

Abstract
Aim:
To compare the anti-cancer activity and cellular uptake of curcumin (Cur) delivered by targeted and non-targeted drug delivery systems in multidrug-resistant cervical cancer cells.
Methods:
Cur was entrapped into poly (DL-lactide-co-glycolide) (PLGA) nanoparticles (Cur-NPs) in the presence of modified-pluronic F127 stabilizer using nano-precipitation technique. On the surface of Cur-NPs, the carboxy-terminal of modified pluronic F127 was conjugated to the amino-terminal of anti-P-glycoprotein (P-gp) (Cur-NPs-APgp). The physical properties of the Cur-NPs, including particle size, zeta potential, particle morphology and Cur release kinetics, were investigated. Cellular uptake and specificity of the Cur-NPs and Cur-NPs-APgp were detected in cervical cancer cell lines KB-V1 (higher expression of P-gp) and KB-3-1 (lower expression of P-gp) using fluorescence microscope and flow cytometry, respectively. Cytotoxicity of the Cur-NPs and Cur-NPs-APgp was determined using MTT assay.
Results:
The particle size of Cur-NPs and Cur-NPs-APgp was 127 and 132 nm, respectively. The entrapment efficiency and actual loading of Cur-NPs-APgp (60% and 5 μg Cur/mg NP) were lower than those of Cur-NPs (99% and 7 μg Cur/mg NP). The specific binding of Cur-NPs-APgp to KB-V1 cells was significantly higher than that to KB-3-1 cells. Cellular uptake of Cur-NPs-APgp into KB-V1 cells was higher, as compared to KB-3-1 cells. However, the cellular uptake of Cur-NPs and Cur-NPs-IgG did not differ between the two types of cells. Besides, the cytotoxicity of Cur-NPs-APgp in KB-V1 cells was higher than those of Cur and Cur-NPs.
Conclusion:
The results demonstrate that Cur-NPs-APgp targeted to P-gp on the cell surface membrane of KB-V1 cells, thus enhancing the cellular uptake and cytotoxicity of Cur.
Keywords: anticancer drug, curcumin, targeting drug delivery, nanoparticle, P-glycoprotein, multidrug resistance, cervical cancer
Introduction
Multidrug resistance (MDR) in cancer cells reduces the cytotoxic effects of various anticancer drugs and enhances the ability of cancer cells to actively efflux drugs, leading to a decrease in cellular drug accumulation below toxicity1. Although several mechanisms are proposed for drug resistance, the best-studied mechanism of MDR is related to the overexpression of P-glycoprotein (P-gp), a 170 kDa ATP dependent membrane transporter that acts as a drug efflux pump2. Overexpression of P-gp in various cancer cells leads to a decrease in the cytotoxicity in a broad spectrum of anticancer drugs including doxorubicin, vincristine, etopoxide and paclitaxel3. There have been many studies investigating the ability of P-gp inhibitors to overcome MDR by inhibiting MDR transporters or by suppressing MDR mechanisms. The compounds that would reverse resistance against anticancer drugs are called MDR inhibitors, MDR modulators or chemosensitizers4. Many pharmacologic agents from diverse structure classes have been identified as MDR modulators. It has been reported that many agents from natural products and dietary plants also modulate MDR phenotype of cancer cells including the extraction from Rosmarinus officinalis, Momordica charantia4, 5, Stemona aphylla6, and including Curcuma longa7, 8.
Curcumin (Cur), a phenolic compound purified from the rhizome of Curcuma longa, has a long history of being used as a spice and in traditional medicine. Several studies demonstrated that Cur has been shown to display antioxidant, anti-inflammation, anti-diabetes, anti-carcinogenic, anti-tumor invasion and anti-angiogenesis activities9. Moreover, our previous findings indicated that Cur down-regulated both MDR1 gene expression and P-gp function10, 11. The most compelling and key rationale for the therapeutic use of Cur is its extremely superior safe profile. Cur has been associated with the regression of premalignant lesions of various organs and shows no toxicity to healthy organs at high doses12. However, low oral bioavailability, poor pharmacokinetics, insolubility in water12 and degradability at natural to basic pH conditions are the limited efficiency factors of Cur in vivo. The development of novel delivery systems could be one strategy to overcome these problems. One possible way to improve the water solubility and stability of Cur is to entrap it into nanoparticles (NPs)12.
NPs can target tumors by either the passive or active process. Passive targeting implies that the enhanced permeability and retention effect (EPR effect), which characterizes malignant tissues, allows the passive accumulation of encapsulated drugs to tumor sites13, 14 . Active targeting involves tagging the drug vehicle with a ligand and allows it to specifically sequester in the targeted tumor15, 16. Among the various ligands, such as peptides, carbohydrates, and polymers, monoclonal antibodies are most widely investigated for selectively targeting nanoparticulate drug delivery systems to tumors17, 18, 19. The P-gp is one such cell-surface target. P-gp-positivity is associated with more aggressive tumor behavior20 and is a biomarker which overexpresses on the plasma membrane of MDR phenotypes of a variety of cancer cell lines21, where Cur could be a promising candidate for cancer targeted therapy.
