

Letter to the Editor
Three main categories of pharmacological inhibitors of kinase activity include: (1) Type I, or “DFG-in” ATP competitive inhibitors (the Asp-Phe-Gly or “DFG” motif is highly conserved in protein kinases and sits near the beginning, or N-terminus, of the activation loop), characterized by competition with ATP in the ATP binding site, (2) Type II, or “DFG-out” ATP competitive inhibitors, which bind to the ATP binding site as well as an adjacent hydrophobic binding site accessible solely when the kinase is in an inactivated configuration, and (3) non-ATP competitive inhibitors that bind at sites outside the ATP binding site that affect kinase activity1. Second generation Abl inhibitors in clinical use for chronic myeloid leukemia (CML), such as the Type I inhibitor, dasatinib, and the Type II inhibitor, nilotinib, show significantly more potency against BCR-ABL than imatinib, and inhibit most imatinib-resistant BCR-ABL mutants2,3 with the exception of the T315I “gatekeeper” mutant.
HG-7-85-01 represents a new class of type II ATP-competitive inhibitors capable of inhibiting T315I-BCR-ABL, as well as gatekeeper mutants of Kit (T670I-KIT) and PDGFRα (T674I/M-PDGFRα) that are clinically observed in gastrointestinal stromal tumor (GIST) and hypereosinophilic syndrome (HEL)6. HG-7-85-01 is distinctive in having the ability to accommodate either a gatekeeper threonine, present in the non-mutated forms of target kinases, or a large hydrophobic amino acid without becoming a promiscuous kinase inhibitor.
The GNF2 & 5 family of inhibitors bind to the myristate binding site of Bcr-Abl and inhibit kinase activity by stabilizing a catalytically less competent conformation of the kinase7,8. GNF-5 exhibits additive inhibitory activity with nilotinib in cellular and in vivo models against both non-mutated and T315I Bcr-Abl.8
The combination of more than one Abl inhibitor in the treatment of imatinib-resistant disease may have beneficial therapeutic value, since clonal resistance could potentially be overcome by combining two agents with different resistance profiles. We investigated the ability of HG-7-85-01, which inhibits T315I6, to positively combine with the allosteric non-ATP competitive inhibitor, GNF-5, which is unable to potently inhibit T315I as a single agent8. We show here that combinations of HG-7-85-01 with GNF-5 have at least additive effects against both non-mutated BCR-ABL and BCR-ABL T315I in vitro and in vivo.
HG-7-85-01 was observed to positively combine with GNF-5 in vitro against Ba/F3.p210 cells (Calcusyn combination indices: ED25: 0.15, (strong synergism); ED50: 0.25, (strong synergism); ED75: 0.40 (synergism); ED90: 0.65(synergism)). The two inhibitors were also shown to positively combine in vivo against 32D.p210-luc+ xenografted cells (Figure 1).
Figure 1. In vivo combination study between HG-7-85-01 and GNF-5 against non-mutant BCR-ABL.
