

Abstract
Recent genome-sequencing studies in human neurodevelopmental and psychiatric disorders have uncovered mutations in many chromatin regulators. These human genetic studies, along with studies in model organisms, are providing insight into chromatin regulatory mechanisms in neural development and how alterations to these mechanisms can cause cognitive deficits, such as intellectual disability. We discuss several implicated chromatin regulators, including BAF (also known as SWI/SNF) and CHD8 chromatin remodellers, HDAC4 and the Polycomb component EZH2. Interestingly, mutations in EZH2 and certain BAF complex components have roles in both neurodevelopmental disorders and cancer, and overlapping point mutations are suggesting functionally important residues and domains. We speculate on the contribution of these similar mutations to disparate disorders.
The specialization that cells must acquire to form an organism from a single zygote is achieved by stepwise changes in gene expression throughout the course of development. These changes occur in response to both extracellular signals and cell-intrinsic genetic circuitries. Chromatin regulators contribute to dynamic changes in gene expression but also maintain cell fates by providing stable, heritable states of gene expression1–3. Many chromatin regulators are essential for developmental processes, including the development of the brain, on which this Review focuses (FIG. 1).
Figure 1. Chromatin regulators have essential roles throughout neural development.
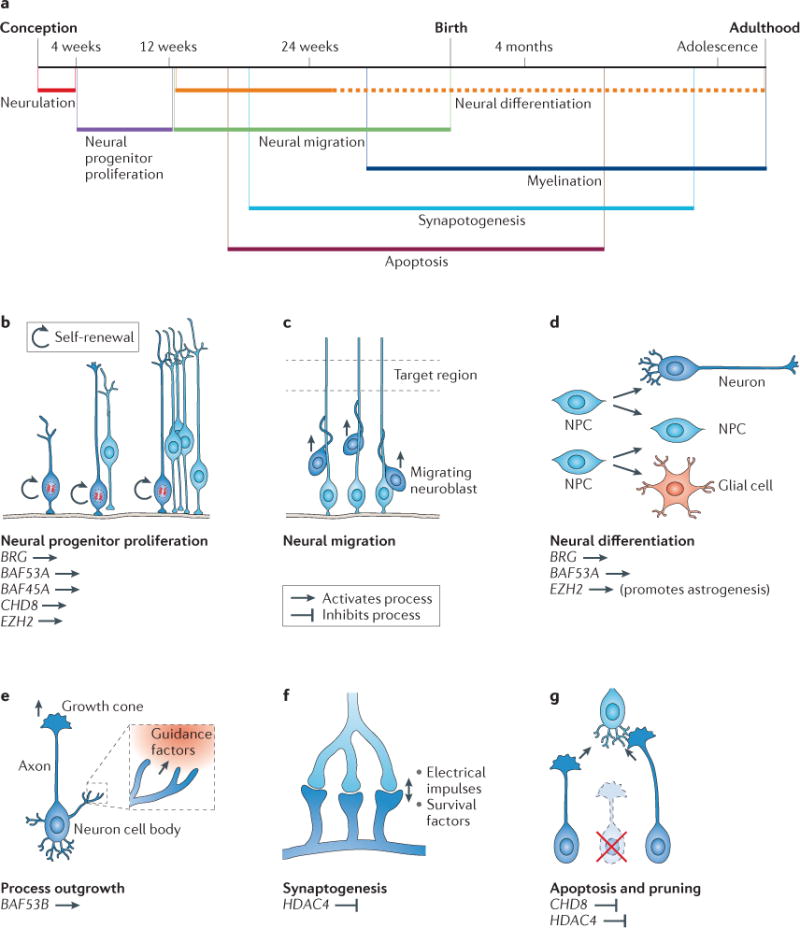
The fundamental processes of neural development are illustrated. Chromatin regulators discussed in this Review are noted under the processes in which they have important roles. The key indicates whether a particular regulator promotes or inhibits each neurodevelopmental process. a | A timeline of human neural development. b | The development of the vertebrate nervous system begins during gastrulation. In the early embryo, neural progenitor cells undergo symmetrical proliferative division. c | With the expansion of the number of cell types and the size of the nervous system, the cell bodies of both neural progenitors and resulting postmitotic neurons migrate away from their birthplace to appropriate regions in response to environmental cues. d | Neural progenitors asymmetrically divide to give rise to neurons, glial cells or intermediate progenitors. Neural differentiation generates enormous numbers of diverse cell types in the nervous system. e | After migrating neurons have reached their destinations, they extend axonal and dendritic processes, which are guided by intricate cellular interactions and guidance molecules to appropriate target regions, where they further elaborate processes to cover receptive fields and innervate targets. f | Mature synapses are formed between neurons that are connected to each other. Synaptogenesis begins during embryonic development, but subsequent synaptic stabilization and plasticity occur throughout life and are adaptive to learning experiences and other activity-dependent environmental inputs. g | Active apoptosis and local degenerative pruning events maintain and refine established neuronal morphologies and neural circuit assembly. NPC, neural progenitor cell. Part a is modified, with permission, from REF. 107 © American Psychiatric Association.
At least three mechanisms regulate the assembly and biological states of chromatin. ATP-dependent chromatin remodellers alter the physical state of chromatin probably by moving nucleosomes in relation to the DNA or exchanging nucleosomes into and out of DNA4,5. Chromatin modifiers — enzymes that alter the tails of histones projecting from nucleosomes — control the accessibility of DNA to regulatory mechanisms6,7. Additionally, modified histones may recruit chromatin remodellers and other proteins to chromatin. Therefore, chromatin remodeller ‘readers’ and histone modification ‘writers’ and ‘erasers’ work in concert to regulate chromatin structure and gene expression. We refer to them collectively as chromatin regulators. DNA methylation also contributes to gene regulation; its involvement in neurodevelopmental disorders has previously been reviewed and is not discussed here8.
Several major events and processes must be precisely orchestrated during normal brain development (FIG. 1); misregulation due to a genetic or environmental insult can result in cognitive deficits and other features of neurodevelopmental disorders. Insights into the role of chromatin in neural development are rapidly emerging from human disease studies using new sequencing and analytical technologies and from more traditional studies of model organisms. A consensus is emerging that chromatin regulatory mechanisms have a key role in many of the major events during neural development9–13 (FIG. 1).
So far, dozens of mutated chromatin regulators have been causally implicated in human neurodevelopmental and psychiatric disorders, such as autism spectrum disorder (ASD) and schizophrenia (TABLE 1 and Supplementary information S1 (table)). Interestingly, mutations of several chromatin regulators discussed here in a neurodevelopmental context are also involved in human cancer; the implication of this coincidence will be speculated on, but it awaits future investigation (BOX 1).
Table 1.