Polymeric nanoparticle-based drugs have been increasingly developed as preferred drug nanocarriers against many diseases. Special focus on the use of particles prepared from poly(DL-lactide-co-glycolic acid) (PLGA)22, is warranted due to their biocompatibility, biodegradability and high stability in biological fluids23. These NPs have proven to be effective carriers for hydrophobic and hydrophilic drugs24. Bisht et al showed that Cur-loaded NPs could suppress constitutive NF-ĸB in pancreatic cancer cells25. The next previous study again found that Cur-loaded NPs were more active than Cur in suppressing NF-ĸB activation induced by TNF26. These differences could have been due to differential Cur uptake.
Several studies have been reported the evaluations of Cur-loaded PLGA NPs such as the effect in improving oral bioavailability of Cur may be associated with improved water solubility, higher release rate in the intestinal juice, enhanced absorption by improved permeability, inhibition of P-gp-mediated efflux, and increased residence time in the intestinal cavity12, 17, 18, 19, 23. Thus, encapsulating hydrophobic drugs or medicinal compounds in PLGA polymer is a promising candidate method for sustained and controlled drug delivery with improved bioavailability of Biopharmaceutics Classification System (BCS) class IV, such as Cur27. In this study we conjugated anti-P-gp antibody to the surface of biodegradable PLGA-NPs to target P-gp on the surface of MDR cancer cells and Cur was encapsulated to PLGA nanospheres by nanoprecipitation technique. The Cur loaded NPs were then characterized for their actual loading, encapsulation efficiency, particle size, morphology, and then evaluated for their in vitro release profiles. Moreover, we compared the cellular uptake and cytotoxicity of targeted and nontargeted Cur-loaded NPs in drug resistant (KB-V1) and drug sensitive (KB-3-1) cervical carcinoma cell lines.
Materials and methods
Materials
Poly(DL-lactide-co-glycolide) (PLGA; lactide to glycolide ratio 50:50), N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC), N-hydroxy succinimide (NHS) and Pluronic® F127 (Poloxamer 407) were purchased from Sigma Aldrich (St Louis, MO, USA). 4-Dimethylaminopyridine (DMAP) and succinic anhydride were purchased from Fluka Chemie GmbH (Buchs, Switzerland). Dulbecco's modified Eagle's medium (DMEM), penicillin-streptomycin, fetal bovine serum (FBS) and 0.05% trypsin-EDTA were provided by Gibco (Grand Island, NY, USA). Triethylamine and D-TubeTM Dialyzer Midi, MWCO 6–8 kDa, Amicon® Uitra-4 centrifugal Filter Devices (30 K) were supplied by Merck KGaA (Darmstadt, Germany). Acetone, tetrahydrofuran (THF), chloroform, dimethyl sulfoxide (DMSO) and diethyl ether were purchased from Lab Scan (Bangkok, Thailand). PTFE membrane syringe filter (Puradisc13, PTFE; 0.45 μm) was purchased from Whatman (UK). Mouse monoclonal antibody (F4) to P-glycoprotein (P-gp) was purchased from Abcam (Cambridge, UK). Normal mouse IgG1 isotype control was perchased from Santa Cruz (CA, USA).
Cell culture
The multidrug resistant (KB-V1) and drug sensitive (KB-3-1) cervical carcinoma cell lines8, 10 were generous gifts from Dr Michael M GOTTESMAN (National Cancer Institute, Bethesda, MD, USA). Both cell lines were cultured in DMEM with 10% FBS, 2 mmol/L L-glutamine, 50 U/mL penicillin and 50 μg/mL streptomycin and 1 μg/mL of vinblastine was added only to the KB-V1 culture medium. These two-cell lines were maintained in a humidified incubator with an atmosphere comprising 5% CO2 at 37 °C. The expression of P-gp in KB-V1 and KB-3-1 cells was confirmed by Western blot analysis as described in our previous reports4, 10.
Preparation of poloxamer-carboxylic (Poloxamer-COOH)
Terminal hydroxyl groups on pluronics were first converted to carboxyl groups with a slight modification of the previous procedure described by Guerrouache et al28 and Chittasupho et al29. Twelve grams of poloxamer (Pluronic® F127) was dissolved in 55 mL tetrahydrofuran (THF), then 122.5 mg of 4-dimethylaminopyridine (DMAP), 135 μL of triethylamine and 1 g of succinic anhydride were added and the mixture was stirred for 48 h at room temperature. The solution was dried by a rotary evaporator, and was then dissolved in 75 mL of chloroform. The excess succinic anhydride was removed by filtration with a 0.45 μm PTFE membrane syringe filter. The poloxamer-COOH was purified by precipitation for 48 h with ice-cold diethyl ether. The precipitate was dissolved in ethanol, filtered and dried. The product was identified by fourier transform infrared spectrophotometer (Nicolet 470 FT-IR spectrometers, USA).
Preparation of curcumin-encapsulated PLGA nanoparticles (Cur-NPs)
PLGA NPs with encapsulated Cur were formulated by the nanoprecipitation technique30. Fifty milligrams of PLGA was dissolved in 7.5 mL acetone containing 500 μg of Cur. One hundred milligrams of poloxamer-COOH was dissolved in 10 mL Deionized (DI) water. Then the PLGA solution was dropwise-added into the poloxamer solution under stirring (400 rounds per minute) overnight. The NPs were then washed twice with DI water, by centrifugation using Amicon filtration device with 30 kDa molecular weight cut off membrane at 4400 rounds per minute for 50 min at 4 °C to obtain Cur-NPs. The resulting NPs were resuspended in 1 mL of sterile DI water.
