

Abstract
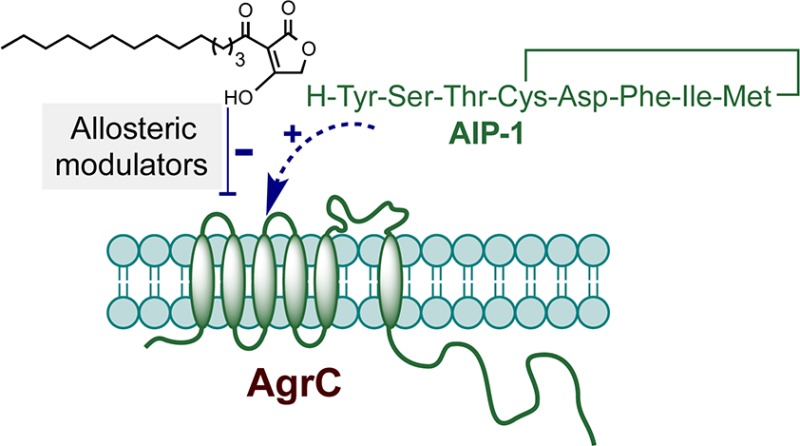
A series of 3-oxo-C12-HSL, tetramic acid, and tetronic acid analogues were synthesized to gain insights into the structural requirements for quorum sensing inhibition in Staphylococcus aureus. Compounds active against agr were noncompetitive inhibitors of the autoinducing peptide (AIP) activated AgrC receptor, by altering the activation efficacy of the cognate AIP-1. They appeared to act as negative allosteric modulators and are exemplified by 3-tetradecanoyltetronic acid 17, which reduced nasal cell colonization and arthritis in a murine infection model.
Introduction
Bacterial pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus employ cell-to-cell communication or “quorum sensing” (QS) systems for coordinating collective activities that depend on the actions of one or more chemically distinct signal molecules.1 Such molecules largely operate as effectors of QS-dependent gene expression through direct activation of membrane associated sensor kinases or via cytoplasmically located DNA-binding proteins. They are also emerging as multifunctional signals that can influence interactions between different bacterial species and impact significantly the outcome of host–pathogen interactions by also acting directly on the host.2
In P. aeruginosa, N-(3-oxododecanoyl)-l-homoserine lactone (3-oxo-C12-HSL, 1; Table 1) activates the transcriptional regulator LasR to drive the expression of multiple QS target genes involved in exotoxin, exoenzyme and secondary metabolite production, as well as biofilm development.2 3-Oxo-C12-HSL also exerts a wide spectrum of other biological activities, for example, acting as a modulator of immune and inflammatory responses, impacting on the cardiovascular system, and disrupting epithelial barriers.2 As yet, the direct target(s) for 3-oxo-C12-HSL in eukaryotic cells has not been identified.3,4 In lymphocytes, optimal immune suppressive activity is dependent on a C11–C13 acyl chain containing a 3-oxo or a 3-hydroxy group. Compounds lacking the l-configuration or with polar substituents were essentially devoid of activity.5 Further modification of the 3-oxo-C12-HSL structure to generate compounds retaining the C12 acyl chain length while replacing the chiral homoserine lactone with more stable achiral heteroaryl moieties yielded compounds that possess similar immune suppressive activity to 3-oxo-C12-HSL but that inhibit rather than activate LasR in P. aeruginosa, thus indicating that at the molecular level it is possible to separate immune suppressive activity from QS activation.6
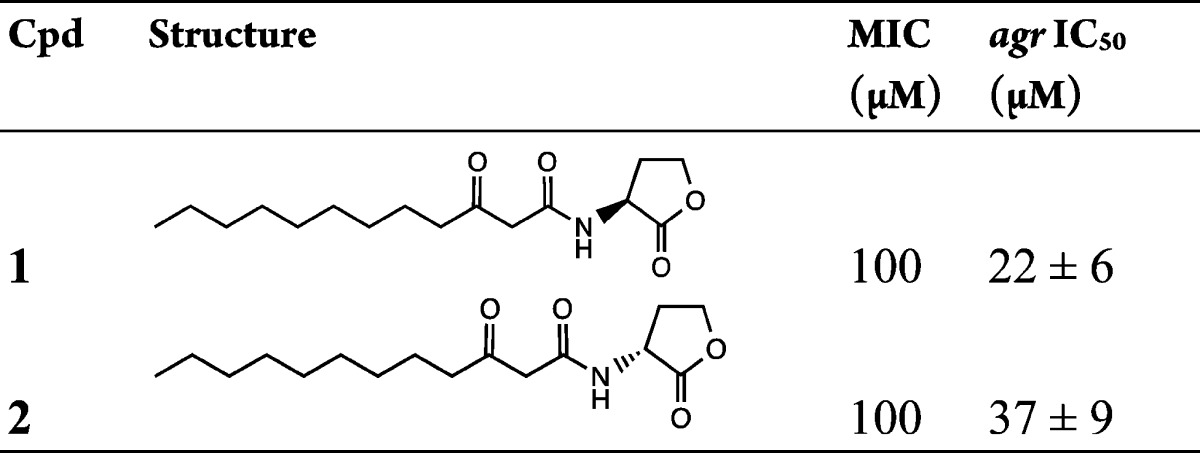
Table 1. QS and Growth Inhibitory Activities of 3-Oxo-C12-HSL and Its Enantiomer.
3-Oxo-C12-HSL also acts on other microorganisms, inhibiting filamentation in Candida albicans(7) and the growth of Gram-positive bacteria.8,9 At subgrowth inhibitory concentrations, 3-oxo-C12-HSL antagonizes the production of S. aureus exotoxins (including α-hemolysin, δ-hemolysin, and toxic shock syndrome toxin) while enhancing cell wall protein biosynthesis including the fibronectin- and immunoglobulin-binding proteins.9 The mode of action of 3-oxo-C12-HSL in S. aureus appears to involve inhibition of agr-dependent QS, which reciprocally regulates exotoxins and cell wall colonization factors.9,10 The agr locus consists of two divergent transcriptional units, the P2 and P3 operons. The P2 operon consists of four genes, agrBDCA, which are required for the activation of transcription from the P2 and P3 promoters, while the untranslated P3 transcript RNAIII is itself the effector for the agr response. AgrA and AgrC constitute a two-component system in which AgrC is the sensor kinase and AgrA is the response regulator. The system is activated by the interaction of AgrC with a 7- to 9-mer macrocyclic-containing peptide termed the autoinducing peptide (AIP) generated from the agrD gene product by AgrB.10 Since 3-oxo-C12-HSL binds to the S. aureus cytoplasmic membrane in a specific saturable manner, such membrane interactions may account for the agr inhibitory properties of 3-oxo-C12-HSL given the membrane localization of the AgrB and AgrC proteins.