Chromatin regulators mutated in human mental disorders
| Chromatin regulator* | Disease (or diseases) | Mutations | Chromatin regulator type | Refs |
|---|---|---|---|---|
| BAF250A (ARID1A; BAF complex) | Coffin–Siris syndrome | Nonsense, frameshift indel | Chromatin-remodelling complex subunit | 29 |
| BAF250B (ARID1B; BAF complex) | Intellectual disability, Coffin–Siris syndrome, autism, schizophrenia | Translocation, frameshift indel, nonsense, missense, microdeletion | Chromatin-remodelling complex member | 32,33,34,28,36 |
| BRM (SMARCA2; BAF complex) | Coffin–Siris syndrome, Nicolaides–Baraitser syndrome, schizophrenia | Partial deletion, missense, intronic alteration | Chromatin-remodelling complex ATPase | 31,30,39,40 |
| BRG1 (SMARCA4; BAF complex) | Coffin–Siris syndrome | Missense | Chromatin-remodelling complex ATPase | 29 |
| BAF47 (SMARCB1; BAF complex) | Coffin–Siris syndrome, Kleefstra’s syndrome phenotypic spectrum | In-frame deletion, missense | Chromatin-remodelling complex subunit | 29,108 |
| BAF155 (SMARCC1; BAF complex) | Autism | Missense | Chromatin-remodelling complex subunit | 35 |
| BAF170 (SMARCC2; BAF complex) | Autism | Splice site mutation | Chromatin-remodelling complex subunit | 35 |
| BAF180 (PBRM; BAF complex) | Autism | Missense | Chromatin-remodelling complex subunit | 36 |
| CHD7 | CHARGE syndrome, autism | Missense | Chromatin remodeller | 36,109 |
| CHD8 | Autism | Nonsense, frameshift indel missense | Chromatin remodeller | 35,36,51,52 |
| ATRX | X-linked α-thalassaemia/mental retardation syndrome | Missense | Chromatin remodeller | 110 |
| p300 | Rubinstein–Taybi syndrome | Large deletions and duplications, misssense | Histone acetyltransferase | 111 |
| CBP | Rubinstein–Taybi syndrome | Microdeletions, nonsense | Histone acetyltransferase | 112 |
| KAT6B (MYST4 or MORF) | Say–Barber–Biesecker–Young–Simpson syndrome (SBBYSS or Ohdo’s syndrome) | Frameshift indel, missense | Histone acetyltransferase | 113 |
| HDAC4 | Brachydactyly mental retardation syndrome | Balanced chromosomal translocation; deletion | Histone deacetylase | 87,88 |
| EZH2 | Weaver’s syndrome (learning disability) | Missense, frameshift indel | Histone methyltransferase | 74,75 |
| EHMT1 | Kleefstra’s syndrome phenotypic spectrum; autism | Microdeletions, nonsense, frameshift, missense | Histone methyltransferase | 36,108,114 |
| MLL | Wiedemann–Steiner syndrome | Nonsense | Histone methyltransferase | 115 |
| MLL2 | Kabuki’s syndrome | Nonsense, frameshift | Histone methyltransferase | 116,117 |
| MLL3 | Autism, Kleefstra’s syndrome | Missense, nonsense | Histone methyltransferase | 35,36,108 |
| KDM5C (JARID1C) | Non syndromic X-linked mental retardation | Missense, frameshift, nonsense, intronic alteration | Histone lysine demethylase | 118,119 |
| PHF8 | X-linked mental retardation | Missense mutation, nonsense | Histone lysine demethylase | 120,121 |
| HUWE1 | XLMR Turner type | Duplications, missense, copy number gains | Histone ubiquitylation | 122 |
| MECP2 | Rett’s syndrome, Angelman’s syndrome, nonsyndromic X-linked mental retardation, autism | Missense, nonsense, frameshift indel, duplication | DNA methylation binding protein | 123,124 |
| MBD5 | Autism, Kleefstra’s syndrome | Frameshift indel, nonsense | DNA methylation binding protein | 35,36,108 |
| MED12 | Lujan–Fryns syndrome, FG syndrome (also known as Opitz–Kaveggia syndrome) | Missense | REST mechanism for disease, Mediator complex subunit | 125,126 |
| MED23 | Non-syndromic intellectual disability | Missense | Mediator complex subunit | 127 |
| PHF21A (BHC80) | Intellectual disability, craniofacial anomalies | Translocation and deletion | Chromatin reader, histone deacetylase complex member | 128 |
Protein aliases are given in brackets; if the protein is a member of a complex, the complex name is also given in brackets. EZH2, enhancer of zeste 2; HDAC4, histone deacetylase 4.
Box 1. Coincidence of mutations in cancer and neurodevelopmental disorders.
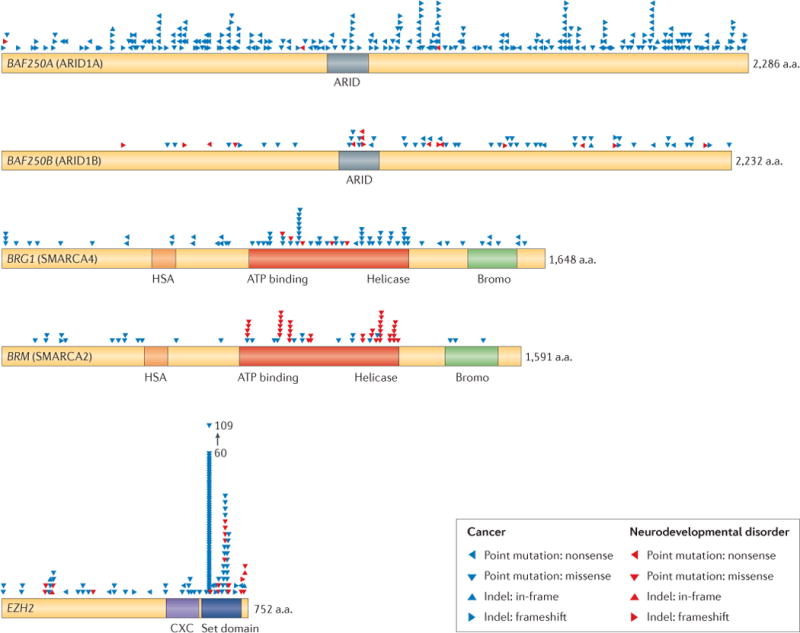
Recently, 44 human genome-sequencing studies have implicated mutations in BAF subunits as potential drivers in human cancer, including some neurological cancers. So far, more than 20% of human cancers are estimated to have mutations in BAF subunits, making BAF complexes the most commonly mutated chromatin regulator in human cancer102,103. Strikingly, many of the mutations are similar to those in neurodevelopmental disorders.
BAF250A is the most commonly mutated BAF subunit in human cancer103–105. Frameshift and nonsense mutations of BAF250A and BAF250B, which occupy the same subunit position, are widespread, indicating that there is probably a loss of function for many alleles (see the figure). However, the reported missense mutations may highlight regions of BAF250A and BAF250B with unknown function and should guide further studies of shared and disparate roles of the two homologues. BAF250 subunits are not necessary for in vitro chromatin remodelling, so novel mechanistic studies may be required to understand the roles of these subunit homologues in neurodevelopment and cancer.
BRG and BRM missense mutations in cancers, Coffin–Siris syndrome (CSS) and Nicolaides–Baraitser syndrome (NBS) highlight the ATPase and helicase domain as a hotspot for genetically dominant, probably dominant-negative, mutations (see the figure). The concentration of mutations in this domain reinforces its fundamental cell biologic role. BRG and BRM are highly homologous and are co-expressed in many tissues, including the brain, yet there are no reported BRG mutations in NBS and no reported BRM mutations in CSS. BRG is more frequently mutated in cancer, including medulloblastoma, which is a brain cancer. It seems likely that BRG- or BRM-containing complexes may mediate different functions in neural development, as very similar mutations in each protein lead to different diseases.
Enhancer of zeste 2 (EZH2) mutations have been implicated as drivers of myeloid malignancies106. Some of the germline EZH2 mutations identified in patients with Weaver’s syndrome are identical to those that are found in somatic cancer mutations. It is not clear whether tumorigenesis is common in Weaver’s syndrome, as it is a rare syndrome, and most reports include only young patients. Analysis of EZH2 missense mutations in cancer and Weaver’s syndrome reveal that the carboxy-terminal SET domain, the methyltransferase, is most highly mutated (see the figure), reinforcing its important biological role.
The group of mutations in the figure highlights the mystery of how mutational context can lead to very different outcomes. The context of mutations, including timing, genetic background and cell type, may contribute to the differing disease phenotypes, despite similar protein defects. More information about the displayed mutations can be found in Supplementary information S2 (table).
In this Review, we highlight several chromatin regulators that were recently implicated in human disorders of brain function and that have not been extensively reviewed elsewhere. Additionally, genetic studies in model organisms have provided insight into their roles in normal development, which allows speculation on associated disease mechanisms. We also selected chromatin regulators for which mechanistic links through regulation of common pathways have been indicated. We first discuss selected chromatin remodellers, including SWI/SNF-like or BRG1- and BRM-associated factor (BAF) complexes and chromodomain helicase DNA-binding protein 8 (CHD8); then we discuss selected chromatin modifiers, including enhancer of zeste 2 (EZH2) and histone deacetylase 4 (HDAC4), speculating on underlying disease-causing mechanisms for these regulators on the basis of their known functions in neural development. Importantly, these four chromatin regulators are united by their genetic dominance in neurodevelopment, meaning that mutation in one allele can confer disease. We conclude our discussion with themes and interesting questions that have arisen from the study of chromatin regulators in neural development and disorders along with ideas for future research directions in this field.
Chromatin remodellers
The canonical role of chromatin remodellers is to alter the placement or position of nucleosomes through a catalytic process powered by ATP hydrolysis, thereby regulating transcription4. In vivo, however, there may be non-canonical roles for chromatin remodellers, such as regulating higher-order chromatin structure14. Recently, exome-sequencing studies have identified causative mutations in subunits of mammalian BAF complexes in patients with various neurodevelopmental and psychiatric disorders, as well as de novo mutations in the chromodomain helicase DNA-binding protein (CHD) remodeller family in patients with non-familial ASD.