Preparation of anti-P-gp antibody-conjugated Cur-NPs (Cur-NPs-APgp)
Cur-NPs were conjugated to anti-P-gp antibody through the carbodiimide reaction. Briefly, the NPs were adjusted to pH 5.8, and then incubated with the appropriate amount of NHS (50 mmol/L) and EDC (100 mmol/L) for 30 min. The resulting NHS-activated particles were then centrifuged at 12 000 rounds per minute for 15 min at 4 °C and the supernatant was removed. The NPs were resuspended in 800 μL DI water and 100 μL PBS (pH 7.4), and then 10 μL of anti-P-gp antibody was added under gentle stirring and the solution was then incubated overnight. Cur-NPs-APgp were collected by centrifugation (12 000 rounds per minute for 15 min at 4 °C) and washed 2 times with DI water.
Characterization of NPs
The NPs were characterized for size, zeta potential, and particle distribution by photon correlation spectroscopy (PCS) (Zetasizer, Malvern Instrument, UK).
Fifteen microliters of nanoparticles was dissolved in 25 μL of DI water (2 μg of nanoparticle/mL) and stained with 25 μL of 2% (w/v) phosphotungstic acid solution. Five microliters of stained-nanoparticles were dropped on a copper grid and dried by air flow. The morphology of NPs was determined by TEM.
Determination of Cur loading and encapsulation efficiency
The encapsulation efficiency of Cur-NPs was determined by analyzing the supernatant of the final suspension. The NPs and supernatant were separated from each other by centrifugation using an Amicon filtration device with 30 kDa molecular weight cut off membrane at 4400 rounds per minute for 50 min. The remaining Cur in the supernatant was measured by UV-visible spectrophotometer at 425 nm. The concentration of Cur was calculated using the standard curve of Cur in methanol (1–10 μg/mL). The Cur loading content and encapsulation efficiency were calculated as follows:
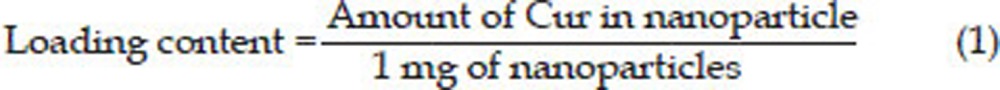 |
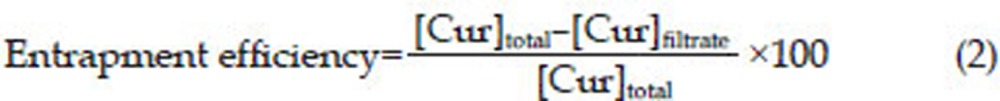 |
In vitro Cur release
The release of Cur from the NPs was carried out by the dialysis method as previously described30 with slight modifications. Briefly, 100 μg/mL of Cur-NPs were added in a dialysis tube with a molecular cut off of 6–8 kDa and suspended in 10 mL of release medium (50% v/v of ethanol) at 37 °C in shaking incubator at 70 rounds per minute. One milliliter from the release medium was withdrawn at predetermined time interval and replaced with 1 mL of the fresh medium. Finally, Cur in the samples was quantified with a spectrophotometer. The percentage of Cur released from the NPs at various time points was calculated as follows:
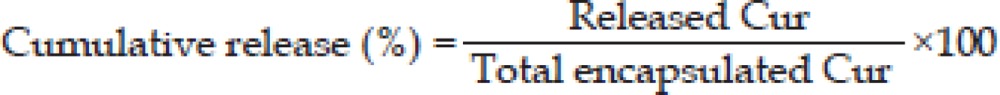 |
Cellular uptake of Cur-NPs-APgp into KB-V1 and KB-3-1 cells
KB-V1 and KB-3-1 (20 000 cells) were seeded on cover slips in 12-well tissue culture plates and incubated at 37 °C, 5% CO2 in DMEM supplement with 10% FBS, overnight. The cells were then exposed to 10 μg/mL concentrations of Cur-NPs-APgp, Cur-NPs, or Cur-NPs-IgG for 30 and 60 min. Free NPs were removed by being washed 3 times with PBS. The cells were fixed with 4% paraformaldehyde. Cellular uptake of Cur-NPs was determined using a fluorescence microscope.
Specific binding of Cur-NPs-APgp to KB-V1 and KB-3-1 cells
The specific binding of Cur-NPs-APgp was studied by using flow cytometry. KB-V1 and KB-3-1 cells (2×105 cells/well) were treated with 5 μmol/L of Cur-NPs-APgp for 5, 30, and 60 min in DMEM without phenol red and incubated at 37 °C. After incubation, the cells were centrifuged at 4400 rounds per minute for 5 min at 4 °C and the cells were washed 2 times with ice cold PBS. The cells were centrifuged at 4400 rounds per minute for 5 min at 4 °C. Then the supernatant was removed and the cell pellet was resuspended with 0.5 mL PBS. The fluorescence intensity was measured by flow cytometer.
