

Abstract
Replication-competent latent HIV-1 proviruses that persist in the genomes of a very small subset of resting memory T cells in infected individuals under life-long antiretroviral therapy present a major barrier towards viral eradication. Multiple molecular mechanisms are required to repress the viral trans-activating factor Tat and disrupt the regulatory Tat feedback circuit leading to the establishment of the latent viral reservoir. In particular, latency is due to a combination of transcriptional silencing of proviruses via host epigenetic mechanisms and restrictions on the expression of P-TEFb, an essential co-factor for Tat. Induction of latent proviruses in the presence of antiretroviral therapy is expected to enable clearance of latently infected cells by viral cytopathic effects and host antiviral immune responses. An in-depth comprehensive understanding of the molecular control of HIV-1 transcription should inform the development of optimal combinatorial reactivation strategies that are intended to purge the latent viral reservoir.
Keywords: HIV latency, epigenetics, Tat, P-TEFb, inducible proviruses, latency reversal, NF-κB, 7SK snRNP, BRD4
Introduction
Potent combination antiretroviral therapy (HAART) can effectively block viral replication in the host and reduce the circulating virus to undetectable levels. Although patients adhering to the HAART regimen have minimal viremia, HIV persists due to the existence of latent but replication-competent proviruses in a very small population of resting memory CD4+ T cells (~1 in 106 cells) in peripheral blood (Finzi et al., 1999) and tissues (Yukl et al., 2010). Viral persistence during antiretroviral therapy has also been attributed to ongoing low-level viral replication in anatomical sanctuary sites (eg. gut-associated lymphoid tissues, other lymphoid tissues, genital tract and the central nervous system) that may be comparatively inaccessible to drugs (Cory et al., 2013; Palmer et al., 2011).
In virtually all patients, when HAART is interrupted circulating HIV levels rebound within 2 to 8 weeks and uncontrolled replication resumes (Chun et al., 1999; Stöhr et al., 2013) although a few rare patients are able to spontaneously control their infections (Sáez-Cirión et al., 2013). However, residual virus recovered from treated patients (Brennan et al., 2009), and the rebounding virus recovered during the short treatment interruptions (Joos et al., 2008), have much less sequence heterogeneity than seen in actively replicating viral populations, suggesting that replenishment of the viral population arises from reactivation of a very restricted set of latently infected cells. HIV-1 subsets in the GALT of patients under HAART initiated at the acute phase have low diversity, suggesting highly restricted viral replication in this anatomical compartment (Evering et al., 2012).
HIV-1 infects CD4+ T lymphocytes, and to a lesser extent, myeloid cells including perivascular macrophages, microglia, astrocytes and dendritic cells in the brain. Both T-cells and myeloid cells can establish latent infections (Marban et al., 2007; Wires et al., 2012). Latently infected T-cells are usually established during the acute phase (Chun et al., 1998), are undetectable by host immune surveillance, are extremely stable (t1/2 ~44 months) (Siliciano et al., 2003; Siliciano et al., 2007), and can be replenished through homeostatic proliferation (Chomont et al., 2009).
The latent viral reservoir has therefore proven to be the major obstacle towards finding a sterilizing cure or even a functional cure for HIV-1 infection. As part of current efforts to identify a broad-based cure for HIV infection, research has focused on identifying drugs capable of inducing latent HIV reservoirs without causing deleterious host systemic inflammatory responses (“shock”). Reactivation of proviruses in the presence of HAART is expected to make them visible to the immune system and lead to the clearance of latently infected cells (“kill”). For instance, a recent promising study by Shan et al. demonstrated that latently infected resting CD4+ T cells treated with the histone deacetylase inhibitor SAHA to reactivate proviral gene expression were susceptible to efficient killing in vitro by HIV antigen-specific CD8+ T cells (Shan et al., 2012).
This article will review our knowledge of these intricate mechanisms involved in the establishment and maintenance of latent HIV infections and how these insights are informing the development of therapies that are intended to purge latent HIV reservoirs. Over the last 20 years numerous factors involved in the regulation of HIV transcription have been identified. To help the reader, Table 1 provides a glossary and overview of the key factors referred to in this review.