BAF complexes in neural development
Mammalian SWI/SNF-like BAF complexes are ATP-dependent chromatin-remodelling complexes that are made up of 15 subunits. The core ATPase subunit can be either of the two homologues BRG1 (also known as SMARCA4) or BRM (also known as SMARCA2), and the rest of the subunits are known as BAFs. Because 9 of the 15 mammalian subunits are not present in the yeast SWI/SNF complex and because some subunits are homologous to those that are exclusively found in other yeast chromatin-remodelling complexes, we refer to them as BAF complexes rather than SWI/SNF complexes to avoid bias and extrapolation. Mammalian BAF complexes have several subunit positions (such as the ATPase position) that can be filled by one member from an expanded gene family. Through incorporation of 29 different human gene family members by combinatorial assembly, hundreds of distinct complexes are predicted to form, and studies have shown unique assemblies and biological specificities in different tissues15,16. Functional specificity is thought to be an emergent feature of the complex, reflecting composite surfaces of adjacent subunits, which may mediate differential BAF genome targeting and/or interaction with distinct partners and signalling pathways.
BAF complexes have important developmental roles in several tissue types outside the nervous system5. This widespread role of BAF complexes could explain the syndromic features of neurodevelopmental disorders associated with BAF subunit mutations. Distinct BAF complexes have been shown to bind to and to coordinate with tissue-specific transcription factors to regulate gene expression in resident cells15,17. For example, in neural progenitors, neural progenitor BAF (npBAF) complexes interact with the repressive transcription factor REST and its co-repressor to facilitate the inhibition of neuronal genes, thereby maintaining neural progenitor identity17.
BAF complexes have essential roles in the development of the mammalian nervous system. Mice that lack Brg18, Baf47 (also known as Smarcb1)19 and Baf155 (also known as Smarcc1)20 die in pre- or peri-implantation stages. Targeted deletion of Brg in the developing nervous system produces mice with smaller brains that lack a cerebellum16. Heterozygous mice with null alleles of Brg or Baf155 show defects in neural tube closure, indicating a dosage-sensitive role for BAF complexes in neural development18,20. Studies suggest that these defects might be due to failure of neural progenitor self-renewal and differentiation16,21 (FIG. 2a). The underlying mechanisms could be similar to those in Caenorhabditis elegans, in which BAF subunits — SWSN-1, SWSN-4, LET-526 and PBRM-1, which are homologues of BRG, BRM, BAF155 and BAF250, respectively — contribute to asymmetric division of precursor-like T blast cells to generate neural cells22,23.
Figure 2. BAF complex roles in neurodevelopment and disorders of brain function.
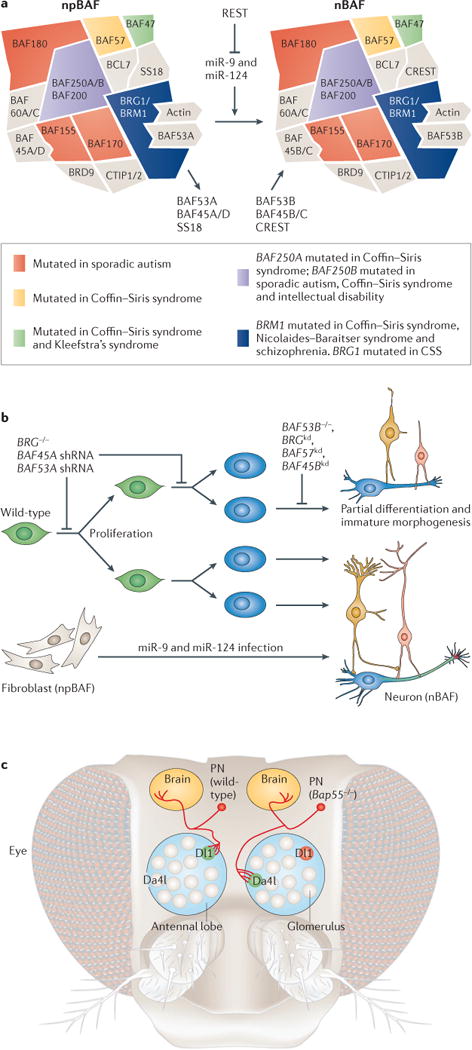
a | The composition of neural progenitor BAF (npBAF) and neuronal BAF (nBAF) complexes is indicated, along with the triple-negative genetic circuit that leads to npBAF to nBAF switching and their involvement in various disorders (right). b | Knockout (−/−) of BRG and knockdown (kd) of npBAF subunits BAF45A and BAF53A in neural progenitors (green cells) impede neural progenitor self-renewal and differentiation into postmitotic neurons (blue cells). Loss-of-function of BRG and BAF57 in the developing nervous system and nBAF subunits BAF45B and BAF53B affects activity-dependent process outgrowth of postmitotic neurons21. MicroRNA (miRNA)-mediated direct conversion of human fibroblasts to neurons recapitulates the switching of npBAF to nBAF complexes during normal neural development. miR-9, miR-9* and miR-124 act together by binding to independent sites in the BAF53A 3′ untranslated regions, functioning as a molecular AND gate for this developmental transition. c | Bap55, the homologue of mammalian BAF53A and BAF52B in Drosophila melanogaster, controls dendritic targeting of olfactory projection neurons (PNs; red). In comparison to wild-type PNs, which target glomerulus Dl1 (green, left), PNs lacking Bap55 are precisely mistargeted to an alternative glomerulus, Da4l (green, right), in the antennal lobe. This dendritic targeting occurs before the axonal patterning of the glomerulus and is thus thought to be mediated through genetic, cell-intrinsic mechanisms and not in response to particular guidance molecules. shRNA, short hairpin RNA.
During the development of the mammalian nervous system, an essential switch of BAF complex subunits occurs as neural progenitors exit mitosis and initiate differentiation16 (FIG. 2a). The mammalian npBAF subunits BAF45A and BAF53A are necessary for neural progenitor proliferation, whereas the neuronal subunits BAF53B and calcium-responsive transactivator (CREST; also known as SS18L1) are required for activity-dependent dendritic outgrowth and axonal development16,21 (FIG. 2b). The function of BAF53B in dendritic morphogenesis cannot be replaced by BAF53A, demonstrating the functional specialization of different BAF complex subunits21.
The switch of npBAF to neuronal BAF (nBAF) subunits is initiated when miR-9, miR-9* and miR-124 repress expression of BAF53A of the npBAF complex24 (FIG. 2b). REST — a transcriptional repressor that is selectively repressed in postmitotic neurons — negatively regulates these microRNAs (FIG. 2a). This triple-negative circuitry, which leads to the npBAF–nBAF switch, appears to occur in all neurons, suggesting that it is a fundamental process of neural development24. Excitingly, recent studies showed that synergistic effects of miR-9, miR-9* and miR-124 can convert human fibroblasts to neurons25 (FIG. 2b). This process recapitulates the BAF subunit switch to nBAF composition, suggesting that subunit switching has an instructive role in neuronal cell fate determination.
Recently, a demanding genetic screen for genes that produce perfect retargeting of olfactory dendritic trees to an incorrect glomerulus uncovered Bap55, the Drosophila melanogaster homologue of mammalian BAF53A and BAF53B. No other genes display this phenotype, and human BAF53B provides near-complete rescue of the phenotype, demonstrating its conserved function in dendritic targeting26 (FIG. 2c). Additionally, subunits of BAF complexes were discovered in an RNA interference (RNAi) screen for genes necessary to form and maintain specific dendritic morphologies in D. melanogaster27. These studies strongly support an evolutionarily conserved role for BAF complexes in dendritic development and also suggest that some of the human diseases produced by mutation of BAF subunits (see below) are related to incorrect dendritic targeting.
BAF mutations in disorders of brain function
The importance of BAF complexes in neural development is further underlined by recent exome-sequencing studies (FIG. 2A; TABLE 1), which identified ~100 mutated BAF subunit alleles affecting human brain development and function. For example, recent studies revealed multiple protein-truncating de novo mutations of BAF250B (also known as ARID1B) as the genetic cause of Coffin–Siris syndrome (CSS), which is a rare autosomal dominant disease that is characterized by intellectual disability with marked language impairment, microcephaly, coarse facial features and hypoplasia of the nail on the fifth finger and/or toe28. Another study identified additional de novo mutations of BRG, BRM, BAF47, BAF57 and BAF250A (also known as ARID1A) as the cause of CSS in 20 out of 23 patients (87%)29. Both BAF250B and BAF250A alleles probably cause haploinsufficiency as only nonsense and frameshift indel mutations were found in these patients. Conversely, BRG-, BRM-, BAF47- and BAF57-mutant alleles might be consistent with a gain-of-function or dominant-negative effect because all mutations are either missense or in-frame deletions.