Cytotoxicity assay
Cytotoxicity of NPs, NPs-APgp, free Cur, Cur-NPs, and Cur-NPs-APgp against KB-V1 and KB-3-1 cells was evaluated using a colorimetric MTT assay as was described before31. Briefly, cells (1×103 cells/well) were seeded in a 96-well plates and incubated at 37 °C, 5% CO2 overnight in DMEM containing 10% FBS. The cells were treated with various concentrations (0–27 μmol/L) of NPs, NPs-APgp, free Cur, Cur-NPs, and Cur-NPs-APgp for 6 h. Then cell culture supernatant was removed and fresh DMEM containing 10% FBS was added and the cell culture was further incubated for 42 h. For the time interval treatment, the cells were treated with 27 μmol/L of free Cur, Cur-NPs, and Cur-NPs-APgp for 1, 6, or 24 h, after that cultured in the drug free medium for another 23, 18, and 0 h, respectively. At the end of time point, MTT dye (15 μL, 5 mg/mL) was added and incubated for an additional 4 h. The MTT-formazan was dissolved in DMSO and absorbance was measured using a microplate reader at 540 nm with a reference wavelength of 630 nm.
Statistical analysis
All data are presented as mean±SD values. Statistical analyses were conducted with Prism version 5.0 using one-way ANOVA, Dunnett's test or Tukey's test. Statistical significance was concluded with: b, c: P<0.05 or 0.01, respectively when compared with control and e, f: P<0.05 or 0.01, respectively when compared with free Cur.
Results
Expression of P-gp in KB-V1 and KB-3-1 cells
Western blot analysis for the level of P-gp in KB-V1 and KB-3-1 cells showed that the KB-V1 cells expressed large amount of P-gp while the expression of P-gp in KB-3-1 cells could not be detected (Figure 1).
Figure 1.

P-gp expression in KB-V1 and KB-3-1 cells determined by Western blot analysis.
Preparation of carboxylated poloxamer (Poloxamer-COOH)
To conjugate anti-P-gp antibody to NPs, the hydroxyl groups of the surfactant and poloxamer were converted to carboxyl groups. The COOH formation was determined by FTIR spectroscopy. The FTIR spectrum showed a strong C=O stretching absorption at 1720 cm−1 and a very broad and strong absorption at 3500 cm−1, which indicated the appearance of the hydroxyl of carboxylic acid (Figure 2). These results confirmed the formation of the carboxylated poloxamer. These results were in line with our previous report32 confirming the partial formation of carboxylated Poloxamer.
Figure 2.

FTIR spectrogram of carboxylated Pluronic F127. Figure A exhibits the carboxylated poloxamer. The FTIR spectrum showed a strong C=O stretching absorption at 1720 cm−1 which indicated the appearance of carboxylic acid. Figure B exhibits native Pluronic F127, a very broad and strong absorption at 3500 cm−1, which indicated the appearance of the hydroxyl group.
Size, particle distribution, zeta potential, entrapment efficiency, and actual loading of NPs, Cur-NPs and Cur-NPs-APgp
PLGA NPs were prepared using a nanoprecipitation method. NPs were formed from PLGA, which served as a hydrophobic core to encapsulate the poorly water-soluble Cur. The size, polydispersity (PDI), zeta potential, and actual loading for Cur-NPs and Cur-NPs-APgp are shown in Table 1. The mean diameters of Cur-NPs and Cur-NPs-APgp were 127 and 132 nm, respectively, which were bigger than NPs (111 nm). Moreover, low polydispersity was observed in both Cur-NPs and Cur-NP-APgp suggesting a narrow size distribution. The zeta potential of the Cur-NPs and Cur-NPs-APgp were -23.1 and -40.3 mV, respectively. The entrapment efficiency and actual loading of Cur in nanoparticles were determined using equations 1 and 2, as described in the method section. The entrapment efficiency and actual loading of Cur-NPs-APgp (60%, 5 μg Cur/mg NP) were lower than that of Cur-NPs (99%, 7 μg Cur/mg NP).
Table 1. Mean size, zeta potential, polydispersity index (PDI), percent entrapment efficiency (%EE) and actual loading for NPs, Cur-NPs, and Cur-NPs-APgp.
| Formation | Size (nm) | Polydispersity | Zeta potential (mV) | EE (%) | Actual loading (μg Cur/mg NP) |
|---|---|---|---|---|---|
| NPs | 111±5.0 | 0.245 | −23.0 | ND | ND |
| Cur-NPs | 127.4±2.2 | 0.105±2.2 | −23.1±1.7 | 99 | 7 |
| Cur-NPs-APgp | 132.4±1.5 | 0.091±1.5 | −40.3±6.1 | 60 | 5 |
ND=Not determined
In addition, with this formulation procedure, the Cur samples encapsulated in NPs were completely dissolved in aqueous solution with no aggregation, unlike those observed in free Cur, which exhibited poor aqueous solubility (Figure 3A).
Figure 3.

Nano-encapsulation renders Cur completely dispersible in aqueous media. The figure shows solubility of Cur compared with Cur-NPs and Cur-NPs-APgp in aqueous solution (A). In vitro Cur release kinetic profile from Cur-NPs in 50% v/v of ethanol at 37 °C in shaking incubator at 70 rounds per minute over a period of 10 d (B). Transmission electron microscope image of NPs, Cur-NPs, and Cur-NPs-Apgp, respectively. TEM scanning shows the formation of spherical and smooth particles (C).