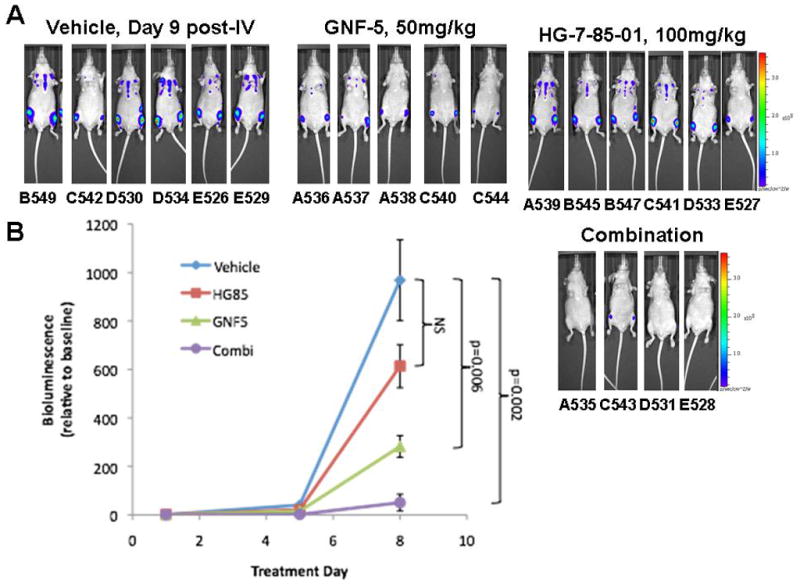
Day 9 post-IV injection of 1,000,000 32D.p210-luc+ cells/mouse. (A) Representative mouse images. (B) Graph of plotted bioluminescence values (relative to baseline bioluminescence values). HG-7-85-01 is the same as the label “HG85” that is shown in the graph. Vehicles (n=6). Treatment mice were administered 1X daily 50mg/kg GNF-5 (n=5), 1X daily 100mg/kg HG-7-85-01 (n=6), or 1X daily a combination of GNF-5 and HG-7-85-01 at the aforementioned doses (n=4). Baseline imaging was performed 2 days post IV injection of 32D.p210-luc+ cells. Mice were treated for a total of 4 consecutive days, prior to imaging on day 6 post-IV (with no drug treatments for that day), and followed by two additional days of drug treatment with imaging on day 9 post-IV. Three mice (32D.p210-luc+ cell-injected) died between Day 6 post-IV injection of cells and Day 9 post-IV injection of cells (B550-combination (found dead on day 9 post-IV day of imaging), B546-GNF5 only (died on day 8 post-IV), D532-combination (died on day 8 post-IV) (p=0.0002, one-way analysis of variance). The difference in tumor burden between vehicle-treated mice and GNF5 only-treated mice was less significant (p=0.006) than the difference in tumor burden between vehicle-treated mice and combination-treated mice (p=0.002).
The combination of HG-7-85-01 plus GNF-5, as compared to either agent alone, effectively killed more T315I-positive cells in vitro (Figure 2). In vivo combination studies were also performed, investigating the effects of HG-7-85-01 plus GNF-5 as compared to each agent alone. The average percent spleen size in HG-7-85-01+GNF-5-treated mice harboring T315I-positive leukemia was smaller than mice treated with either single agent or mice treated with vehicle (Figure 3).
Figure 2. Combination studies between GNF-5+HG-7-85-01 against T315I-BCR-ABL-expressing cells.
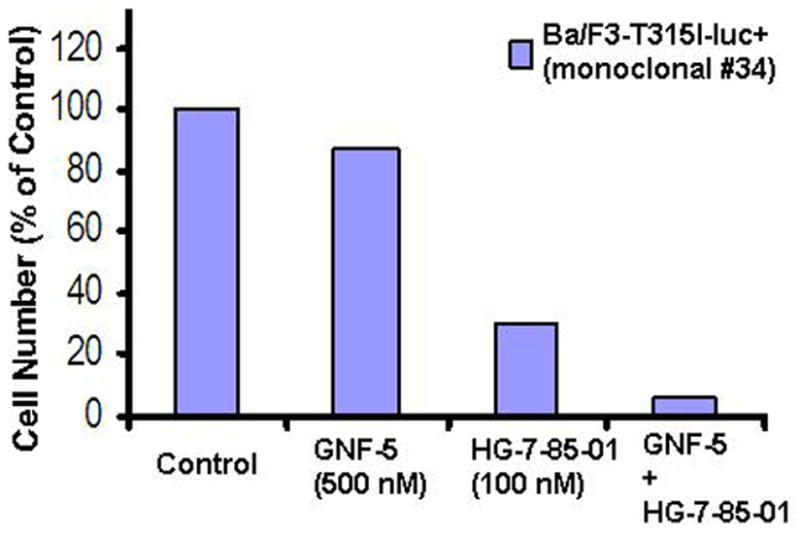
Proliferation study showing results of treatment of Ba/F3-T315I-luc+ cells (monoclonal #34) with GNF-5 and HG-7-85-01, alone and in combination.
Figure 3. In vivo combination studies between HG-7-85-01 and GNF-5 against BCR-ABL-T315I.