Under aqueous alkaline conditions, 3-oxo-C12-HSL undergoes lactonolysis to form the corresponding ring-opened homoserine compound11 or an intramolecular rearrangement reaction to afford a vinylogous acid product, 3-(1-hydroxydecylidene)-5-(2-hydroxyethyl)pyrrolidine-2,4-dione [(S)-5-hydroxyethyl-3-decanoyltetramic acid;8 5-HE-C10-TMA, 5] (see Table 2). This compound belongs to the TMA class of compounds, such as reutericycline which display antibacterial properties.8,12 It is primarily active against Gram-positive bacteria including Bacillus, Clostridium, and Staphylococcus(8,13) where growth inhibition involves the dissipation of bacterial membrane potential and pH gradient.14 The physiological function of 5 in P. aeruginosa is not known, but it is capable of weakly inhibiting the LasR/3-oxo-C12-HSL-dependent activation of the elastase (lasB) gene6 and reducing P. aeruginosa viability.15 In contrast to 3-oxo-C12-HSL, 5 is also a ferric ion chelator.8 However, while it does not function as a siderophore for P. aeruginosa,16 iron was reported to abolish the antibacterial activity of 5 toward P. aeruginosa(15) and Clostridium difficile.13 In common with 3-oxo-C12-HSL, 5 also exhibits immune suppressive activity in a murine peripheral blood lymphocyte proliferation assay and is cytotoxic toward Jurkat6 but not bone-marrow-derived macrophage cells.14
Table 2. QS and Growth Inhibitory Activities of 3-Acyltetramic Acidsa.
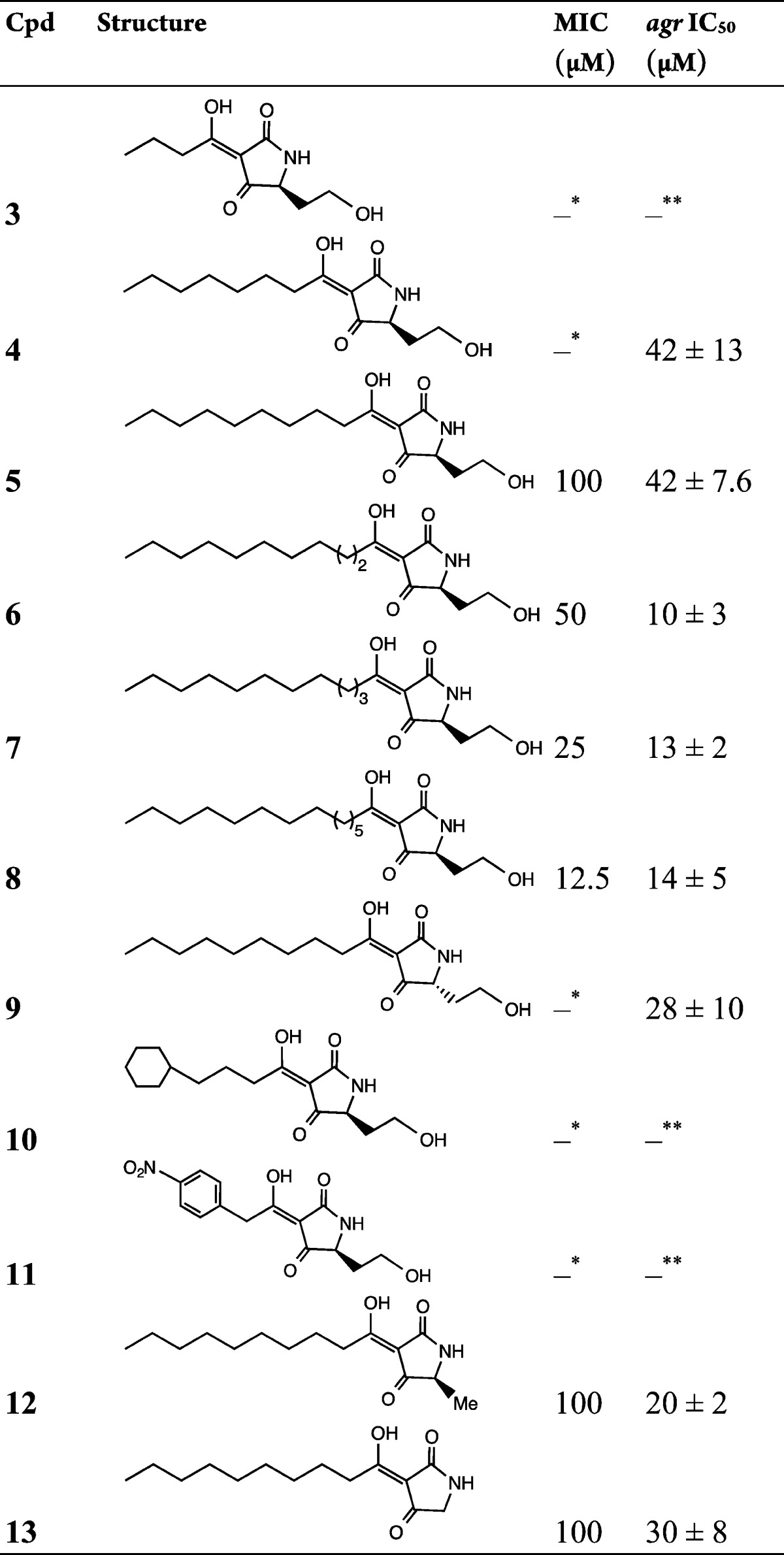
The asterisks indicate the following: ∗, no growth inhibition up to 100 μM; ∗∗, no inhibition of agr observed at concentrations up to 100 μM.
Given the threat posed by the emergence of bacterial strains resistant to conventional growth inhibitory antibiotics, there is renewed interest in the discovery of agents that control infection through the attenuation of bacterial virulence.10,17 This latter strategy has the potential advantage of expanding the repertoire of bacterial targets and exerting reduced selective pressure which, in turn, may retard the development of resistance.10 In this context, the development of strategies for inhibiting QS-dependent regulation of virulence has received significant attention particularly for problematic multiantibiotic resistant human pathogens such as S. aureus.10 Here major efforts have been directed toward the inhibition of AIP/AgrC interactions to attenuate staphylococcal virulence largely through the synthesis and evaluation of AIP mimetics.10,18 However, such AIPs have been reported to be susceptible to inactivation by host innate defenses.19 Given our desire to discover new nonpeptidic agents that are capable of modulating the AIP/AgrC interaction, we have considered simple heterocyclic compounds. In this context, although a number of small molecules have been reported to cause blockade of the staphylococcal agr system, none of these compounds are known to directly modulate ligand/cognate receptor interactions.10 Since we have previously shown that 3-oxo-C12-HSL can inhibit agr-dependent QS in S. aureus,9 a series of 3-oxo-C12-HSL, TMA, and related TOA analogues were synthesized and evaluated for their agr- and growth-inhibitory properties, structure–activity relationships, and mechanism of action. In addition, the efficacy of the most potent TOA analogue 17 was investigated in a mouse staphylococcal infection model.