Table 1.
Host factors involved in the regulation of HIV proviral transcription
| Factor or complex | Description | Reference |
|---|---|---|
| Initiation | ||
| Sp1 | Required for assembly of the pre- initiation complex at the LTR TATA box. Co-operatively interacts with NF-κB p65/p50. | (Perkins et al., 1993) |
| TFIIH | Component of the pre-initiation complex whose recruitment to the HIV LTR is NF-κB-dependent. Stimulates RNAPII to clear the promoter and initiate transcription. | (Kim et al., 2006) |
| NF-kB (p65/p50) | Central to the signal-dependent activation of HIV transcription initiation. Enables recruitment of histone acetyltransferases (HATs) to the LTR. | (Nabel and Baltimore, 1987) (Gerritsen et al., 1997) |
| NFAT | Calcium-dependent activator of HIV transcription initiation. NFAT recognition sequences on the viral promoter overlap with NF-κB elements. | (Chan et al., 2013) (Giffin et al., 2003) |
| AP-1 | Nuclear complex of c-fos and c-jun that is formed upon T-cell activation and can directly bind the HIV LTR and enhance NF-κB activity. | (Yang et al., 1999) |
| Repression | ||
| LSF/YY1 | Cooperative recruitment of LSF and YY1 to the LTR enables repression via recruitment of HDAC1. | (He and Margolis, 2002) |
| CBF-1 | CBF1 interacts with the LTR and represses proviral transcription by facilitating recruitment of HDAC1 and its co-repressor subunits CIR and mSin3A, | (Tyagi and Karn, 2007) (Tyagi et al., 2010) |
| p50/p50 | Binding of p50 homodimers to the κB elements permits recruitment of HDAC1. | (Williams et al., 2006) |
| E2F1 | Can partner with p50 to bind κB elements and suppress transcription initiation, probably by recruiting BRD2. | (Kundu et al., 1997) (Karn, 2013) |
| CTIP2 | Suppresses HIV transcription initiation in microglial cells in cooperation with COUP-TF and Sp1. Recently identified as a regulatory component of 7SK snRNP in T-cells. | (Marban et al., 2007) (Cherrier et al., 2013) |
| BRD2 | Present at the promoter of latent proviruses. Suppresses proviral transcription initiation, possibly through an interaction with E2F1. | (Boehm et al., 2013) (Karn, 2013) |
| Chromatin Modification | ||
| HDACs | Histone deacetylases HDAC1, HDAC2, and HDAC3 are each involved in establishment and maintenance of HIV latency. | (Keedy et al., 2009) |
| HATs | The histone acetyltransferases p300/CBP and P/CAF reverse chromatin blocks and are recruited to the viral promoter by NF-κB and NFAT. | (Gerritsen et al., 1997) |
| SUV39H1 | Trimethylates histone H3 Lys9 (H3K9me3), an epigenetic marker of HIV latency. | (du Chene et al., 2007) |
| G9a | Dimethylates histone H3 Lys9 (H3K9me2). | (Imai et al., 2010) |
| EZH2 | Histone methyltransferase component of PRC2. Responsible for histone H3 Lys27 trimethylation (H3K27me3) found on silenced proviral templates. | (Friedman et al., 2011) |
| DNMT1 | Methylates CpG islands at the HIV LTR. | (Blazkova et al., 2012) |
| Chromatin Remodeling | ||
| BAF | SWI/SNF chromatin remodeling complex which positions proviral Nuc-1 to repress transcription. | (Rafati et al., 2011) |
| PBAF | Replaces BAF at the viral LTR to remodel Nuc-1 and enable transcription. | (Rafati et al., 2011) |