In vitro release kinetic profile
Cumulative percentage of Cur released from Cur-NPs is shown in Figure 3B. On the first day, only 40% of Cur was released from the nanoparticles and increased up to 80% on the third days and reached 90% in 5 d, which was a maximum. After that, the sustained-Cur was released at about 80% up to 10 d.
Surface morphology of NPs
The surface morphology of the nanosphere encapsulated Cur was determined by TEM. Figure 3C illustrates a TEM scanning and shows the formation of spherical and smooth particles. The average size of Cur-NPs and Cur-NPs-APgp were 125 nm and 130 nm, respectively, which correlated to that which was observed using photon correlation spectroscopy (Zeta Sizer).
Specific binding of Cur-NPs-APgp to KB-V1 and KB-3-1cells
To determine the specific binding of anti-P-gp antibody conjugated NPs on KB-V1 cells (high expression of P-gp), KB-V1 and KB-3-1 cells were incubated with Cur-NPs-APgp for various time points and then fluorescence intensity was determined by flow cytometry. Figure 4 shows that the interaction of Cur-NPs-APgp to KB-V1 cells was substantially greater than to KB-3-1 cells. The fluorescence intensity in KB-V1 cells was higher than that in the KB-3-1 cells when the cells were treated with Cur-NPs-APgp for 5, 30 and 60 min.
Figure 4.

Specific binding of Cur-NPs-APgp to KB-V1 cells determined by FACS. Gray and blank histograms represent Cur-NPs-APgp targeting to KB-V1 and KB-3-1 cells, respectively (A) and show the fluorescence intensity in bar graph (B). All assays have been demonstrated in duplicate and the mean±standard deviations are shown. bP<0.05 vs KB-3-1 cells.
Cellular uptake of NPs by KB-V1 and KB-3-1 cells
In order to investigate the cellular uptake of Cur encapsulation in NPs, KB-V1 and KB-3-1 were treated with Cur-NPs, Cur-NPs-APgp, and Cur-NPs-IgG for 30 and 60 min. As shown in Figure 5, the fluorescence intensity of Cur in Cur-NPs-APgp-treated KB-V1 cells was higher than Cur-NPs-APgp-treated KB-3-1 cells at 60 min. In contrast to Cur-NPs-APgp-treated cells, there was no difference in the fluorescence intensity between Cur-NPs and Cur-NPs-IgG treated-KB-V1 and KB-3-1 cells at 60 min (Figure 5B).
Figure 5.

The cellular uptake of Cur-NPs-APgp and Cur-NPs and Cur-NPs-IgG in KB-V1 and KB-3-1 cells. Cells were incubated with 10 μg/mL Cur-NPs-APgp or Cur-NPs and Cur-NPs-IgG for 30 (A) and 60 min (B), and the fluorescence levels were determined by a fluorescence microscope.
Cytotoxicity of nanoparticle to KB-V1 and KB-3-1 cells
To determine the cytotoxicity of NPs, NPs-APgp, free Cur, Cur-NPs and Cur-NPs-APgp, KB-V1 and KB-3-1 cells were treated with NPs, NPs-APgp, free Cur, Cur-NPs, or Cur-NPs-APgp for 6 h and cell viability was then determined by MTT assay. In KB-V1 cells (Figure 6A), Cur-NPs-APgp was found to be the most effective in inducing cancer cell death among three different Cur formations. Free Cur, Cur-NPs, and Cur-NPs-APgp reduced KB-3-1 cell viability in a dose-dependent manner, but the difference did not differ between treatment groups (Figure 6B) and IC50 of free Cur, Cur-NPs, and Cur-NPs-APgp in KB-3-1 and KB-V1 was more than 27 μmol/L, while NPs and NPs-APgp did not reduce cell viability when compared with the control (data not shown). For time interval treatment, the cells were treated with 27 μmol/L of free Cur, Cur-NPs, and Cur-NPs-APgp for 1, 6, or 24 h, after that cultured in the drug free medium for another 23, 18 and 0 h, respectively. The result showed that Cur-NPs-APgp had the most potential to induce cancer cell death among the three different Cur formations. Cur-NPs-APgp at 1 and 6 h showed a significant ability to induce cancer cell death when compared with free Cur and control in KB-V1 cell, while Cur-NPs showed significant ability to induce cancer cell death when compared only with the control, whereas at 24 h both the Cur-NPs and Cur-NPs-APgp showed significant ability to induce cancer cell death when compared with free Cur and the control (Figure 7).
Figure 6.

Cytotoxicity of Cur, Cur-NPs, and Cur-NPs-APgp in KB-V1 (A) and KB-3-1 (B) cells. The cells were incubated with the compounds for 6 h, and the cytotoxicity was then determined by MTT assay. All assays have been demonstrated in triplicate and the mean±standard deviations are shown.
Figure 7.

Cytotoxicity of 27 μmol/L Cur, Cur-NPs, and Cur-NPs-APgp in KB-V1 cells. The cells were incubated with the compounds for 1, 6, or 24 h, after that cultured in the drug free medium for another 23, 18, and 0 h, respectively. The cytotoxicity was determined by MTT assay at the end of time point. All assays have been demonstrated in triplicate and the mean±standard deviations are shown. cP<0.01 vs control; eP<0.05, fP<0.01 vs Cur.
