. doi: 10.1152/ajpregu.00117.2007. [DOI] [PubMed] [Google Scholar]
- 35.Samuelsson AM, Matthews PA, Argenton M, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 36.Jones AP, Simson EL, Friedman MI. Gestational undernutrition and the development of obesity in rats. J Nutr. 1984;114:1484–1492. doi: 10.1093/jn/114.8.1484. [DOI] [PubMed] [Google Scholar]
- 37.Desai M, Li T, Han G, Guberman C, Ross MG. Maternal obesity and increased risk of offspring metabolic syndrome. Reprod Sci Suppl. 17:104A–139. [Google Scholar]
- 38.Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci. 2004;24:2797–2805. doi: 10.1523/JNEUROSCI.5369-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouret SG, Simerly RB. Developmental programming of hypothalamic feeding circuits. Clin Genet. 2006;70:295–301. doi: 10.1111/j.1399-0004.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- 40.Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouret SG, Simerly RB. Minireview: Leptin and development of hypothalamic feeding circuits. Endocrinol. 2004;145:2621–2626. doi: 10.1210/en.2004-0231. [DOI] [PubMed] [Google Scholar]
- 42.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 43.Coupe B, Amarger V, Grit I, Benani A, Parnet P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinol. 2010;151:702–713. doi: 10.1210/en.2009-0893. [DOI] [PubMed] [Google Scholar]
- 44.Yousheng J, Nguyen T, Desai M, Ross MG. Programmed alterations in hypothalamic neuronal orexigenic responses to ghrelin following gestational nutrient restriction. Reprod Sci. 2008;15:702–709. doi: 10.1177/1933719108316982. [DOI] [PubMed] [Google Scholar]
- 45.Desai M, Gayle D, Han G, Ross MG. Programmed hyperphagia due to reduced anorexigenic mechanisms in intrauterine growth-restricted offspring. Reprod Sci. 2007;14:329–337. doi: 10.1177/1933719107303983. [DOI] [PubMed] [Google Scholar]
- 46.Desai M, Ti L, Ross MG. Fetal hypothalamic neuroprogenitor cell culture: Preferential differentiation paths induced by leptin and insulin. J Neurosci. doi: 10.1210/en.2010-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desai M, Li T, Ross MG. Hypothalamic neurosphere progenitor cells in low birth weight rat newborns: Neurotrophic effects of leptin and insulin. J Clin Invest. doi: 10.1016/j.brainres.2010.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirk SL, Samuelsson AM, Argenton M, et al. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE. 2009;4:e5870. doi: 10.1371/journal.pone.0005870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wabitsch M. The acquisition of obesity: insights from cellular and genetic research. Proc Nutr Soc. 2000;59:325–330. doi: 10.1017/s0029665100000367. [DOI] [PubMed] [Google Scholar]
- 50.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 51.Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev. 2001;2:239–254. doi: 10.1046/j.1467-789x.2001.00042.x. [DOI] [PubMed] [Google Scholar]
- 52.Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 53.Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 54.Auwerx J, Martin G, Guerre-Millo M, Staels B. Transcription, adipocyte differentiation, and obesity. J Mol Med. 1996;74:347–352. doi: 10.1007/BF00210629. [DOI] [PubMed] [Google Scholar]
- 55.Fajas L, Fruchart JC, Auwerx J. Transcriptional control of adipogenesis. Curr Opin Cell Biol. 1998;10:165–173. doi: 10.1016/s0955-0674(98)80138-5. [DOI] [PubMed] [Google Scholar]
- 56.Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 57.Morrison RF, Farmer SR. Insights into the transcriptional control of adipocyte differentiation. J Cell Biochem. 1999;(Suppl 32–33):59–67. doi: 10.1002/(sici)1097-4644(1999)75:32+<59::aid-jcb8>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 58.Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) 2005;29 (Suppl 1):S13–S16. doi: 10.1038/sj.ijo.0802907. [DOI] [PubMed] [Google Scholar]