| Promoter-proximal pausing | ||
| DSIF | Heterodimer of hSpt4 and hSpt5 which interacts with RNAPII, mRNA capping enzyme, and nascent RNA. Stimulates 5′ RNA capping and recruitment of NELF. Phosphorylation of hSpt5 by P-TEFb transforms DSIF into a positive elongation factor. | (Yamaguchi et al., 2002) |
| NELF | Heterotetramer that binds TAR RNA and a preformed DSIF/RNAPII complex. Induces premature termination and formation of a stable RNAPII paused complex. | (Pagano et al., 2014) (Yamaguchi et al., 2002) |
| Microprocessor | Processes TAR RNA leading to recruitment of Setx, Xrn2, and Rrp6. These factors cooperatively induce RNAPII pausing and abortive transcription. | (Wagschal et al., 2012) |
| Elongation | ||
| P-TEFb | Heterodimer of CDK9 and cyclin T1. Recruited by Tat to paused RNAPII at TAR RNA. Stimulates processive transcription by phosphorylating RNAPII, E subunit of NELF and hSpt5 subunit E subunit of NELF and hSpt5 subunit of DSIF. | (Yamada et al., 2006) (Fujinaga et al., 2004) (Czudnochowski et al., 2012) |
| Super Elongation Complex (SEC) | Complex comprising the factors shown in Fig. 3A. AFF4 and AFF1 organize the assembly of Tat/P-TEFb into the SEC. ELL proteins of Tat/P- TEFb into the SEC. ELL proteins stimulate efficient elongation by preventing RNAPII pausing and backtracking. AF9 and ENL link the SEC to RNAPII. | (He et al., 2011) (Chou et al., 2013) |
| Tat modifiers | Histone modifying enzymes that regulate the trans-activating function of Tat by modifying its P-TEFb or Arg-rich RNA binding regions. These include P/CAF, SIRT1 and LSD1/CoREST. | (Ott et al., 2011) (Lu et al., 2013) |
| Co-transcriptional processing | ||
| SKIP | Splicing factor that is recruited by Tat/P-TEFb to the HIV LTR. Associates with activated spliceosomes within the transcribed region of the proviral gene. | (Bres et al., 2005) |
| P-TEFb regulation | ||
| 7SK snRNP | Provides an exchangeable pool of P-TEFb to support transcription of proviral and cellular genes. Contains 7SK snRNA, HEXIM, LARP7 and MEPCE. | (Nguyen et al., 2001) (Yang et al., 2001) |
| CDK7 | Kinase component of TFIIH. Recently identified as the CDK9-activating kinase. | (Larochelle et al., 2012) |
| PPM1A | Inactivates CDK9 via dephosphorylation of its activation loop. | (Budhiraja et al., 2012b) |
| BRD4 | Extracts P-TEFb from 7SK snRNP and directs it to cellular genes. Competes with Tat for P-TEFb binding. | (Jang et al., 2005) (Bisgrove et al., 2007) |
Regulation of HIV transcription by Tat
Control of latency by the Tat feedback mechanism
There are no specific repressors of proviral transcription encoded by HIV. Instead, the entry into latency is a consequence of multiple cellular restrictions at the promoter and elongation phase of viral transcription that ultimately repress expression of the viral trans-activating factor Tat and disrupt the positive regulatory Tat feedback circuit (Fig. 1) (Karn, 2011). In particular, transcriptional silencing of proviruses via epigenetic and non-epigenetic mechanisms, and regulation of the essential Tat-cofactor P-TEFb, are critical for the establishment and maintenance of latent infections.
Fig. 1.