Discussion
Curcumin (Cur), a naturally occurring product, has exhibited potent anti-cancer activities in various models which can be used in cancer therapy33. Although Cur showed multiple medicinal benefits, but revealed a low oral bioavailability of Cur34, which continues to be highlighted as a major challenge in developing its formation for clinical efficacy. Cur encapsulated to nanoparticles is one of the possible ways to increase its solubility and bioavailability30. In this direction, various nanocurcumin including liposome, micelles, and biodegradable polymer NPs have been developed and most of them involve passive targeting35, 36. However, actively targeting nanocurcumin has not yet been demonstrated. Targeted drug delivery represents a potential approach to further enhance anti-tumor efficacy. P-gp is minimally expressed on normal tissue, but significantly upregulated on MDR cancer cells37. Targeted drug delivery to MDR cancer cells is considered a strategy for the increased drug sensitivity of the cancer cells via enhancement of drug accumulation and cellular uptake.
In the current report, we prepared Cur-encapsulated PLGA NPs based on the nanoprecipitation technique. Pluronic is a widely known stabilizer for PLGA-NPs and many other pharmaceutical formations. To conjugate the antibody to the surface of NPs, we converted the hydroxyl groups of pluronic to carboxyl groups and conjugated the anti-P-gp antibody to the surface of NPs using the EDC-NHS carbodiimide reaction. Particles in the 100 to 200 nm range avoid premature clearance by the reticuloendothelial system16. We have prepared Cur-NPs and Cur-NPs-APgp with a mean diameter of less than 200 nm and the polydispersity index (PDI) of both was near 0.1. When the PDI value is less than 0.3, this indicated that the distribution of the sample particle size was narrow. These finding demonstrated that the small mean particle size and the uniform particle distribution were observed in Cur-NPs and Cur-NPs-APgp. Our NPs preparation (Cur-NPs) provided smaller size (127 nm) and higher entrapment efficiency (99%) compared to previous study30 which used emulsion evaporation method. The mean size of the Cur-NPs and Cur-NPs-APgp was confirmed by TEM. The diameter of the particles, observed in TEM (125 and 130 nm, respectively), was smaller than those that were detected by the PCS method. This is due to the fact that the TEM observation of the NPs was done in their dry form (air dried particles). The zeta potential values of Cur-NPs and Cur-NPs-APgp was negative, which is typical of the presence of PLGA terminal carboxylic residues. However, the value of this negative charge is much lower than that of the classical PLGA-NPs, which has been attributed to the presence of the carboxylic group of the pluronic on the surface of the NPs. The entrapment efficiency was lower in Cur-NP-APgp compared to that in Cur-NPs, probably because of the washing of NPs suspension during the antibody conjugation process, which was consistent with the literature that targeted NPs via carbodiimide reaction38 and the percent conjugation of anti-P-gp antibody on NPs surface was 22% as described in our previous study32. Unlike free Cur, Cur-NPs formulations have shown improved solubility in aqueous solution. The prepared particles were further evaluated for in vitro Cur releasing. In the present case, considering the solubility of Cur in ethanol, a 50% v/v of ethanol was used as the release medium. The release of Cur from Cur-NPs displayed a rapid release in the early phase, and reached a maximum at 5 d of up to 90%, and then gradually slowed down. The sustained Cur release was about 80%, until 10 d.
In order to obtain the antibody conjugated NPs, which are able to target desired cells, the conjugation procedure must preserve the biological activity of targeting ligand. We performed flow cytometry to evaluate the recognition properties of Cur-NPs-APgp to P-gp antigen, and found that the anti-P-gp immunoparticles bound specifically to KB-V1 cells, but exhibited less binding to KB-3-1 negative control cells.
It has been demonstrated that the therapeutic effect of drug-loaded NPs is depended on the internalization and sustained retention of the NPs. By combining the tumor targeting properties of the antibody with NPs, immunoparticles offer the promise of selective drug delivery to tumor cells, increased internalization and an intracellular drug release within targeted cells. Herein, we used Cur as a fluorescence probe to follow the NPs in the cellular uptake experiment. The intensity of Cur-NPs-APgp in KB-V1 was higher than in KB-3-1 cells, while the fluorescence intensity of Cur-NPs and Cur-NPs-IgG between both the cells did not differ. The higher intensity of fluorescence demonstrated the higher amount of Cur-NPs bound or taken up by the cells. To confirm the specificity of NPs conjugated with anti-P-gp antibody, another control using random IgG modified NPs in study of the cellular uptake (Figure 5) showed that IgG-modified nanoparticles did not demonstrate any differences of cellular uptake of Cur in KB-V1 cells compared to KB-3-1 cells. This finding suggested that Cur-NPs-APgp specifically bound to P-gp on KB-V1 cell surface, and enhanced the cellular uptake of Cur-NPs into MDR cancer cells (P-gp overexpressing cancer cells).