- 59.Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J Biol Chem. 1998;273:30057–30060. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 60.Lane MD, Lin FT, MacDougald OA, Vasseur-Cognet M. Control of adipocyte differentiation by CCAAT/enhancer binding protein alpha (C/EBP alpha) Int J Obes Relat Metab Disord. 1996;20 (Suppl 3):S91–S96. [PubMed] [Google Scholar]
- 61.Fajas L, Schoonjans K, Gelman L, et al. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19:5495–5503. doi: 10.1128/mcb.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- 63.Walczak R, Tontonoz P. PPARadigms and PPARadoxes: expanding roles for PPARgamma in the control of lipid metabolism. J Lipid Res. 2002;43:177–186. [PubMed] [Google Scholar]
- 64.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim JB, Sarraf P, Wright M, et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998;101:1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boizard M, Le LX, Lemarchand P, et al. Obesity-related overexpression of fatty-acid synthase gene in adipose tissue involves sterol regulatory element-binding protein transcription factors. J Biol Chem. 1998;273:29164–29171. doi: 10.1074/jbc.273.44.29164. [DOI] [PubMed] [Google Scholar]
- 67.Samra JS. Sir David Cuthbertson Medal Lecture. Regulation of lipid metabolism in adipose tissue. Proc Nutr Soc. 2000;59:441–446. doi: 10.1017/s0029665100000604. [DOI] [PubMed] [Google Scholar]
- 68.Vazquez-Vela ME, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Arch Med Res. 2008;39:715–728. doi: 10.1016/j.arcmed.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 69.Bulcao C, Ferreira SR, Giuffrida FM, Ribeiro-Filho FF. The new adipose tissue and adipocytokines. Curr Diabetes Rev. 2006;2:19–28. doi: 10.2174/157339906775473617. [DOI] [PubMed] [Google Scholar]
- 70.Bahceci M, Gokalp D, Bahceci S, et al. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J Endocrinol Invest. 2007;30:210–214. doi: 10.1007/BF03347427. [DOI] [PubMed] [Google Scholar]
- 71.Lefebvre AM, Laville M, Vega N, et al. Depot-specific differences in adipose tissue gene expression in lean and obese subjects. Diabetes. 1998;47:98–103. doi: 10.2337/diab.47.1.98. [DOI] [PubMed] [Google Scholar]
- 72.Freedland ES. Role of a critical visceral adipose tissue threshold (CVATT) in metabolic syndrome: implications for controlling dietary carbohydrates: a review. Nutr Metab (Lond) 2004;1:12. doi: 10.1186/1743-7075-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi H, Clegg DJ. Sex differences in the regulation of body weight. Physiol Behav. 2009;97:199–204. doi: 10.1016/j.physbeh.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jensen MD. Lipolysis: contribution from regional fat. Annu Rev Nutr. 1997;17:127–139. doi: 10.1146/annurev.nutr.17.1.127. [DOI] [PubMed] [Google Scholar]
- 75.Sewter CP, Blows F, Vidal-Puig A, O’Rahilly S. Regional differences in the response of human pre-adipocytes to PPARgamma and RXRalpha agonists. Diabetes. 2002;51:718–723. doi: 10.2337/diabetes.51.3.718. [DOI] [PubMed] [Google Scholar]
- 76.Adams M, Montague CT, Prins JB, et al. Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J Clin Invest. 1997;100:3149–3153. doi: 10.1172/JCI119870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hauner H, Wabitsch M, Pfeiffer EF. Differentiation of adipocyte precursor cells from obese and nonobese adult women and from different adipose tissue sites. Horm Metab Res Suppl. 1988;19:35–39. [PubMed] [Google Scholar]
- 78.Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 79.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45:1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 80.Shepherd PR, Crowther NJ, Desai M, Hales CN, Ozanne SE. Altered adipocyte properties in the offspring of protein malnourished rats. Br J Nutr. 1997;78:121–129. doi: 10.1079/bjn19970124. [DOI] [PubMed] [Google Scholar]