Results and Discussion
N-Acyl-l-homoserine lactones (AHLs) (Table 1, 1 and 2; Supporting Information Table S1, S1–S16) were synthesized using well-established methodologies. The synthesis of 3-oxo-C12-HSL 1 and its analogues has been previously reported by Chhabra et al.5,20 and Jadhav et al.6 Similarly, the 3-acyl-5-(2-hydroxyethyl)tetramic acids 3–13 listed in Table 2 were prepared from their respective N-3-oxoacyl-l-homoserine lactones by the method previously described by Kaufmann et al.8
(S)-3-Decanoyl-5-methyltetramic acid 12 and 3-decanoyltetramic acid 13 were constructed by applying the strategy outlined in Scheme S1. Thus, the appropriate N-Boc-α-aminoacylated Meldrum’s acid, prepared by C-acylation of Meldrum’s acid with carbodiimide-activated N-Boc-amino acid, was subjected to thermal cyclative elimination of Me2CO and CO2 to yield the required 5-substituted N-Boc-tetramic acid.21 Subsequent acylation with activated decanoic acid followed by trifluoroacetic acid mediated acidolysis delivered (S)-3-acyl-5-methyltetramic acid 12. The 3-acyltetramic acid 13 was similarly obtained by using N-Boc-glycine as the starting reagent.
The 3-acyltetronic acids 14–18 listed in Table 3 were prepared by the general method of C-acylation of commercially available tetronic acid.22
Table 3. QS and Growth Inhibitory Activities of 3-Acyltetronic Acidsa.
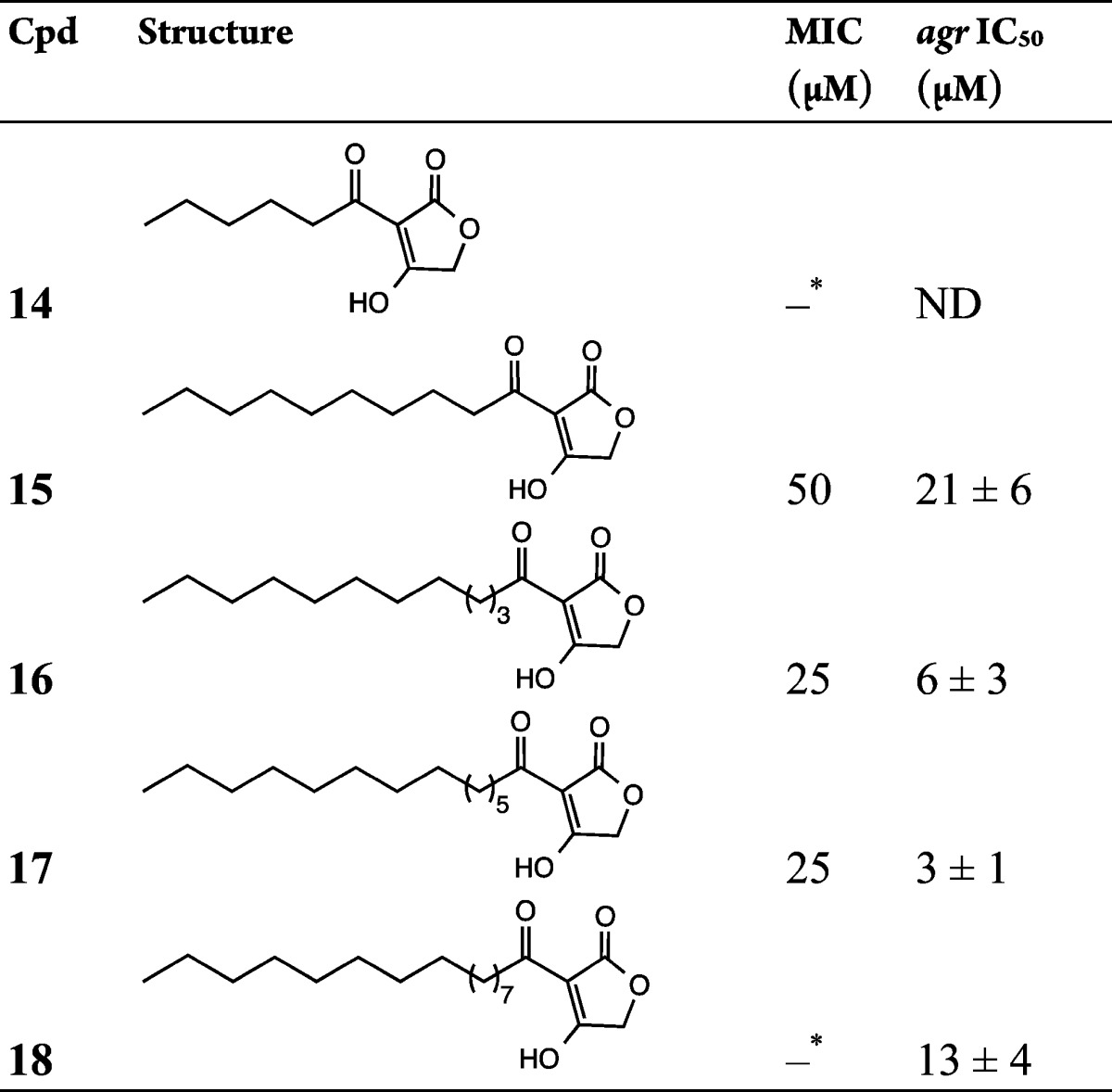
The asterisk (∗) indicates no growth inhibition up to 100 μM. ND, not determined.