The finding that mutations in multiple BAF subunits result in the same congenital syndrome underscores the fact that different subunits of BAF complexes coordinately regulate chromatin and gene expression as a functional unit. Because these mutations occur in common subunits of both npBAF and nBAF complexes, it could be speculated that some CSS phenotypes, such as microcephaly, may be attributable to the roles of npBAF complexes in neural progenitor proliferation and brain size observed in mouse models. In addition, the role of nBAF in dendritic morphogenesis and neural circuit wiring may underlie intellectual disabilities in these patients (see above). The systemic symptoms are probably attributable to some of the above-mentioned BAF functions outside the nervous system.
In addition to CSS, multiple de novo missense mutations and in-frame deletions of BRM were found in 36 of 44 individuals with Nicolaides–Baraitser syndrome (NBS), which includes features of intellectual disability, again with marked language impairment, microcephaly, epilepsy and morphological defects30,31. As mutations of BRM in both CSS and NBS occur in conserved domains that are thought to be responsible for nucleotide binding or ATP hydrolysis, it is possible that these mutations generate structurally unchanged but functionally defective BAF complexes, which might functionally compete with wild-type complexes. Furthermore, as BRM is a core ATPase of the npBAF and nBAF complexes, its mutation may contribute to microcephaly and intellectual disability as described for CSS above.
Mutations of BAF250B can also cause intellectual disability with a lower burden of syndromic features in other tissues32,33,34. Two balanced chromosomal translocations that cause truncating mutations of BAF250B occur in patients with agenesis of the corpus callosum, intellectual disability and language defects32,33. In addition, various de novo nonsense mutations, gene deletion and exon duplication alleles of BAF250B that are predicted to cause loss of function were discovered as frequent causes of sporadic intellectual disability34. Patients in this study showed moderate to severe intellectual disability and speech impairment but no agenesis of the corpus callosum. These studies suggest that haploinsufficiency of BAF250B underlies the aetiology of intellectual disability. The differing disease outcome of BAF250B haploinsufficiency in these patients compared to the syndromic intellectual disabilities discussed above could be due to genetic background and environmental factors.
De novo mutations of BAF155, BAF170 (also known as SMARCC2), BAF180 (also known as PBRM1) and BAF250B were noted in non-familial ASD35,36. ASD is characterized by impaired social interaction or communication and repetitive behaviours, sometimes accompanied by intellectual disability, with symptoms appearing in early childhood. Although these mutations each appeared only once in the ASD cohorts35,36, because the subunits are a part of one functional unit, BAF complex mutation is important. Mutations of REST, which is a part of the triple-negative circuitry leading to the switch of npBAF to nBAF subunits, were also identified in the same study, suggesting that the switching of BAF complexes in neural development contributes to autism.
Some cases of ASD are associated with symptoms that are apparent from birth, indicating that prenatal neurodevelopmental abnormalities have occurred, possibly through defective functions of npBAF complexes in neural progenitors. Another contributor to ASD characteristics might be postnatal synaptic dysfunction37,38, possibly caused by dysfunctional nBAF complexes, which regulate dendritic targeting and neural circuit formation21,26. Additionally, a potential connection between BAF complexes and CHD family proteins in modulating the WNT/β-catenin signalling pathway might be implicated in ASD (see ‘The role of CHD8 in ASD’ section below).
Two BRM risk alleles were identified in a genome-wide association study of schizophrenia single-nucleotide polymorphisms (SNPs) in a Japanese cohort39. Schizophrenia is a thought disorder that usually appears post-adolescence and is characterized by hallucinations, paranoia, delusions and disorganized speech. Reducing BRM levels in mouse cortical neurons leads to abnormal dendritic spine morphology40, a function that is controlled by nBAF complexes in postmitotic neurons. Brm-knockout mice showed impaired social interaction and prepulse inhibition39. These are both features of schizophrenia in humans that are thought to result from abnormalities in synaptic maturation, connectivity and plasticity40, which are processes that are influenced by nBAF function. Functional interactome studies using bioinformatics showed that Brm forms a network with the eight other genome-wide-supported schizophrenia-associated genes, further suggesting the potential involvement of Brm in schizophrenia40. These studies suggest that large-scale screening of schizophrenic populations for mutations in BAF subunits would be informative.
The discovery of human genetically dominant alleles of BAF subunits aligns with the heterozygous defects in mouse neural development discussed above and demonstrates a dosage-sensitive mechanism for BAF complex function in uncharacterized but crucial aspects of chromatin regulation during neural development. Perhaps the most interesting route to genetic dominance would be that these mutations are haploinsufficient by virtue of being involved in a rate-limiting process in neural chromatin regulation. The observation that directing the assembly of neuron-specific nBAF complexes can convert fibroblasts to functional neurons is consistent with their possible rate-limiting role in neuronal fate determination25, because rate-limiting roles often define decision points and hubs of regulation in biochemical cascades. Because several of the subunits implicated in neurodevelopmental disorders are not required for in vitro nucleosome remodelling, other functions, such as genomic targeting or opposition of Polycomb-mediated repression (see ‘The role of EZH2 in development’ section below), might contribute to these diseases.
The role of CHD8 during development
Another ATP-dependent chromatin remodeller for which genetic and biochemical studies have led to recent insights is CHD8. Chd8-knockout mice die around embryonic day 5.5 owing to widespread p53-induced apoptosis41, whereas in a WNT-responsive human colorectal carcinoma cell line, Chd8 knockdown leads to derepression of WNT/β-catenin pathway target genes42. Mammalian CHD8 is thought to repress β-catenin and p53 target genes by recruiting linker histone H1 (REFS 41,43,44).
In Xenopus laevis embryos, the CHD8 homologue Duplin negatively regulates WNT/β-catenin signalling, especially in the context of head and brain development43,45. The WNT/β-catenin signalling pathway also has important roles in mammalian neural development: increased dosage and knockout of the β-catenin protein in the brain lead to central nervous system defects46–48. There are not yet any genetic models to test the role of CHD8 in WNT/β-catenin signalling directly in mammalian brain development, although the above studies indicate that it may play an important part. Peptides from CHD8 were discovered in a proteomic analysis of mouse embryonic stem cell (ESC) BAF complexes, so the two may functionally interact15. Interestingly, BAF complexes interact with β-catenin and are important for activating β-catenin target genes49. Thus, BAF and CHD8 appear to regulate WNT/β-catenin signalling reciprocally, perhaps providing important modulation of this essential neural development signalling pathway48,50.
The role of CHD8 in ASD
Recent large-scale sequencing studies have uncovered a handful of genetic risk factors for autism35,36,51–53, many of which were present just once among the sequenced populations, making it difficult to determine whether they have any link to autism. Mutations in CHD8 and a few other factors, however, appeared in multiple cases from the patient cohorts. In addition to 13 mutated alleles of CHD8, a single mutant allele of CHD1, CHD2, CHD3, CHD5 and CHD7 each has been found in the same studies35,36,51 (TABLE 1 and Supplementary information S1 (table)). This number of de novo CHD8 mutations is highly unlikely by chance and means that mutation in one copy of CHD8 and maybe other members of the CHD family contribute to the risk for autism. So far, most of the mutations found in CHD8 are nonsense or frameshift indels that are thought to cause loss of function, suggesting haploinsufficiency to be the pathogenic mechanism. Macrocephaly was a common phenotype among the 12 CHD8 patients with ASD who had reported head circumference measurements36,51,52. Macrocephaly is estimated to occur in 15–35% of autistic children54, so this commonality could not be coincidental. If CHD8 function is impaired in these autism patients, one might expect an upregulation of β-catenin-regulated genes. When occurring in certain areas of the brain and/or in neural precursors, this might lead to a brain overgrowth phenotype that is similar to expanded cortex caused by expression of stabilized β-catenin in mouse brain46. Indeed, WNT/β-catenin-signalling pathways were previously implicated in the aetiology of autism owing to the prevalence of macrocephaly and genetic linkage of some common variants in the signalling pathway to autism50. As mentioned above, BAF complexes are also required for WNT/β-catenin signalling49, suggesting that CHD8 and BAF might be mechanistically linked in their contribution to ASD development.