- 81.Bieswal F, Ahn MT, Reusens B, et al. The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity (Silver Spring) 2006;14:1330–1343. doi: 10.1038/oby.2006.151. [DOI] [PubMed] [Google Scholar]
- 82.Guan H, Arany E, van Beek JP, et al. Adipose tissue gene expression profiling reveals distinct molecular pathways that define visceral adiposity in offspring of maternal protein-restricted rats. Am J Physiol Endocrinol Metab. 2005;288:E663–E673. doi: 10.1152/ajpendo.00461.2004. [DOI] [PubMed] [Google Scholar]
- 83.Desai M, Guang H, Ferelli M, Kallichanda N, Lane RH. Programmed upregulation of adipogenic transcription factors in intrauterine growth-restricted offspring. Reprod Sci. 2008;15:785–796. doi: 10.1177/1933719108318597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yee JK, Lee PW, Ross MG, Desai M. Enhancement of de novo fatty acid synthesis in IUGR adipose tissue prior to onset of programmed obesity. Reprod Sci. (Suppl 17):104A–137. [Google Scholar]
- 85.Yang T, Fu M, Pestell R, Sauve AA. SIRT1 and endocrine signaling. Trends Endocrinol Metab. 2006;17:186–191. doi: 10.1016/j.tem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 86.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kloting N, Bluher M. Extended longevity and insulin signaling in adipose tissue. Exp Gerontol. 2005;40:878–883. doi: 10.1016/j.exger.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 88.Powell E, Kuhn P, Xu W. Nuclear Receptor Cofactors in PPARgamma-Mediated Adipogenesis and Adipocyte Energy Metabolism. PPAR Res. 2006;2007:53843. doi: 10.1155/2007/53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 90.Desai M, Lane RH, Han G, Ross MG. Failure to suppress adipogenic transcription factor (PPARy) activity leads to programmed obesity in IUGR offspring. Reprod Sci. (Suppl 15):76A–54. [Google Scholar]
- 91.Desai M, Han G, Joss-Moore L, Ross MG, Lane RH. (SIRT1) mediated mechanims for programmed enhanced adipogenesis in intrauterine growth restricted newborns. PAS Annual Meeting; pp. 4045–10. [Google Scholar]
- 92.Desai M, Lane RH, Han G, Ross MG. Response of IUGR primary cell culture adipocytes to PPARy activator-ligand and repressor-ligand mechanism of programmed obesity. Reprod Sci. (Suppl 15):194A–474. [Google Scholar]
- 93.Choy L, Skillington J, Derynck R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J Cell Biol. 2000;149:667–682. doi: 10.1083/jcb.149.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Desai M, Lane RH, Han G, Ross MG. Mechanism of enhanced adipogenesis in intrauterine growth restricted offspring: dysregulation of adipogenic function and reduced response to PPARγ modulators. Early Hum Dev. 83:S48. [Google Scholar]
- 95.Plagemann A, Harder T, Dudenhausen JW. The diabetic pregnancy, macrosomia, and perinatal nutritional programming. Nestle Nutr Workshop Ser Pediatr Program. 2008;61:91–102. doi: 10.1159/000113179. [DOI] [PubMed] [Google Scholar]
- 96.Desai M, Han G, Li T, Ross MG. Transcriptional regulation of adipogenesis in newborns exposed to maternal obesity. Reprod Sci. (Suppl 17):217A–532. [Google Scholar]
- 97.Wakabayashi K, Okamura M, Tsutsumi S, et al. The peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha heterodimer targets the histone modification enzyme PR-Set7/Setd8 gene and regulates adipogenesis through a positive feedback loop. Mol Cell Biol. 2009;29:3544–3555. doi: 10.1128/MCB.01856-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wright HM, Clish CB, Mikami T, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem. 2000;275:1873–1877. doi: 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]