We observed significantly higher cytotoxicity of Cur-NPs-APgp treatment in KB-V1 cells when compared to free-Cur, while all three forms of Cur did not show significant cytotoxicity in KB-3-1 cells. When KB-V1 cells were treated with Cur-NPs-APgp at 1 and 6 h, cell death was induced at a greater rate than free Cur and Cur-NPs. These results were consistent with cellular uptake results. The superior cytoxicity of Cur-NPs-APgp in MDR-cancer cells was apparently due to anti-P-gp on the surface of NPs, and could help Cur-NPs-APgp to target KB-V1 cells while rapidly providing an increase of cell internalization efficiency and cytotoxicity in KB-V1 cells. However, the cytotoxicity of Cur-NPs-APgp and Cur-NPs was not markedly different that might be because of the multiple-NH2 groups in an antibody and the conjugation of anti-P-gp antibody is not specific to amino-terminus thus has no control on orientation of antibody after the antibody was linked to the NPs. Therefore the accessibility of anti-P-gp antibody on NPs to P-gp probably was reduced. Nevertheless, our study revealed the specificity of Cur-NPs-APgp to KB-V1, P-gp-overexpressed multidrug resistance cell line, and the conjugation of anti-P-gp antibody to Cur-NPs did not decrease their cellular uptake and also cytotoxicity.
The development of the P-gp targeted drug delivery system, Cur-NPs-APgp has shown efficacy in specific binding and internalization to the multidrug resistant cell line (KB-V1), and therefore provide the way to deliver Cur or anti-cancer drugs to targeted cells. Moreover, the formation of Cur-NPs could improve the solubility of Cur, sustain and prolong the release from Cur-NPs in vitro. An effective drug delivery system renders Cur induced target cell death, even when used at low doses. For further study, the combination of anti-cancer drugs and nanocurcumin against cancer cells has to be investigated. Additionally, in vivo studies are required to determine whether Cur-NPs-APgp are suitable as drug delivery agents, and could be a promising alternative to administered drug delivery systems for cancer therapy.
Author contribution
Wanisa PUNFA carried out all experiments, acquisition and analysis of data and wrote the manuscript; Supachai YODKEEREE and Pornsiri PITCHAKARN provided technical support; Chadarat AMPASAVATE provided technical support and some instruments; and Pornngarm LIMTRAKUL designed and supervised the study, and wrote and revised the manuscript. All authors have read and approved the final manuscript.
Acknowledgments
This work was funded by the Royal Golden Jubilee PhD Program of Thailand and the National Nanotechnology Center, National Science and Technology Development Agency of Thailand.
References
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Fujita T, Washio K, Takabatake D, Takahashi H, Yoshitomi S, Tsukuda K, et al. Proteasome inhibitors can alter the signaling pathways and attenuate the P-glycoprotein-mediated multidrug resistance. Int J Cancer. 2005;117:670–82. doi: 10.1002/ijc.21063. [DOI] [PubMed] [Google Scholar]
- Limtrakul P, Khantamat O, Pintha K. Inhibition of P-glycoprotein activity and reversal of cancer multidrug resistance by Momordica charantia extract. Cancer Chemother Pharmacol. 2004;54:525–30. doi: 10.1007/s00280-004-0848-4. [DOI] [PubMed] [Google Scholar]
- Pitchakarn P, Ohnuma S, Pintha K, Pompimon W, Ambudkar SV, Limtrakul P. Kuguacin J isolated from Momordica charantia leaves inhibits P-glycoprotein (ABCB1)-mediated multidrug resistance. J Nutr Biochem. 2012;23:76–84. doi: 10.1016/j.jnutbio.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanmahasathien W, Ampasavate C, Greger H, Limtrakul P. Stemona alkaloids, from traditional Thai medicine, increase chemosensitivity via P-glycoprotein-mediated multidrug resistance. Phytomedicine. 2011;18:199–204. doi: 10.1016/j.phymed.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Romiti N, Tongiani R, Cervelli F, Chieli E. Effects of curcumin on P-glycoprotein in primary cultures of rat hepatocytes. Life Sci. 1998;62:2349–58. doi: 10.1016/s0024-3205(98)00216-1. [DOI] [PubMed] [Google Scholar]
- Anuchapreeda S, Leechanachai P, Smith MM, Ambudkar SV, Limtrakul PN. Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem Pharmacol. 2002;64:573–82. doi: 10.1016/s0006-2952(02)01224-8. [DOI] [PubMed] [Google Scholar]
- Singh S, Khar A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med Chem. 2006;6:259–70. doi: 10.2174/187152006776930918. [DOI] [PubMed] [Google Scholar]
- Chearwae W, Anuchapreeda S, Nandigama K, Ambudkar SV, Limtrakul P. Biochemical mechanism of modulation of human P-glycoprotein (ABCB1) by curcumin I, II, and III purified from Turmeric powder. Biochem Pharmacol. 2004;68:2043–52. doi: 10.1016/j.bcp.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351:19–29. doi: 10.1016/j.jcis.2010.05.022. [DOI] [PubMed] [Google Scholar]