The level of contribution of CHD8 to these particular cases of autism is unclear, as CHD8 mutations may be dominant and causal or a part of a polygenic contribution to this complex disorder. More genome-sequencing studies, in combination with bioinformatics and mathematical analyses, will allow the identification of epistatic interactions and genetic networks underlying ASD. However, these mutations suggest that chromatin remodellers are important for normal neural development owing to their ability to regulate the downstream targets of developmental signalling pathways. It is also possible that after biochemical characterization of CHD8-containing complexes, associated proteins and subunits will emerge as contributors to autism.
Chromatin modifiers
Chromatin modifiers are the ‘writer’ and ‘eraser’ enzymes that post-translationally alter histone proteins, mainly at their unstructured tails, by adding or removing acetylation, methylation, ubiquitylation and phosphorylation6,7,55. Certain modifications are associated with repressive chromatin structure (for example, histone H3 at lysine 27 (H3K27) and H3K9 methylation), whereas others are associated with greater chromatin accessibility (for example, acetylated histones and H3K4 methylation). The discovery of histone deacetylases56 and demethylases57 began a transition in chromatin biology from a view that chromatin modifications were epigenetic by virtue of largely being irreversible to the present concept that epigenetic states are actively maintained. This view is supported by the discovery that histone acetylation and methylation are rapidly reversible in living cells and require active maintenance58,59. It is important to note that histone modification enzymes have also been reported to modify non-histone proteins, so the complete identification of substrates will be important.
Many chromatin modifiers have been implicated in human neurodevelopmental and psychiatric disorders (TABLE 1 and Supplementary information S1 (table)), including the histone acetyltransferases CBP and p300, and the methyl-CpG-binding repressor MeCP2 in Rubinstein–Taybi syndrome and Rett’s syndrome, respectively; their mechanistic roles have been well-reviewed60–62. For many modifiers, however, studies are lagging in terms of understanding their functional involvement in neurodevelopmental processes. Here, we will focus on two chromatin modifiers associated with gene repression: EZH2 and HDAC4. We chose these modifiers because emerging evidence of their functional roles is allowing speculation on their mechanistic roles in human mental disorders. Additionally, EZH2 and BAF complexes have opposing roles in chromatin regulation and are both linked to WNT/β-catenin signalling, like CHD8, so comparing their involvement in neurodevelopmental mechanisms will be informative. In addition, studies have shown that blocking HDAC activity with inhibitors facilitated improved learning and memory in mice, and as mentioned before, HDACs can contribute to dynamic chromatin changes, so speculating on the role of an HDAC in cognitive aspects of neurodevelopmental disease is timely and will help to illuminate the role of dynamic chromatin in brain function.
The role of EZH2 in development
The EZH2 enzyme is a Polycomb group protein subunit of the Polycomb repressive complex 2 (PRC2), which methylates H3K27. This methylation mediates recruitment of additional PRC2 and in some cases PRC1 (which ubiquitylates histone H2A), resulting in propagation of repressive chromatin both spatially along the chromosome and through cellular generations63,64. Thus, these two complexes can function together to provide stable, heritable gene repression, and the PRC1 complex may also create a more compact chromatin structure65.
The gene repression mediated by Polycomb complexes is important in many developmental processes, including embryonic mouse development and maintenance of stem cell identity and execution of differentiation programmes66–68. In one conditional knockout model that provides almost complete EZH2 deletion by the onset of neurogenesis around day 12.5, there was an acceleration of neurogenesis in the cerebral cortex: more rapidly cycling neural precursors, an early increase in neuron numbers and precocious production of astrocytes69. However, this led to a decreased number of neurons at birth, possibly owing to early exhaustion of neural precursor cells. By contrast, when EZH2 is deleted at a later time in neural precursors, close to the shift from neurogenesis to astrogenesis, precursors continued to produce neurons rather than shift to astrocyte production. This was posited to result from lack of repression of the WNT/β-catenin target neurogenin 1, which is normally repressed by EZH2 in neural precursors70. Thus, EZH2 has a timing-dependent role in neurogenesis. It is not known whether EZH2 plays an important part in postmitotic neurons. Another link between Polycomb repression and WNT/β-catenin targets was found in the self-renewal of D. melanogaster ovary follicle stem cells (FSCs). In the FSCs, D. melanogaster Polycomb complex components antagonize the WNT/β-catenin signalling pathway to prevent tumour-like growths of self-renewing FSCs at ectopic sites71.
Whereas Polycomb complexes have been shown to repress WNT/β-catenin targets70,71, BAF complexes are co-activators of this pathway and directly interact with β-catenin49. Polycomb and BAF complexes have antagonistic roles in relation to WNT/β-catenin signalling in D. melanogaster, paralleling their direct antagonistic relationship described in ESCs at leukaemia inhibitory factor (LIF) signal-activated genes72. Similar antagonistic relationships between these complexes probably exist in the developing nervous system, where WNT/β-catenin activity drives neurogenesis46.
The role of EZH2 in Weaver’s syndrome
Weaver’s syndrome is an autosomal dominant disease characterized by learning disabilities, dysmorphic facial features and general overgrowth, which can include tall stature, obesity and macrocephaly73. Recent exome-sequencing studies identified numerous de novo and familial missense mutations and in-frame deletions of one copy of EZH2 in all seven sequenced patients with Weaver’s syndrome74,75. In a larger cohort of 300 patients, some diagnosed with Weaver’s syndrome and others with a nonspecific overgrowth syndrome, targeted EZH2 sequencing revealed another 15 likely pathogenic mutations75. As most mutations occur at conserved residues in the catalytic SET domain, it is likely that these mutations are dominant-negative, although the mutations could also result in loss of function and haploinsufficiency if there is a crucial dosage requirement for EZH2. As EZH2 mutations were not uncovered in all overgrowth patients from the larger cohort, other Polycomb components or other genes may eventually be implicated in causing this syndrome.
The macrocephaly phenotype in Weaver’s syndrome patients with EZH2 mutations is especially interesting in light of the microcephaly in NBS and CSS patients with different BAF subunit mutations owing to the antagonistic relationship between the BAF and PRC2 complexes. Reciprocal regulation of WNT/β-catenin targets, especially those that are crucial for the proliferation of neural progenitors, is one likely mechanism for these opposing phenotypes (FIG. 3B). Indeed, as macrocephaly was also noted in 12 ASD patients with CHD8 mutations, and as CHD8 is known to repress WNT/β-catenin targets, it seems likely that imbalance of WNT/β-catenin signalling is a key mechanism leading to the opposing micro- versus macrocephaly phenotypes in NBS, CSS and Weaver’s syndrome29,30,48. This speculation will require much validation but would be a fertile area for future work.
Figure 3. Repressive chromatin modifiers involved in disorders of brain function.
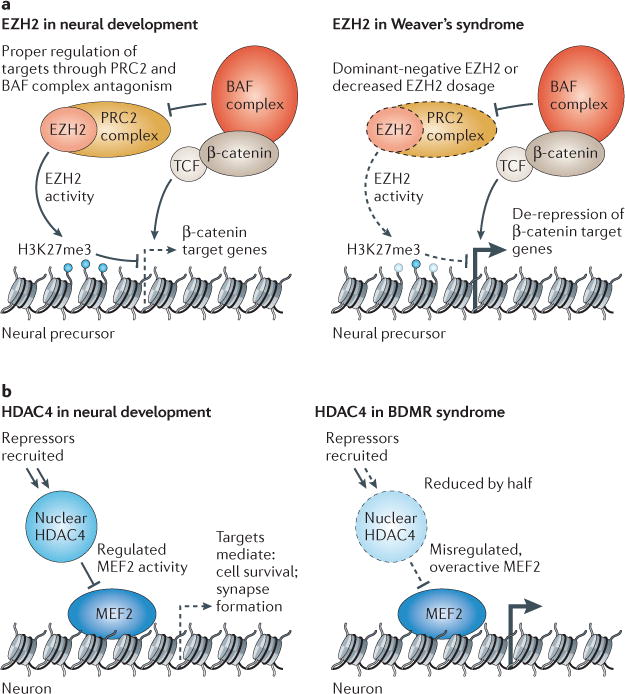
a | During later stages of neurogenesis, enhancer of zeste 2 (EZH2) has been shown to repress particular β-catenin target genes in neural progenitors in order to mediate proper cell fate transitions. It is also likely that EZH2 and BAF complexes have antagonistic roles in these cells as they do in embryonic stem cells and Drosophila melanogaster. Decreased functional EZH2 dosage (owing to haploinsufficiency or altered function of mutant proteins) will lead to de-repression or over-expression of its targets, leading to altered developmental pathways. Patients with Weaver’s syndrome have macrocephaly and learning disabilities of varying severity. b | In normal development, histone deacetylase 4 (HDAC4) is dynamically regulated in the cell, moving into and out of the nucleus in response to physiological signals. When localized in the nucleus, HDAC4 binds myocyte-specific enhancer factor 2 (MEF2) transcription factors and recruits repressors such as class I HDACs and heterochromatin protein 1 (HP1) to MEF2 targets. HDAC4 dosage or nuclear residence is critically affected in patients with brachydactyly mental retardation (BDMR) syndrome, probably leading to misregulated MEF2 target gene expression in particular temporal and cellular contexts. H3K27me3, histone H3 trimethylated at lysine 27; PRC2, Polycomb repressive complex 2.