To gain insights into the 3-oxo-C12-HSL structural requirements for staphylococcal agr inhibition and to discover quorum sensing inhibitors that do not impact on staphylococcal growth, systematic modification of 3-oxo-C12-HSL was carried out, focusing initially on the homoserine lactone (I), 3-oxo substituent (II), acyl side chain (III), and amide (IV) structural units of the molecule (Figure S1). Seventeen analogues of 3-oxo-C12-HSL were synthesized and evaluated for inhibition of agr and bacterial growth (Table 1 and Table S1). While the l-isomer of 3-oxo-C12-HSL 1 inhibited agr with an IC50 of 22 ± 6 μM, the d-isomer 2 was approximately 2-fold less active (IC50 of 37 ± 9 μM). However, neither the corresponding ring opened N-(3-oxododecanoyl)-l-homoserine S1 nor the heteroring truncated 3-oxododecanamide S2 inhibited agr. Moreover, analogues in which the homoserine lactone moiety was modified by substitution with different heteroatoms or replaced with alternative heterocyclic ring systems failed to inhibit agr. Removal or reduction of the 3-oxo substituent also abolished agr inhibitory activity (Table S1). Modification of the acyl chain by the incorporation of a double bond or partial replacement with phenyl or cyclohexyl substituents all resulted in the loss of agr inhibitory properties (Table S1). Apart from the two 3-oxo-C12-HSL isomers 1 and 2, none of the other analogues examined inhibited bacterial growth at 100 μM. Taken together, these data show that subtle changes in 3-oxo-C12-HSL are sufficient to abolish QS and growth inhibitory properties.
Since 1 undergoes a base-catalyzed rearrangement to the TMA 5, we explored the agr inhibitory activities of TMA analogues 3–13 (Table 2) by varying the 3-acyl chain length 3–8, stereochemistry 9, and substitution at the 5-position of the heterocyclic ring 12 and 13. Each of the TMA analogues examined apart from 3, 10, and 11 inhibited agr (Table 2), with the most active compound being 6 (IC50 = 10 ± 3 μM). Switching the C5 stereochemistry from (S)-5 to (R)-9 improved agr inhibitory activity by 1.5-fold, and replacement with Me (12) or removal (13) of the 5-(2-hydroxyethyl) substituent in 5 also resulted in enhanced agr inhibitory activity (Table 2), thus indicating that the 5-position can withstand alteration.
Since TMAs such as 5 are known to inhibit bacterial growth14 and since growth inhibition is an undesirable property for agents that attenuate virulence,10 we evaluated the growth inhibitory properties of each of the TMA compounds synthesized. Table 2 shows that the MIC for the 3-oxo-C12-HSL-derived TMA 5 was 100 μM.
While analogues with shorter 3-acyl chains (3 and 4) did not inhibit growth at this concentration, extension of the chain by one, two, or four carbons, 6–8, respectively, substantially increased potency such that the C14 analogue 8 exhibited an 8-fold lower MIC (12.5 μM). These data are broadly in agreement with the staphylococcal MICs for 5 and 7 reported by Lowery et al.14 Interestingly, the shorter chain compound 5-HE-C4-TMA 3, which we found was not active against S. aureus, has been reported to kill P. aeruginosa(15) but not C. difficile.13 TMAs with 3-acyl chains of 10–14 carbons inhibited growth and agr with differences between the MIC and IC50 values ranging from little different (e.g., 5-HE-C14-TMA 8) to 5-fold (e.g., 5-HE-C11-TMA 6). Compound 4, 5-HE-C8-TMA, retained reasonable agr inhibitory activity (42 ± 13 μM) and completely inhibited production of the agr-dependent exotoxin α-hemolysin at 100 μM without affecting staphylococcal growth (Figure S2). It therefore offers a platform for the further improvement of agr antagonism independent of growth inhibition.
TMAs such as 5 are ferric ion chelators,8,16 and so it is possible that the ability to inhibit staphylococcal growth is, at least in part, a consequence of its ability to restrict the availability of an essential bacterial nutrient.23 Since iron has been reported to abolish the antibacterial activity of 5 toward C. difficile and P. aeruginosa,13,15 we synthesized TOA (Table 3, 14–18; Table S2, S17–S24) variants of the TMAs in which the ring nitrogen is replaced with oxygen. We predicted that the tetronic acid structure would not chelate iron, as it largely exists as a 4-enolic tautomer24 as opposed to the TMA structure where the 3-exoenolic tautomer in (Z)-configuration predominates. The latter is in syn orientation with the 2-oxo group and thus can serve as a bidentate ligand for Fe(III) binding.8 This was confirmed experimentally, and Figure 1a shows that in contrast to the TMA 5, the TOAs 14–16 with acyl chains ranging from C6 to C12 do not chelate ferric iron.
Figure 1.
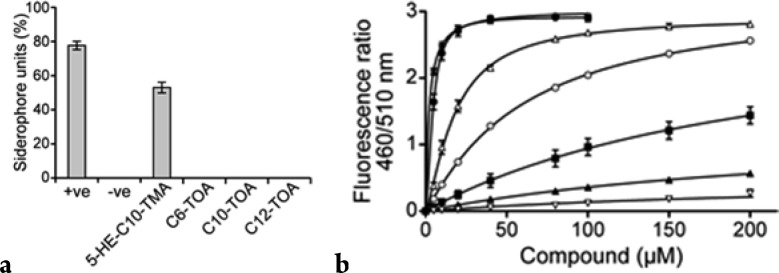
(a) Iron-chelating properties of 5-HE-C10-TMA 5 and the C6-, C10-, and C12-TOAs 14, 15, 16 (at 50 μM) as determined using the CAS assay: positive control, desferrioxamine (10 μM); solvent control, MeCN. (b) 5-HE-TMAs and TOAs disturb the membrane dipole potential. Changes in dipole potential were determined using di-8-ANEPPS to measure the variation in the fluorescence ratio R(460/520) as a function of concentration. The binding profiles for 5-HE-C10-TMA 5 (■), C10-TOA 15 (▲), 5-HE-C12-TMA 7 (△), C12-TOA 16 (○), 5-HE-C14-TMA 8 (∗), and C14-TOA 17 (●) compared with 3-oxo-C12-HSL 1 (▽) are shown.
When assayed for staphylococcal agr and growth inhibitory properties (Table 3 and Table S2), each of the TOAs apart from the C6-TOA 14 exhibited similar activities to the TMAs with potency increasing as a function of acyl chain length. For agr inhibition, C14-TOA 17 was the most potent compound with an IC50 of 3 ± 1 μM, approximately 8 times lower than the MIC (25 μM). In fact, when S. aureus USA300 was exposed to 10 μM C14-TOA 17, production of the agr-dependent exotoxin α-hemolysin was substantially reduced but growth was unaffected (Figure S3). This TOA was also the most active agr inhibitor among the 3-oxo-C12-HSL, TMA, and TOA compounds evaluated in the present investigation. Two additional TOA analogues S18 and S19 incorporating an unsaturated acyl chain were also synthesized. While the presence of a trans double bond in S18 had little effect on activity (cf., 15), it was almost abolished in the cis analogue S19 (cf., 16). Further modifications of the 3-acyl chain by introducing aryl substituents or 3-oxo substituent did not yield active analogues (Table S2). Taken together, these data demonstrate that TOAs can also be effective agr inhibitors and that iron chelation is not required for growth or agr inhibition.