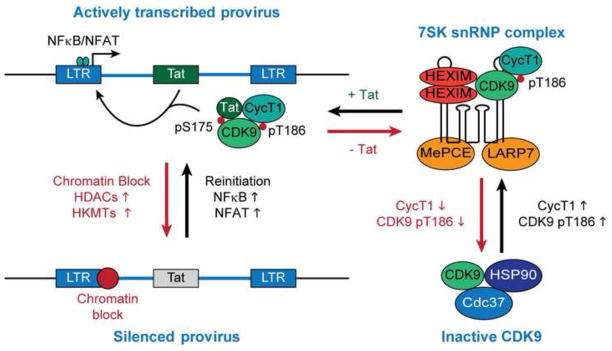
Autoregulation of HIV transcription by Tat via recruitment of P-TEFb from 7SK snRNP. Inactivation of the HIV promoter, mainly due to epigenetic silencing, drives down Tat to below its threshold levels leading to entry of the provirus into latency, whereas stimulation of transcription initiation induces Tat production. Tat is able to extract the P-TEFb enzyme complex from the inhibitory 7SK snRNP. Competitive binding of Tat to P-TEFb is favored by signal-dependent modification of the activation loop of the CDK9 subunit.
The vast majority of productive HIV infections occur in activated, or at least partially activated, T-cells. Activated effector CD4+ T cells have an intracellular environment that makes them highly permissive for productive infection by HIV (Siliciano and Greene, 2011), however, the transition of infected effector cells to a resting memory phenotype substantially diminishes transcription initiation at the HIV LTR which enables the establishment of transcriptionally silent proviruses. Following re-stimulation, latently infected resting memory CD4+ T-cells convert to a permissive intracellular environment with high levels of P-TEFb and transcription initiation factors in the nucleus. The central role of the Tat feedback regulatory mechanism to reactivate HIV from latency is clearly demonstrated by two observations: First, expression of Tat in trans from an ectopic promoter prevents viral entry into latency (Pearson et al., 2008); and second, latent proviruses encoding a functionally defective Tat mutant (C22G) can initiate transcription following T-cell activation but are absolutely defective for viral gene expression (Kim et al., 2011).
Structure and function of the proviral long terminal repeat
The U3 and R regions of the proviral 5′ long terminal repeat (LTR) act as the viral promoter. Although both the 5′ LTR and 3′ LTR have identical sequences, concomitant transcription from the 3′ LTR is highly inefficient due to interference of its U3 region by positive-sense transcribing complexes that terminate RNA synthesis at the R/U5 junction (Cullen et al., 1984; Gallastegui et al., 2011).
The viral core promoter comprises three tandem Sp1 binding sites, a TATA box, and an initiator element at the transcription start site. The enhancer and modulatory regions of the LTR contain cis-acting recognition elements for regulatory cellular transcription factors. Specifically, NF-κB p65/p50 heterodimers, NFAT, STAT5 and AP-1 have been identified as signal-dependent positive regulators of HIV transcription initiation (Kinoshita et al., 1997; Nabel and Baltimore, 1987; Selliah et al., 2006; Yang et al., 1999).
Immediately downstream of the transcription start site is the TAR sequence which encodes an RNA stem-loop structure that is directly bound by Tat in order to mediate recruitment of a cellular elongation factor complex that stimulates efficient transcription elongation (Dingwall et al., 1990; Kao et al., 1987).
Transcriptional pausing of RNAPII at the proviral LTR
One of the most insightful recent advances in studies of transcriptional control is that the majority of mammalian protein-encoding genes carry poised RNAP II in the promoter proximal region (Core and Lis, 2008). The paused RNAP II permits rapid responses to transcription signals. Not surprisingly, recent studies from our laboratory have shown that latent HIV proviruses is also always primed for rapid transcriptional responses due to promoter proximal pausing of RNAP II (Jadlowsky et al., 2014) (Fig. 2A). Promoter proximal pausing accounts for the characteristic formation of short transcripts of ~60 nucleotides that accumulate following processing of the nascent transcripts produced by abortive RNAP II escape (Laspia et al., 1989) and is seen in both actively transcribing and latently infected cells. Recently, Wagschal et al. (2012) have proposed that the short transcripts are generated by processing of TAR RNA by microprocessor, the termination factors Setx and Xrn2, and the 3′ to 5′ exoribonuclease, Rrp6.
Fig. 2.