The role of HDAC4 in learning and memory
HDAC4, which is highly expressed in early postnatal mouse brain, is a class IIa HDAC that shuttles between the cytoplasm and nucleus in response to calcium-regulated physiological signals76,77. The measured histone deacetylase activity of vertebrate class IIa HDACs is very weak compared to invertebrate forms and to class I HDACs owing to a tyrosine to histidine substitution in the enzyme active site78. HDAC4 and other class IIa HDACs may have a non-classical enzymatic activity, but specific targets remain unknown. Despite low histone deacetylase activity, HDAC4 associates with chromatin and developmental transcription factors and is able to mediate gene repression possibly by recruiting co-repressors such as class I HDACs and heterochromatin protein 1 (HP1)79,80.
Conditional deletion of HDAC4 in mouse forebrain neurons leads to impaired learning, memory and long-term synaptic plasticity81, in contrast to global HDAC inhibition or HDAC2 deficiency during embryonic development, which led to improved memory and synaptic plasticity82,83. Transgenic mice that express a truncated HDAC4 allele lacking the deacetylase domain and nuclear export signal exhibited defects similar to conditional deletion84.
Myocyte enhancer factor 2 (MEF2), which is a transcription factor that promotes neuronal survival and synaptogenesis in response to activity-dependent and neurotrophin stimulation85, may be involved in the phenotypes produced by HDAC4 deletion in the central nervous system (FIG. 3). MEF2 directly interacts with HDAC4 and is also highly expressed in the mammalian brain. In response to presynaptic glutamatergic inputs, calcium-regulated kinases phosphorylate HDAC4, leading to its nuclear export. MEF2 target genes are repressed by HDAC4 binding, so the calcium-stimulated HDAC4 nuclear export enables MEF2 to activate transcription77. HDAC4 and MEF2 antagonistically regulate a common set of genes in cortical neurons that are crucial for synaptogenesis and plasticity of synapses84,86. The HDAC4 catalytic domain is dispensable for this repression, which may be mediated instead by its recruitment of co-repressors such as class I HDACs and HP1 (REFS 79,80). In vitro electrophysiological tests of cortical neurons indicate that constitutively nuclear HDAC4, with or without the HDAC domain, causes decreased amplitudes of excitatory postsynaptic currents owing to decreased synaptic strength rather than synapse number84. Additionally, HDAC4 was shown to have a neuroprotective function in cortical cultures that was not dependent on its deacetylase domain or nuclear gene repression but rather on its presence in the cytoplasm84.
The role of HDAC4 mutations in BDMR syndrome
Recently, mutations and deletions in one allele of HDAC4 were found to be responsible for the brachydactyly mental retardation (BDMR) syndrome, which is characterized by intellectual disability, ASD, developmental delay, behavioural abnormalities, sleep disturbance, skeletal abnormalities, dysmorphic features, cardiac defects and obesity87. Overlapping de novo 2q37.3 chromosomal deletions in multiple patients causing ~50% HDAC4 expression refined the crucial region for BDMR syndrome to the HDAC4 gene. Targeted sequencing of the gene in two further patients revealed that smaller truncating mutations also gave rise to BDMR syndrome87,88.
Another report investigated an instance of inherited BDMR syndrome in which a mother with HDAC4 levels at 67% of normal controls and mild BDMR syndrome symptoms gave birth to a son with 23% HDAC4 levels and a more severe phenotype88. This lends support to the idea that HDAC4 has dosage-sensitive roles and that haploinsufficiency can be a cause of BDMR syndrome. At least one of the truncating mutations identified by targeted HDAC4 sequencing, however, did not cause reduced HDAC4 expression. Instead, this allele produced a constitutively nuclear HDAC4 that lacked the deacetylase domain87. In mice, expression of a similar truncated allele caused learning, memory and long-term potentiation (LTP) defects84. It seems that reduced HDAC4 dosage or constitutive nuclear activity can result in similar phenotypes — a theme noted previously for other genes involved in intellectual disability89. Altered HDAC4 activity or dosage may result in BDMR syndrome through misregulation of MEF2 proteins in the developing and adult brain, giving rise to specific defects in neuronal survival, synaptogenesis and synapse strength, as discussed above (FIG. 3a). Interestingly, MEF2 and HDAC4 are also co-expressed outside the brain, in skeletal and cardiac tissues, where major non-neurological features of BDMR syndrome originate.
Conclusion
Human genetic studies have discovered mutations in chromatin regulators in a wide variety of human mental disorders, raising the question of why mutation of these often ubiquitous proteins can lead to selective defects in the nervous system. Although the answer to this question is not clear, it may be that this selective requirement arises from the need to generate hundreds of different cell types in the nervous system. The myriad functions of this array of neuronal cell types are tightly linked to the stability of their morphologies. Hard-wired neuronal circuitry derived from enormous numbers of cells, distinct morphologies and complex connections of neurons almost certainly require stable neuron-specific gene expression. Thus, the frequency of chromatin regulatory mutations that underlie mental disorders may point to an unappreciated role in producing stable levels of gene expression and reducing transcriptional noise that is necessary to maintain stable morphologies over many years. However, the functional tasks of learning and memory require flexible fine-tuning of neuronal circuitries and synaptic plasticity, which might rely on dynamic changes in chromatin structure90–94. Thus, creating a balance between flexibility and stability might be a feature of chromatin regulation that becomes particularly important in the development of the nervous system.
Many of the recently uncovered mutations in chromatin regulators appear to be genetically dominant. Genetic dominance can have many different mechanistic underpinnings, including: the production of dominant-negative proteins that can block wild-type protein function; abnormal polymerization processes in which one mutated protein blocks polymer extension; and haploinsufficiency due to mutation of a gene encoding a protein that is involved in a rate-limiting process. ATP-dependent chromatin remodelling is well suited to a rate-limiting role in biological processes, as the rate of chromatin alteration may be limited by the intrinsic rate of energy use by these complexes rather than by ATP levels, which are generally not rate-limiting. The ability of nBAF complex reconstitution to reprogram fibroblasts into human neurons is consistent with a rate-limiting role for BAF complexes, but how this role is involved in development and human disease needs further investigation.
The fact that EZH2 mutations also give rise to genetically dominant diseases suggests that the crucial mechanism might be balancing the antagonism between BAF and Polycomb complexes. The opposition was first noted in flies at the homeobox (Hox) genes95, and recent studies in mice have confirmed the conservation of this relationship72. Although the opposing roles of Polycomb and BAF are emerging from studies of both neurodevelopment and cancer96, we have very little understanding of the underlying biochemical mechanisms. Understanding the balance between these complexes will be crucial to understand fully the growing number of human diseases that arise owing to the genetic disruption of these complexes.
In BOX 1, the known human mutations in BRG, BRM, BAF250A, BAF250B and EZH2 are shown. These mutations demonstrate that BAF complexes based on BRM and BAF250B have crucial roles in human neural development, whereas BAF complexes based on BRG and BAF250A may have more important roles in tumour suppression. As mutations in EZH2, BRG, BRM, BAF250A, and BAF250B can result in both neurodevelopmental disorders and cancer, it is likely that dosage sensitivity of these proteins is resulting in disparate effects in the context of different developmental stages and cellular environments. The ability of similar mutations in the same protein to produce different diseases probably arises from different genetic backgrounds. For example, we speculate that BAF250A mutations in the context of additional mutations in cell cycle regulatory or checkpoint genes might lead to a loss of tumour suppression, whereas the same or similar BAF250A mutations in the presence of weak alleles of proteins that encode synaptic or dendritic components might lead to intellectual disability. Thus, searching the exome-sequencing databases for these second site mutations may prove useful.