Since two of the major proteins involved in agr-dependent QS (AgrC and AgrB) are located in the cytoplasmic membrane, we sought to determine whether the TMAs and TOAs bind directly to staphylococcal membranes. This was achieved using the fluorescent probe di-8-ANEPPS in a dual-wavelength ratiometric method that detects changes in membrane dipole potential and yields binding information, such as affinity and overall binding capacity.25 Figure 1b shows the binding profiles for the 5-HE-C10-, 5-HE-C12-, and 5-HE-C14-TMA and the C10-, C12, and C14-TOA compared with 3-oxo-C12-HSL 1. For each of the 5-HE-TMAs 5, 7, 8 and TOAs 15–17, the data fit a hyperbolic function that is consistent with a noncooperative, single-site binding model (Figure 1b). The TMAs and TOAs bind to the staphylococcal membrane with higher affinity than 3-oxo-C12-HSL with affinity increasing as chain length increases. In these experiments, Kd values of 222, 23, and 2 μM were estimated for the 5-HE-C10 5, 5-HE-C12 7, and 5-HE-C14 8 TMAs, respectively, compared with 270 μM for 3-oxo-C12-HSL 1. For the C10-, C12-, and C14-TOAs 15–17, the dissociation constants were similar to those of the 5-HE-TMAs, i.e., 258, 72, and 4 μM, respectively. From these TMAs and TOAs, it is clear that the higher the membrane affinity, the more active is the compound against agr. Hence, these data are consistent with AgrB or AgrC as the target.
Consequently, we sought to determine whether 3-oxo-C12-HSL 1, 5-HE-C10-TMA 5, and C12-TOA 16 are competitive inhibitors of the interaction between the S. aureus AIP-1 and its cognate receptor AgrC-1. Figure 2 shows that in contrast to the rightward shift in the concentration–response curve for AIP-1 (i.e., the EC50 of AIP-1 increased by about 200-fold, from 5 nM to 1.10 μM) in the presence of increasing concentrations of the competitive agr inhibitor, (Ala5)AIP-1,26 neither 3-oxo-C12-HSL 1, 5-HE-C10-TMA 5, nor C12-TOA 16 competitively inhibits the activation of AgrC-1 by AIP-1. Consequently, agr inhibition by these compounds is likely to involve an allosteric interaction with AgrC, preventing efficient activation by the AIP signal molecule. It is worth noting that C12-TOA 16 is at least 5-fold more potent than 3-oxo-C12-HSL 1 and 5-HE-C10-TMA 5 in preventing the AIP-mediated activation of AgrC (Figure 2). Significantly, the three classes of negative allosteric modulators appeared to act by altering the efficacy of the natural ligand AIP-1 with no noticeable effect on the affinity of AIP-1 to cognate AgrC receptor. For example, in the presence of increasing concentrations of 5-HE-C10-TMA 5 (from 0 to 100 μM), the maximum level of activation was decreased by over 10-fold while the EC50 of AIP-1 remained broadly in the 2–4 nM range (Figure 2b). The consequence(s) of the allosteric interaction mediated by these agr inhibitors could be prevention of AgrC receptor dimerization or interference with the interactions between the AgrC cytoplasmic domain and the response regulator protein AgrA which activates the agr P2 and P3 promoters.27 To date, we have not been able to express and purify functional recombinant AgrC protein, and therefore, further elucidation of this inhibitory mechanism and/or binding mode awaits resolution of this technical hurdle.
Figure 2.

3-Oxo-C12-HSL 1, 5-HE-C10-TMA 5, and C12-TOA 16 do not compete with AIP-1 for cognate AgrC. Dose–response curves showing the inhibition of a blaZ-based agrP3 reporter by (a) 3-oxo-C12-HSL 1, (b) 5-HE-C10-TMA 5, (c) C12-TOA 16, and (d) the competitive antagonist (Ala5)AIP-1.
To determine whether the TMAs and TOAs have potential as antistaphylococcal agents, we first examined the ability of the naturally produced P. aeruginosa TMA, 5-HE-C10-TMA 5, and the most potent TOA C14-TOA 17 to reduce the adherence of S. aureus to human nasal squamous cells, since a key risk factor for staphylococcal disease is nasal carriage.28 Figure 3a shows that C14-TOA 17 and 5-HE-C10-TMA 5 at 1 and 10 μM, respectively, substantially reduced attachment to human squamous cells.
Figure 3.

Impact of C14-TOA 17 on adherence of S. aureus to desquamated human nasal epithelial cells and experimental infection. (a) Binding of S. aureus to squamous epithelial cells before or after treatment with 5-HE-C10-TMA 5 (A, 10 μM) or C14-TOA 17 (B, 1 μM). Counts represent the number of bacterial cells adhered to 100 nasal cells. Results are expressed as a mean of three experiments performed in duplicate. (b) Frequency of arthritis and (c) arthritic index of mice treated with 17 and challenged with S. aureus. White bars represent data from the control animals (PBS treated) and gray bars animals treated with 17 (10 mg/kg body weight). Data are presented as median (horizontal lines), interquartile ranges (bars), and ranges (error bars). An arthritic index was calculated by scoring all four limbs of each animal. Comparisons of groups for weight change and arthritis score were done by Mann–Whitney U test (∗, p < 0.05). Fischer exact probability test was used to calculate statistical differences in the frequency of arthritis.
To investigate the in vivo activity of C14-TOA 17 and 5-HE-C10-TMA 5, we used the established murine arthritis infection model.29 In these experiments, mice treated with C14-TOA 17 were challenged with S. aureus. The frequency and severity of induced arthritis were assessed over a 10 day period. The compound C14-TOA 17 significantly reduced the frequency of arthritis and the arthritic index compared with the control group over the first 3–7 days (Figure 3b,c). While a reduction in synovitis and joint destruction was noted (Figure S4), treatment with C14-TOA 17 did not reduce weight loss or the numbers of viable staphylococci in the kidneys (Figure S4). These results suggest that although 17 did not reduce bacterial growth in vivo at the dose tested, it exhibits antivirulence and/or anti-inflammatory activities in vivo which impact the course of staphylococcal disease. In contrast, 5-HE-C10-TMA 5 was inactive in this murine infection model (data not shown).