- Cirstoiu-Hapca A, Buchegger F, Bossy L, Kosinski M, Gurny R, Delie F. Nanomedicines for active targeting: physico-chemical characterization of paclitaxel-loaded anti-HER2 immunonanoparticles and in vitro functional studies on target cells. Eur J Pharm Sci. 2009;38:230–7. doi: 10.1016/j.ejps.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Cheng Y, C Samia A, Meyers JD, Panagopoulos I, Fei B, Burda C. Highly efficient drug delivery with gold nanoparticle vectors for in vivo photodynamic therapy of cancer. J Am Chem Soc. 2008;130:10643–7. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, et al. Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets. 2004;8:335–53. doi: 10.1517/14728222.8.4.335. [DOI] [PubMed] [Google Scholar]
- Lei T, Srinivasan S, Tang Y, Manchanda R, Nagesetti A, Fernandez-Fernandez A, et al. Comparing cellular uptake and cytotoxicity of targeted drug carriers in cancer cell lines with different drug resistance mechanisms. Nanomedicine. 2011;7:324–32. doi: 10.1016/j.nano.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartlick H, Michaelis K, Balthasar S, Strebhardt K, Kreuter J, Langer K. Highly specific HER2-mediated cellular uptake of antibody-modified nanoparticles in tumour cells. J Drug Target. 2004;12:461–71. doi: 10.1080/10611860400010697. [DOI] [PubMed] [Google Scholar]
- Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120:18–26. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Debotton N, Zer H, Parnes M, Harush-Frenkel O, Kadouche J, Benita S. A quantitative evaluation of the molecular binding affinity between a monoclonal antibody conjugated to a nanoparticle and an antigen by surface plasmon resonance. Eur J Pharm Biopharm. 2010;74:148–56. doi: 10.1016/j.ejpb.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Ng IO, Liu CL, Fan ST, Ng M. Expression of P-glycoprotein in hepatocellular carcinoma. A determinant of chemotherapy response. Am J Clin Pathol. 2000;113:355–63. doi: 10.1309/AC1M-4TY4-U0TN-EN7T. [DOI] [PubMed] [Google Scholar]
- Abolhoda A, Wilson AE, Ross H, Danenberg PV, Burt M, Scotto KW. Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin Cancer Res. 1999;5:3352–6. [PubMed] [Google Scholar]
- Wang G, Uludag H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv. 2008;5:499–515. doi: 10.1517/17425247.5.5.499. [DOI] [PubMed] [Google Scholar]
- Mukerjee A, Vishwanatha JK. Formulation, characterization and evaluation of curcumin-loaded PLGA nanospheres for cancer therapy. Anticancer Res. 2009;29:3867–75. [PubMed] [Google Scholar]
- Bennewitz MF, Saltzman WM. Nanotechnology for delivery of drugs to the brain for epilepsy. Neurotherapeutics. 2009;6:323–36. doi: 10.1016/j.nurt.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht S, Feldmann G, Soni S, Ravi R, Karikar C, Maitra A. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): a novel strategy for human cancer therapy. J Nanobiotechnology. 2007;5:3. doi: 10.1186/1477-3155-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Nair HB, Sung B, Kunnumakkara AB, Yadav VR, Tekmal RR, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79:330–8. doi: 10.1016/j.bcp.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xie X, Tao Q, Zou Y, Zhang F, Guo M, Wang Y, et al. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: characterizations and mechanisms. J Agric Food Chem. 2011;59:9280–9. doi: 10.1021/jf202135j. [DOI] [PubMed] [Google Scholar]
- Guerrouache M, Karakasyan C, Gaillet C, Canva M, Millot MC. Immobilization of a functionalized poly(ethylene glycol) onto beta-cyclodextrin-coated surfaces by formation of inclusion complexes: Application to the coupling of proteins. J Appl Polym Sci. 2006;100:2362–70. [Google Scholar]
- Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37:141–50. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh J, Ankola DD, Beniwal V, Singh D, Kumar MN. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci. 2009;37:223–30. doi: 10.1016/j.ejps.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Wieder ME, Hone DC, Cook MJ, Handsley MM, Gavrilovic J, Russell DA. Intracellular photodynamic therapy with photosensitizer-nanoparticle conjugates: cancer therapy using a 'Trojan horse'. Photochem Photobiol Sci. 2006;5:727–34. doi: 10.1039/b602830f. [DOI] [PubMed] [Google Scholar]
- Iangcharoen P, Punfa W, Yodkeeree S, Kasinrerk W, Ampasavate C, Anuchapreeda S, et al. Anti-P-glycoprotein conjugated nanoparticles for targeting drug delivery in cancer treatment. Arch Pharm Res. 2011;34:1679–89. doi: 10.1007/s12272-011-1012-4. [DOI] [PubMed] [Google Scholar]
- Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell Div. 2008;3:14. doi: 10.1186/1747-1028-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–18. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- Singh R, Lillard JW Jr. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86:215–23. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou M, Men K, Shi H, Xiang M, Zhang J, Song J, et al. Curcumin-loaded biodegradable polymeric micelles for colon cancer therapy in vitro and in vivo. Nanoscale. 2011;3:1558–67. doi: 10.1039/c0nr00758g. [DOI] [PubMed] [Google Scholar]
- Matheny CJ, Lamb MW, Brouwer KR, Pollack GM. Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Pharmacotherapy. 2001;21:778–96. doi: 10.1592/phco.21.9.778.34558. [DOI] [PubMed] [Google Scholar]
- Cirstoiu-Hapca A, Bossy-Nobs L, Buchegger F, Gurny R, Delie F. Differential tumor cell targeting of anti-HER2 (Herceptin) and anti-CD20 (Mabthera) coupled nanoparticles. Int J Pharm. 2007;331:190–6. doi: 10.1016/j.ijpharm.2006.12.002. [DOI] [PubMed] [Google Scholar]