If it is assumed that chromatin remodellers provide balance to systems that are characterized by processes such as acetylation–deacetylation or methylation–demethylation, then further genetic definition of crucial chromatin regulators in human disease could reveal potential therapeutic targets as their activity can be modulated by small molecules (reviewed in REF. 11; see especially Table 2 in this reference). It could be envisioned that these small molecules be used to reinstate the proper balance between chromatin regulatory activities. Furthermore, a full mechanistic understanding of chromatin regulators in neurodevelopment is necessary to provide effective ways of making human neurons from other cell types. The creation of patient-specific neural cells as therapeutic models and the development of effective neural replacement strategies are two possible goals97–101.
As the feasibility of large genomic sequencing studies rapidly increases, along with our abilities to analyse, process and store the corresponding data, we should expect that more mutated chromatin regulators, and the genetic background in which they exist, will be defined. This approaching era should provide insight into actual human disease mechanisms, an area in which animal models, although immensely helpful, are not sufficient; genome sequencing will uncover the proteins and domains therein that are most relevant to human disease. As human genomic efforts reveal important functions for new genes in neural development, basic genetic and biochemical research will need to keep pace to elucidate the underlying mechanisms. We are hopeful that these two lines of research will lead to new insights into the development and function of the human brain.
Supplementary Material
Acknowledgments
We apologize to colleagues whose work we did not cite owing to lack of space or unintentional oversight. This work was supported by the Howard Hughes Medical Foundation, a grant from the California Institute for Regenerative Medicine and US National Institutes of Health grants NS046789 and CA163915 to G.R.C.
Glossary
- Exome sequencing
Targeted sequencing of protein-coding regions of the genome (that is, exons). It is a cheaper yet still effective alternative to whole-genome sequencing to identify clinically relevant gene variations that are responsible for both Mendelian and common diseases but does not detect mutations in non-coding regions of the genome
- Glomerulus
The glomerulus is a spherical structure located in the olfactory bulb. Each glomerulus is thought to receive input from olfactory receptor neurons (expressing only one type of olfactory receptor). They relay this information into higher brain structure through projection neurons
- Autosomal dominant disease
Refers to a disease arising from mutations on non-sex chromosomes that are genetically dominant
- Microcephaly
Microcephaly is a significantly smaller head measurement. It can be caused by an abnormal brain size due to loss of any number of cell types or brain features and can even be caused by abnormal ventricular spaces or cerebral fluid
- Haploinsufficiency
When the product of one normal allele of a gene is not sufficient to allow the normal function of a gene to be executed. This is another possible cause of genetic dominant diseases
- Frameshift indel
An insertion and/or deletion mutation that changes the reading frame of a protein and creates an altered gene product
- Dominant-negative
Interference with the function of the normal allele of a gene by a mutated allele. This usually occurs when the mutant product can still interact with the same elements as the wild-type product, but block some aspect of its function. This is another possible cause of genetic dominant diseases
- Corpus callosum
A wide flat bundle of neural fibres that connects the left and right cerebral hemispheres and facilitates interhemispheric communication
- De novo mutations
Alterations in genes that are present for the first time in one family member as a result of a mutation in a germ cell (that is, an egg or sperm) of one of the parents or in the fertilized egg itself
- WNT/β-catenin signaling
The binding of WNT ligand to receptor Frizzled leads to a cascade of events allowing for β-catenin (an integral cell–cell adhesion adaptor protein as well as transcriptional co-regulator) stabilization, nuclear translocation and transcriptional activation. The WNT/β-catenin pathway integrates signals from many other pathways, including retinoic acid, FGF, TGFβ and BMP in many different cell types and tissues
- Single-nucleotide polymorphisms
(SNPs). DNA sequence variations that occur when a single nucleotide (A, T, C or G) in the genome or other shared sequence differs between members of a biological species or paired chromosomes in an individual
- Linker histone
The linker histone H1 binds the nucleosome and the entry or exit sites of the DNA, allowing for the formation of higher-order chromatin structures that are thought to lead to chromatin compaction and gene repression
- Macrocephaly
Macrocephaly is a significantly larger than average head circumference measurement. It can be caused by an abnormal brain size due to gain of any number of cell types or brain features and can even be caused by abnormal ventricular spaces or cerebral fluid
- Polycomb group proteins
These conserved proteins form multimeric complexes that exert their functions by modifying chromatin structure and by regulating the deposition and recognition of multiple post-translational histone modifications. Their epigenetic role appears to arise from their ability to propagate a repressive chromatin modification over several kilobases of DNA
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Gerald R. Crabtree’s homepage: http://crablab.stanford.edu
SUPPLEMENTARY INFORMATION
See online article: S1 (table) | S2 (table)
References
- 1.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein BE, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Allis CD, Jenuwein T, Reinberg D. Epigenetics. Cold Spring Harbor Laboratory Press; 2007. [Google Scholar]
- 8.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 9.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- 11.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nature Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehler MF. Epigenetics and the nervous system. Ann Neurol. 2008;64:602–617. doi: 10.1002/ana.21595. [DOI] [PubMed] [Google Scholar]
- 13.Yoo AS, Crabtree GR. ATP-dependent chromatin remodeling in neural development. Curr Opin Neurobiol. 2009;19:120–126. doi: 10.1016/j.conb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corona DF, et al. ISWI regulates higher-order chromatin structure and histone H1 assembly in vivo. PLoS Biol. 2007;5:e232. doi: 10.1371/journal.pbio.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho L, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci USA. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lessard J, et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. This paper describes a neuron-specific chromatin-remodelling complex, nBAF, which has three subunits expressed only in the nervous system. The frequency with which BAF complexes are implicated in neurodevelopmental and psychiatric disorders might relate to the specialized functions of the nBAF complex during neural development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ooi L, Belyaev ND, Miyake K, Wood IC, Buckley NJ. BRG1 chromatin remodeling activity is required for efficient chromatin binding by repressor element 1-silencing transcription factor (REST) and facilitates REST-mediated repression. J Biol Chem. 2006;281:38974–38980. doi: 10.1074/jbc.M605370200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bultman S, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 19.Klochendler-Yeivin A, et al. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1:500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JK, et al. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol. 2001;21:7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu JI, et al. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron. 2007;56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Sawa H, Kouike H, Okano H. Components of the SWI/SNF complex are required for asymmetric cell division in C. elegans. Mol Cell. 2000;6:617–624. doi: 10.1016/s1097-2765(00)00060-5. [DOI] [PubMed] [Google Scholar]
- 23.Shibata Y, Uchida M, Takeshita H, Nishiwaki K, Sawa H. Multiple functions of PBRM-1/Polybromo-and LET-526/Osa-containing chromatin remodeling complexes in C. elegans development. Dev Biol. 2012;361:349–357. doi: 10.1016/j.ydbio.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 24.Yoo AS, Staahl BT, Chen L, Crabtree GR. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature. 2009;460:642–646. doi: 10.1038/nature08139. This paper defines the regulatory circuitry that underlies the switch of BAF subunits as neural progenitors exit mitosis and differentiate into neurons. Later studies by this group found that recapitulation of this switch would change human fibroblasts into neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoo AS, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476:228–231. doi: 10.1038/nature10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tea JS, Luo L. The chromatin remodeling factor Bap55 functions through the TIP60 complex to regulate olfactory projection neuron dendrite targeting. Neural Dev. 2011;65 doi: 10.1186/1749-8104-6-5. The authors conducted a daring screen for mutations that would result in the perfect retargeting of the olfactory neuron dendritic tree to a different glomerulus. Surprisingly, the only gene in which a mutation produces perfect retargeting is the homologue of BAF53, which switches during the course of mammalian neural development from BAF53A to BAF53B. These studies suggest that the pathogenic mechanisms involved in diseases associated with mutations in BAF complexes might be due to defects in dendritic targeting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parrish JZ, Kim MD, Jan LY, Jan YN. Genome-wide analyses identify transcription factors required for proper morphogenesis of Drosophila sensory neuron dendrites. Genes Dev. 2006;20:820–835. doi: 10.1101/gad.1391006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santen GW, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin–Siris syndrome. Nature Genet. 2012;44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 29.Tsurusaki Y, et al. Mutations affecting components of the SWI/SNF complex cause Coffin–Siris syndrome. Nature Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- 30.Van Houdt JK, et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides–Baraitser syndrome. Nature Genet. 2012;44:445–449. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- 31.Wolff D, et al. In-frame deletion and missense mutations of the C-terminal helicase domain of SMARCA2 in three patients with Nicolaides–Baraitser syndrome. Mol Syndromol. 2012;2:237–244. doi: 10.1159/000337323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halgren C, et al. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet. 2011;82:248–255. doi: 10.1111/j.1399-0004.2011.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backx L, Seuntjens E, Devriendt K, Vermeesch J, Van Esch H. A balanced translocation t(6;14) (q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet Genome Res. 2011;132:135–143. doi: 10.1159/000321577. [DOI] [PubMed] [Google Scholar]