Conclusions
Given the global health threat posed by multiantibiotic resistant bacteria, there is considerable interest in the discovery of antibacterial compounds that attenuate bacterial virulence rather than growth. In S. aureus, cyclic peptide mimetics of the native QS signal molecules have shown promise as agr-inhibitory virulence attenuators. These findings stimulated our search for simplified chemical structures capable of blocking agr-dependent QS. Previously, we discovered that the P. aeruginosa signal molecule 3-oxo-C12-HSL 1 inhibited agr, and so we utilized this compound as a starting point to investigate the key structural features involved, mechanism of action, and in vivo efficacy.
Intriguingly, changes in 1 resulted mostly in the complete loss of activity. These subtle structural changes in 1 that resulted in the loss of QS inhibitory properties require further investigation and may involve differential access via the cell wall to the cytoplasmic membrane. Fatty acids such as oleate (nontoxic) and palmitoleate (growth inhibitor), for example, show structure-specific antistaphylococcal activity that depends on the cell wall teichoic acids.30
Furthermore, we have identified two new related nonpeptidic classes of compounds, the TMAs and TOAs, as noncompetitive AgrC inhibitors that are more potent than 3-oxo-C12-HSL 1. The TMAs are rearrangement products of their corresponding 3-oxoacylhomoserine lactones. These compounds act as negative allosteric modulators by affecting cognate ligand efficacy. The most potent compound identified, C14-TOA 17, reduced colonization of human nasal epithelial cells and showed efficacy in a staphylococcal experimental mouse infection model without obvious toxicity. Consequently, our findings offer additional opportunities to exploit this chemical architecture for the discovery of more potent analogues for probing agr-dependent QS and for evaluation as antivirulent agents.
Experimental Section
Compounds and Chemistry
Synthesis procedures are in Supporting Information. The chemical structures of the presented compounds were verified by 1H and 13C NMR and high-resolution mass spectrometry. The purity (>95%) was established by RP-HPLC and 1H NMR spectrometry. 1H NMR spectra were recorded in CDCl3 or DMSO-d6 on a Bruker Avance-400 operating at 400 MHz. 13C NMR spectra were recorded on a Bruker Avance-400 or Bruker Avance(III)-500 operating at 100 or 125 MHz, respectively. High-resolution electrospray (ES) mass spectra were recorded using Waters Micromass LCT spectrometer. Analytical RP-HPLC was used to establish purity and was performed using a Waters setup comprising two 510 pumps, a 2487 dual λ absorbance detector, and Millennium software. The separation was performed using a Phenomenex Onyx monolithic C18 column (4.6 mm × 100 mm) and a linear gradient of 30–70% solvent B in 16.0 min, then 70–100% B in 1.0 min at a flow rate of 3.0 mL/min. Solvent A was 0.06% TFA in H2O, and solvent B was 0.06% TFA in MeCN/H2O (90:10). UV detection was at 214 and 254 nm.
General Procedure for the Synthesis of (S)-3-Acyl-5-(2-hydroxyethyl)tetramic Acids
A solution of sodium methoxide in MeOH (0.5 M, 2.0 mL, 1.0 mmol) was added to a stirred solution of N-(3-oxoacyl)-l-homoserine lactone (1.0 mmol) in MeOH (3 mL) under nitrogen.8 The mixture was stirred for 3 h at 55 °C and then overnight at 50 °C. The mixture was cooled to rt and then passed through an acidic ion-exchange resin (Dowex 50 WX2-200). The resin was eluted with MeOH (30 mL). The eluents were combined and concentrated in vacuo to afford the tetramic acids as solids.
(S)-3-Decanoyl-5-(2-hydroxyethyl)tetramic Acid (5-HE-C10-TMA, 5)
Using the above general procedure and N-(3-oxododecanoyl)-l-homoserine lactone gave the desired compound as a cream solid (68%). 1H NMR (CDCl3) δ 0.90 (3H, t, Me), 1.28–1.41 (12H, m, (CH2)6Me), 1.68 (2H, m, CH2(CH2)6Me), 1.81 and 2.14 (2H, 2m, CH2CH2OH), 2.87 (2H, m, CH2(CH2)7Me), 3.84–4.01 (3H, m, ring CH and CH2CH2OH), 6.33 (1H, b, NH). 13C NMR (CDCl3) δ 14.11, 22.66, 25.91, 29.25, 29.27, 29.33, 29.40, 31.85, 33.00, 34.11, 60.91, 61.81, 100.53, 175.13, 189.98, 195.51. ES-MS m/z 298.2002 [M + H], C16H28NO4+ requires 298.2018. RP-HPLC tR = 9.13 min.
(S)-3-Dodecanoyl-5-(2-hydroxyethyl)tetramic Acid (5-HE-C12-TMA, 7)
Using the above procedure and N-(3-oxotetradecanoyl)-l-homoserine lactone gave 7 as an off-white solid (75%). [α]D −7.1 (c 1.42, CHCl3). 1H NMR (CDCl3) δ 0.90 (3H, t, Me), 1.28–1.40 (16H, m, (CH2)8Me), 1.68 (2H, m, CH2(CH2)8Me), 1.83 and 2.15 (2H, 2m, CH2CH2OH), 2.86 (2H, m, CH2(CH2)9Me), 3.84–4.01 (3H, m, ring CH and CH2CH2OH), 6.33 (1H, b, NH). 13C NMR (CDCl3) δ 14.12, 22.69, 23.36, 25.91, 28.99, 29.28, 29.33, 29.44, 29.53, 29.60, 29.80, 31.91, 32.96, 34.09, 60.96, 61.85, 65.95, 100.57, 175.10, 189.94, 195.47. ES-MS m/z 326.2345 [M + H], C18H32NO4+ requires 326.2331. RP-HPLC tR = 13.17 min.
General Procedure for Synthesis of 3-Acyltetronic Acids
N,N′-Dicyclohexylcarbodiimide (2.3 mmol) was added to a stirred solution of an alkanoic acid (2.0 mmol) and 4-(dimethylamino)pyridine (3.0 mmol) in dry CH2Cl2 (20 mL) at rt under nitrogen atmosphere. Tetronic acid (2.1 mmol) was then added, and the mixture was stirred overnight at rt. The mixture was filtered, and the filtrate was extracted with 1 M aq HCl (2 × 10 mL). The organic extract was dried over MgSO4 and concentrated in vacuo to dryness to give, following crystallization, the desired 3-acyltetronic acids.