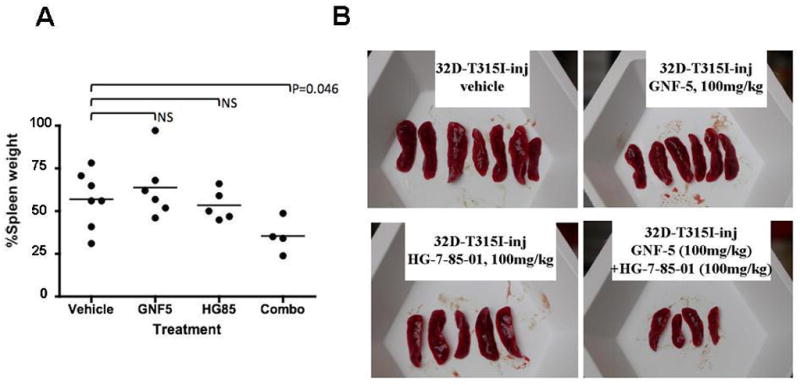
Day 9 post-IV injection of 1,000,000 32D-T315I-luc+ cells/mouse. (A) Percent spleen weights are shown for vehicles (n=7) and treatment mice, the latter administered 1X daily 100mg/kg GNF-5 (n=6), 1X daily 100mg/kg HG-7-85-01 (n=5), or 1X daily a combination of GNF-5 and HG-7-85-01 at the aforementioned doses (n=4). Baseline imaging was performed 2 days post IV injection of 32D.p210-luc+ cells. Mice were treated for a total of 4 consecutive days, with 1 day of no drug treatment, and followed by two additional days of drug treatment with imaging on day 9 post-IV. Three mice that were initially injected with 32D-T315I-luc+ died during the course of the study prematurely and were not included in the final spleen weight analysis (I507- HG-7-85-01 (found dead on day 6 post-IV), I508- combination (found dead on day 6 post-IV), J523-combination (died on day 7 post-IV). There was a statistically significant difference between percent spleen size observed in combination-treated mice and vehicle-treated mice (p=0.046). This study is representative of three independent studies in which similar results were observed. HG-7-85-01 is the same as the label “HG85” that is shown in the graph. (B) Spleens dissected from mice tested in this study.
In conclusion, the non-ATP competitive inhibitor, GNF-5, was observed to positively combine with the type II inhibitor, HG-7-85-01, and the type II inhibitor, nilotinib, respectively, against BCR-ABL in vitro and in a bioluminescent mouse model of BCR-ABL-positive leukemia. Similarly, the combination of GNF-5 and HG-7-85-01 was observed to inhibit BCR-ABL-T315I-positive leukemia cell growth in vitro to a greater extent than either drug alone. Tumor burden in mice harboring 32D.p210-T315I-luc+ cells treated with a combination of HG-7-85-01 and GNF-5 was suppressed to a higher extent than mice treated with vehicle or either agent alone. The ability of two such structurally distinct, targeted Abl inhibitors to positively combine introduces a novel approach to overriding imatinib-resistant leukemia, including that caused by the highly prevalent and strongly imatinib-resistant gatekeeper mutant, T315I.
Acknowledgments
Grant Support: J.D.G is supported by NIH grant CA66996, and a Specialized Center of Research Award from the Leukemia and Lymphoma Society. J.D.G. is also supported by NIH grants CA36167 and DK50654. J.D.G., N.G. and A.L.K. have a financial interest with Novartis Pharma AG.
References
- 1.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 2.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 5.Zhou T, Parillon L, Li F, Wang Y, Keats J, Lamore S, Xu Q, Shakespeare W, Dalgarno D, Zhu X. Crystal structure of the T315I mutant of the Abl kinase. Chem Biol Drug Des. 2007;70:171–181. doi: 10.1111/j.1747-0285.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- 6.Weisberg E, Choi HG, Ray A, Barrett R, Zhang J, Taebo S, Sim T, Zhou W, Seeliger M, Cameron M, Azam M, Fletcher JA, Debiec-Rychter M, Mayeda M, Moreno D, Kung AL, Janne PA, Khosravi-Far R, Melo JV, Manley P, Adamia S, Wu C, Gray N, Griffin JD. Discovery of a small molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFRα, Kit, and Src kinases. Blood. 2010 doi: 10.1182/blood-2009-11-251751. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, Mestan J, Fabbro D, Gray NS. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Woiciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vaipai N, Grzesiek S, Tuntland T, Liu Y, Bursulava B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]