- 34.Hoyer J, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-A chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 3012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 38.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012 Jan 18; doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koga M, et al. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet. 2009;18:2483–2494. doi: 10.1093/hmg/ddp166. [DOI] [PubMed] [Google Scholar]
- 40.Loe-Mie Y, et al. SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum Mol Genet. 2010;19:2841–2857. doi: 10.1093/hmg/ddq184. [DOI] [PubMed] [Google Scholar]
- 41.Nishiyama M, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nature Cell Biol. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol Cell Biol. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakamoto I, et al. A novel β-catenin-binding protein inhibits β-catenin-dependent TCF activation and axis formation. J Biol Chem. 2000;275:32871–32878. doi: 10.1074/jbc.M004089200. [DOI] [PubMed] [Google Scholar]
- 44.Nishiyama M, Skoultchi AI, Nakayama KI. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol Cell Biol. 2012;32:501–512. doi: 10.1128/MCB.06409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heasman J, Kofron M, Wylie C. β-catenin signaling activity dissected in the early Xenopus embryo: a novel antisense approach. Dev Biol. 2000;222:124–134. doi: 10.1006/dbio.2000.9720. [DOI] [PubMed] [Google Scholar]
- 46.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 47.Brault V, et al. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 48.Ille F, Sommer L. Wnt signaling: multiple functions in neural development. Cell Mol Life Sci. 2005;62:1100–1108. doi: 10.1007/s00018-005-4552-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barker N, et al. The chromatin remodelling factor Brg-1 interacts with β-catenin to promote target gene activation. EMBO J. 2001;20:4935–4943. doi: 10.1093/emboj/20.17.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Ferrari GV, Moon RT. The ups and downs of WNT signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–7553. doi: 10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- 51.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Talkowski ME, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams CA, Dagli A, Battaglia A. Genetic disorders associated with macrocephaly. Am J Med Genet A. 2008;146:2023–2037. doi: 10.1002/ajmg.a.32434. [DOI] [PubMed] [Google Scholar]
- 55.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 56.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. The authors describe the first evidence that epigenetic modifications are reversible by enzymes that remove or erase the modification. This paper describes the discovery of HDAC and initiated the idea that epigenetic modification was a dynamic state rather than an irreversible process. [DOI] [PubMed] [Google Scholar]
- 57.Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. The authors use the chromatin immunoprecipitation followed by high-throughput sequencing (ChIP–seq) method, which they invented, to demonstrate that histone acetylases and deacetylases bind to the same sites over the genome indicating that these modifications are in dynamic equilibrium and hence subject to regulatory mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hathaway NA, et al. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 61.Cross NC. Histone modification defects in developmental disorders and cancer. Oncotarget. 2012;3:3–4. doi: 10.18632/oncotarget.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kramer JM, van Bokhoven H. Genetic and epigenetic defects in mental retardation. Int J Biochem Cell Biol. 2009;41:96–107. doi: 10.1016/j.biocel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 63.Margueron R, et al. Role of the Polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nature Cell Biol. 2008;10:1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 65.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a Polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 66.O’Carroll D, et al. The Polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 68.Chamberlain SJ, Yee D, Magnuson T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells. 2008;26:1496–1505. doi: 10.1634/stemcells.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pereira JD, et al. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci USA. 2010;107:15957–15962. doi: 10.1073/pnas.1002530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hirabayashi Y, et al. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63:600–613. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 71.Li X, Han Y, Xi R. Polycomb group genes Psc and Su(z)2 restrict follicle stem cell self-renewal and extrusion by controlling canonical and noncanonical Wnt signaling. Genes Dev. 2010;24:933–946. doi: 10.1101/gad.1901510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ho L, et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nature Cell Biol. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cohen MM., Jr Mental deficiency, alterations in performance, and CNS abnormalities in overgrowth syndromes. Am J Med Genet C. 2003;117:49–56. doi: 10.1002/ajmg.c.10013. [DOI] [PubMed] [Google Scholar]
- 74.Gibson WT, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110–118. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tatton-Brown K, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127–1133. doi: 10.18632/oncotarget.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang AH, Yang XJ. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol Cell Biol. 2001;21:5992–6005. doi: 10.1128/MCB.21.17.5992-6005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lahm A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chan JK, Sun L, Yang XJ, Zhu G, Wu Z. Functional characterization of an amino-terminal region of HDAC4 that possesses MEF2 binding and transcriptional repressive activity. J Biol Chem. 2003;278:23515–23521. doi: 10.1074/jbc.M301922200. [DOI] [PubMed] [Google Scholar]
- 80.Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22:7302–7312. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim MS, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32:10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vecsey CG, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sando R, et al. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell. 2012;151:821–834. doi: 10.1016/j.cell.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shalizi AK, Bonni A. Brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- 86.Flavell SW, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Williams SR, et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morris B, et al. Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am J Med Genet A. 2012;158:2015–2020. doi: 10.1002/ajmg.a.35463. [DOI] [PubMed] [Google Scholar]
- 89.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nature Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 92.Guan Z, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- 93.Gupta S, et al. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nature Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 95.Tamkun JW, et al. Brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. The authors define the opposition between Polycomb and BAF or BAP complexes, which is likely to be a crucial underlying mechanism of both human malignancy and neurodevelopmental disorders. [DOI] [PubMed] [Google Scholar]
- 96.Wilson BG, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim JE, et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc Natl Acad Sci USA. 2011;108:3005–3010. doi: 10.1073/pnas.1007753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brennand KJ, Simone A, Tran N, Gage FH. Modeling psychiatric disorders at the cellular and network levels. Mol Psychiatry. 2012;17:1239–1253. doi: 10.1038/mp.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kriks S, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caiazzo M, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature. 2011;476:224–227. doi: 10.1038/nature10284. [DOI] [PubMed] [Google Scholar]
- 102.Kadoch C, Hargreaves D, Hodges C, Crabtree GR. Nature Genet. (in the press) [Google Scholar]
- 103.Shane AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2012;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 107.Gied J. Brain development IX: human brain growth. Am J Psychiatry. 1999;156:4. doi: 10.1176/ajp.156.1.4. [DOI] [PubMed] [Google Scholar]
- 108.Kleefstra T, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91:73–82. doi: 10.1016/j.ajhg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 110.Iwase S, et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nature Struct Mol Biol. 2011;18:769–776. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roelfsema JH, et al. Genetic heterogeneity in Rubinstein–Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–580. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Petrij F, et al. Rubinstein–Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 113.Clayton-Smith J, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say–Barber–Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kleefstra T, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jones WD, et al. De novo mutations in MLL cause Wiedemann–Steiner syndrome. Am J Hum Genet. 2012;91:358–364. doi: 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hannibal MC, et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155:1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ng SB, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jensen LR, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Najmabadi H, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 120.Koivisto AM, et al. Screening of mutations in the PHF8 gene and identification of a novel mutation in a Finnish family with XLMR and cleft lip/cleft palate. Clin Genet. 2007;72:145–149. doi: 10.1111/j.1399-0004.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- 121.Laumonnier F, et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet. 2005;42:780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Froyen G, et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet. 2008;82:432–443. doi: 10.1016/j.ajhg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 124.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 125.Risheg H, et al. A recurrent mutation in MED12 leading to R961W causes Opitz–Kaveggia syndrome. Nature Genet. 2007;39:451–453. doi: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- 126.Schwartz CE, et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet. 2007;44:472–477. doi: 10.1136/jmg.2006.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hashimoto S, et al. MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science. 2011;333:1161–1163. doi: 10.1126/science.1206638. [DOI] [PubMed] [Google Scholar]
- 128.Kim HG, et al. Translocations disrupting PHF21A in the Potocki–Shaffer-syndrome region are associated with intellectual disability and craniofacial anomalies. Am J Hum Genet. 2012;91:56–72. doi: 10.1016/j.ajhg.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.