3-Dodecanoyltetronic Acid (C12-TOA, 16)
The use of dodecanoic acid in the above procedure gave 16 as a cream solid in 64% yield. 1H NMR (CDCl3) δ 0.90 (3H, t, Me), 1.28–1.41 (16H, m, (CH2)8Me), 1.72 (2H, m, CH2(CH2)8Me), 2.95 (2H, m, CH2(CH2)9Me), 4.58 and 4.70 (2H, 2s, ring CH2). 13C NMR (CDCl3) δ 14.11, 22.65, 25.22, 29.19, 29.32, 29.38, 29.49, 29.56, 29.58, 31.90, 49.25, 68.80, 99.96, 168.34, 192.67, 197.99. ES-MS m/z 281.1743 [M – H], C16H25O4– requires 281.1753. RP-HPLC tR = 11.71 min.
3-Tetradecanoyltetronic Acid (C14-TOA, 17)
The use of tetradecanoic acid in the above procedure gave 17 as a pale gray solid in 73% yield. 1H NMR (CDCl3) δ 0.90 (3H, t, Me), 1.28–1.41 (20H, m, (CH2)10Me), 1.72 (2H, m, CH2(CH2)10Me), 2.95 (2H, m, CH2(CH2)11Me), 4.58 and 4.70 (2H, 2s, ring CH2). 13C NMR (CDCl3) δ 14.12, 22.69, 24.92, 25.59, 29.19, 29.22, 29.35, 29.39, 29.57, 29.64, 29.66, 31.92, 38.73, 68.16, 128.81, 177.17, 192.31, 197.88. ES-MS m/z 309.2061 [M – H], C18H29O4– requires 309.2066. RP-HPLC tR = 16.78 min.
Bacterial Strains and Growth
Staphylococci were cultured in LB or Mueller–Hinton (MH) or CYGP broth26 and incubated with shaking at 37 °C. For agr inhibition assays, the agrP3::blaZ reporter strain S. aureus 6390B (pRN6683) was employed. Growth curves in the presence or absence of test compounds were derived by determining optical density at 600 nm (OD600) over 18 h.
S. aureus agrP3::blaZ Reporter Assays
The agr inhibition assays were carried out as described previously.9,26 Briefly, S. aureus RN6390B (pRN6683) was grown at 37 °C overnight in CYGP containing 5 μg/mL chloramphenicol. The culture was diluted 1/100 into fresh CYGP and grown at 37 °C with shaking to log phase (OD600 ≈ 0.4) and used to inoculate a 96-well microtiter plate containing a range of concentrations of the agr activator AIP-1 (0.01 nM to 10 μM) and the test compounds (0–100 μM). Plates were incubated at 37 °C for 55 min, and the reaction was quenched by the addition of 50 μL of a 5 mM sodium azide solution in CYGP broth. β-Lactamase activity was determined by adding 50 μL of a 125 μg/mL solution of the chromogenic nitrocefin. Experiments were carried out in triplicate on at least two independent occasions. Data were analyzed using the PRISM2 (GraphPad) program to obtain IC50 values.
Antibacterial Activity
MICs were determined in MH broth. Bacteria were grown overnight with shaking (200 rpm) at 37 °C, diluted with fresh MH broth to ∼1 × 106 cells/mL, and dispensed into 96-well microtiter plates. Each compound was evaluated in triplicate at concentrations from 0 to 100 μM and each experiment repeated at least three times. After incubation at 37 °C overnight, MICs were determined by visual inspection and by measurement of OD600.
Acknowledgments
This work was funded by the Medical Research Council UK (Grant G9219778), which is gratefully acknowledged. We are grateful to Dr. Stephan Heeb for his assistance with figures preparation.
Glossary
Abbreviations Used
- agr
accessory gene regulator
- AIP
autoinducing peptide
- CAS
chrome azurol S
- MIC
minimum inhibitory concentration
- 3-oxo-C12-HSL
N-(3-oxododecanoyl)-l-homoserine lactone
- QS
quorum sensing
- SAR
structure–activity relationship
- di-8-ANEPPS
1-(3-sulfonatopropyl)-4-[β-2-(di-n-octylamino)-6-naphthylvinyl]pyridinium betaine
- TMA
tetramic acid
- TOA
tetronic acid
Supporting Information Available
Scheme S1, Figures S1–S4, Tables S1 and S2, and experimental procedures for the chemical synthesis and characterization of N-acyl-l-homoserine lactone, TMA, and TOA analogues and the biochemical, biological, and in vivo infection model tests. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ R.C.C.: Department of Microbiology, Cornell University, Wing Hall, Ithaca, NY, U.S.
Author Present Address
# S.R.C.: School of Biological Sciences, University of Reading, Whiteknights, Reading, RG6 6AJ, U.K.
Author Contributions
∞ E.J.M. and R.C.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Williams P.; Winzer K.; Chan W. C.; Cámara M. Look who’s talking: communication and quorum sensing in the bacterial world. Phil. Trans. R. Soc., B 2007, 362, 1119–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P.; Cámara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [DOI] [PubMed] [Google Scholar]
- Kravchenko V. V.; Kaufmann G. F.; Mathison J. C.; Scott D. A.; Katz A. Z.; Grauer D. C.; Lehmann M.; Meijler M. M.; Janda K. D.; Ulevitch R. J. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science 2008, 321, 259–263. [DOI] [PubMed] [Google Scholar]
- Jahoor A.; Patel R.; Bryan A.; Do C.; Krier J.; Watters C.; Wahli W.; Li G.; Williams S. C.; Rumbaugh K. P. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J. Bacteriol. 2008, 190, 4408–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S. R.; Harty C.; Hooi D. S. W.; Daykin M.; Williams P.; Pritchard D. I.; Bycroft B. W. Synthetic analogues of bacterial quorum sensing molecules as immune modulators. J. Med. Chem. 2003, 46, 97–104. [DOI] [PubMed] [Google Scholar]
- Jadhav G. P.; Chhabra S. R.; Telford G.; Hooi D. S.; Righetti K.; Williams P.; Kellam B.; Pritchard D. I.; Fischer P. M. Immunosuppressive but non-LasR-inducing analogues of the Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone. J. Med. Chem. 2011, 54, 3348–3359. [DOI] [PubMed] [Google Scholar]
- Hogan D. A.; Vik A.; Kolter R. A Pseudomonas aeruginosa quorum sensing molecule influences Candida albicans morphology. Mol. Microbiol. 2004, 54, 1212–1223. [DOI] [PubMed] [Google Scholar]
- Kaufmann G. F.; Sartorio R.; Lee S.-Y.; Rogers C. J.; Meijler M. M.; Moss J. A.; Clapham B.; Brogan A. P.; Dickerson T. J.; Janda K. D. Revisiting quorum sensing: discovery of additional chemical and biological functions for 3-oxo-N-acylhomoserine lactones. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi S.; Middleton B.; Muharram S. H.; Cockayne A.; Hill P.; O’Shea P.; Chhabra S. R.; Cámara M.; Williams P. N-Acylhomoserine lactones antagonize virulence gene expression and quorum sensing in Staphylococcus aureus. Infect. Immun. 2006, 74, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C. P.; Williams P.; Chan W. C. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J. Med. Chem. 2013, 56, 1389–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates E. A.; Philipp B.; Buckley C.; Atkinson S.; Chhabra S. R.; Sockett R. E.; Goldner M.; Dessaux Y.; Cámara M.; Smith H.; Williams P. N-Acylhomoserine lactones undergo lactonolysis in a pH-, temperature-, and acyl chain length-dependent manner during growth of Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Infect. Immun. 2002, 70, 5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert R.; Schlenk A. Tetramic and tetronic acids: an update on new derivatives and biological aspects. Bioorg. Med. Chem. 2008, 16, 4203–4221. [DOI] [PubMed] [Google Scholar]
- Ueda C.; Tateda K.; Horikawa M.; Kimura S.; Ishii Y.; Nomura K.; Yamada K.; Suematsu T.; Inoue Y.; Ishiguro M.; Miyairi S.; Yamaguchi K. Anti-Clostridium difficile potential of tetramic acid derivatives from Pseudomonas aeruginosa quorum-sensing autoinducers. Antimicrob. Agents Chemother. 2010, 54, 683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery C. A.; Park J.; Gloeckner C.; Meijler M. M.; Mueller R. S.; Boshoff H. I.; Ulrich R. L.; Barry C. E. 3rd; Bartlett D. H.; Kravchenko V. V.; Kaufmann G. F.; Janda K. D. Defining the mode of action of tetramic acid antibacterials derived from Pseudomonas aeruginosa quorum sensing signals. J. Am. Chem. Soc. 2009, 131, 14473–14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda N. H.; Kimura S.; Tateda K.; Horikawa M.; Ueda C.; Ishii Y.; Ishiguro M.; Miyairi S.; Yamaguchi K. Roles of Pseudomonas aeruginosa autoinducers and their degradation products, tetramic acids, in bacterial survival and behavior in ecological niches. Microbes Environ. 2011, 26, 160–164. [DOI] [PubMed] [Google Scholar]
- Romano A. A.; Hahn T.; Davis N.; Lowery C. A.; Struss A. K.; Janda K. D.; Böttger L. H.; Matzanke B. F.; Carrano C. J. The Fe(III) and Ga(III) coordination chemistry of 3-(1-hydroxymethylidene) and 3-(1-hydroxydecylidene)-5-(2-hydroxyethyl)pyrrolidine-2,4-dione: novel tetramic acid degradation products of homoserine lactone bacterial quorum sensing molecules. J. Inorg. Biochem. 2012, 107, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [DOI] [PubMed] [Google Scholar]
- Tal-Gan Y.; Stacy D. M.; Foegen M. K.; Koenig D. W.; Blackwell H. E. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group III autoinducing peptide. J. Am. Chem. Soc. 2013, 135, 7869–7882. [DOI] [PubMed] [Google Scholar]
- Hall P. R.; Elmore B. O.; Spang C. H.; Alexander S. M.; Manifold-Wheeler B. C.; Castleman M. J.; Daly S. M.; Peterson M. M.; Sully E. K.; Femling J. K.; Otto M.; Horswill A. R.; Timmins G. S.; Gresham H. D. Nox2 modification of LDL is essential for optimal apolipoprotein B-mediated control of agr type III Staphylococcus aureus quorum-sensing. PLoS Pathog. 2013, 9, e1003166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S. R.; Stead P.; Bainton N. J.; Salmond G. P. C.; Stewart G. S. A. B.; Williams P.; Bycroft B. W. Autoregulation of carbapenem biosynthesis in Erwinia carotovora by analogues of N-(3-oxohexanoyl)-l-homoserine lactone. J. Antibiot. 1993, 46, 441–454. [DOI] [PubMed] [Google Scholar]
- Ivanov A. S. Meldrum’s acid and related compounds in the synthesis of natural products and analogs. Chem. Soc. Rev. 2008, 37, 789–811. [DOI] [PubMed] [Google Scholar]
- Nomura K.; Hori K.; Arai M.; Yoshii E. An efficient method for 3(C)-acylation of tetronic acids. Chem. Pharm. Bull. 1986, 34, 5188–90. [Google Scholar]
- Schaible U. E.; Kaufmann S. H. E. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953. [DOI] [PubMed] [Google Scholar]
- Nomura K.; Iida T.; Hori K.; Yoshii E. Synthesis of γ-unsubstituted-α-acyl-β-tetronic acids from aldehydes. J. Org. Chem. 1994, 59, 488–490. [Google Scholar]
- Davis B. M.; Jensen R.; Williams P.; O’Shea P. The interaction of N-acylhomoserine lactone quorum sensing signaling molecules with biological membranes: implications for inter-kingdom signaling. PLoS One 2010, 5, e13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell P.; Affas Z.; Reynolds C.; Holden M. T. G.; Wood S. J.; Saint S.; Cockayne A.; Hill P. J.; Dodd C. E. R.; Bycroft B. W.; Chan W. C.; Williams P. Structure, activity and evolution of the group I thiolactone peptide quorum sensing system of Staphylococcus aureus. Mol. Microbiol. 2001, 41, 503–512. [DOI] [PubMed] [Google Scholar]
- George Cisar E. A.; Geisinger E.; Muir T. W.; Novick R. P. Symmetric signalling within asymmetric dimers of the Staphylococcus aureus receptor histidine kinase AgrC. Mol. Microbiol. 2009, 74, 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien L. M.; Walsh E. J.; Massey R. C.; Peacock S. J.; Foster T. J. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell. Microbiol. 2002, 4, 759–770. [DOI] [PubMed] [Google Scholar]
- Gjertsson I.; Jonsson I.-M.; Peschel A.; Tarkowski A.; Lindholm C. Formylated peptides are important virulence factors in Staphylococcus aureus arthritis in mice. J. Infect. Dis. 2012, 205, 305–311. [DOI] [PubMed] [Google Scholar]
- Parsons J. B.; Yao J.; Frank M. W.; Jackson P.; Rock C. O. Membrane disruption by antimicrobial fatty acids releases low molecular weight proteins from Staphylococcus aureus. J. Bacteriol. 2012, 194, 5294–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.