Epigenetic regulation and Tat transactivation of proviral transcription. (A) Latent proviruses are characterized by the presence of histone deacetylases (HDACs), histone methyltransferases (HMTs), and their associated modifications at Nuc-1 situated around the transcription start site. The deacetylated proviral chromatin becomes a target for additional silencing via recruitment of polycomb repressive complex-2 (PRC2) which mediates histone methylation. Viral transcripts are inefficiently synthesized but over time there is a build-up of transcriptionally paused polymerase complexes due to the absence of Tat. Escaping RNAP II complexes are forced into abortive transcription due to NELF. (B) Productive transcription. Efficient assembly of pre-initiation complexes due to chromatin remodeling at the HIV LTR is a signal-dependent process that is driven by the activity of NF-κB/NFAT and associated co-activators such as p300. The transitioning of paused polymerase into productive elongation requires kinase activity of P-TEFb. Tat efficiently transactivates HIV transcription elongation by recruiting P-TEFb to the paused complex at the TAR hairpin.
A striking feature of elongation control of the HIV promoter is that both the restriction of elongation in the absence of Tat and the efficiency of elongation in the presence of Tat is much stronger than typically seen in cellular promoters (Kim et al., 2011). Combined surveillance by the negative elongation factor (NELF) and the DRB sensitivity-inducing factor (DSIF) is used to block productive elongation in the absence of Tat (Fig. 2). Human DSIF, a two-subunit complex of hSpt4 and hSpt5, is recruited to newly initiated transcription complexes and stimulates capping (Wen and Shatkin, 1999). Spt5 forms direct contact with nascent RNA as it emerges from the RNAPII exit site (Cheng and Price, 2008; Missra and Gilmour, 2010) and facilitates the recruitment of NELF (Yamaguchi et al., 2002). NELF is able to force premature termination over a range of several hundred nucleotides (Renner et al., 2001), limiting the escape of transcription complexes from the promoter proximal pause site. Pagano et al. (2014) have recently characterized a consensus NELF subunit E binding element (CUGAGGA) on nascent cellular RNAs associated with transcriptionally paused genes, which is homologus to a NBE-like element (CUGGGA) present within the loop region of TAR RNA, and may lead to enhanced NELF recruitment to the HIV LTR. Thus, knockdown of NELF by shRNA leads to enhanced basal HIV transcription and escape from latency (Jadlowsky et al., 2014; Natarajan et al., 2013; Zhang et al., 2007).
Stimulation of proviral transcription by Tat/P-TEFb
As summarized diagrammatically in Fig. 2B, the switch from promoter proximal pausing to productive elongation is mediated by Tat and the positive transcription elongation factor (P-TEFb) (Herrmann and Rice, 1995; Wei et al., 1998). P-TEFb contains the CDK9 serine/threonine kinase and cyclin T. Human cyclin T1 is the only cyclin T that can interact with Tat due to the formation of Zn2+-coordinated interactions with the N-terminal cysteine-rich region of Tat (Cys22-Cys37) (Bieniasz et al., 1998; Fujinaga et al., 1998; Garber et al., 1998). Crystallographic studies (Tahirov et al., 2010) have shown that the binding of Tat to CDK9/cyclin T1 (P-TEFb) allows the viral factor to establish partial helical secondary structure and induces significant conformational changes in P-TEFb, allowing co-operative recognition and stable binding of Tat and the cyclin T1 subunit of P-TEFb to TAR (Wei et al., 1998).
Recruitment of P-TEFb at TAR enables CDK9 to phosphorylate NELF-E and force its dissociation from TAR RNA and the paused polymerase complex (Fujinaga et al., 2004; Jadlowsky et al., 2014; Yamaguchi et al., 2002; Zhang et al., 2007). CDK9 also extensively phosphorylates the CTD of RNAPII mainly at Ser2 and Ser 5 residues of the heptad repeats (Czudnochowski et al., 2012), and homologous heptapeptide repeats (G-S-Q/R-T-P) of hSpt5 at Thr4 residues which converts it into a stimulatory elongation factor (Bourgeois et al., 2002; Yamada et al., 2006). Therefore, the overall effect Tat and P-TEFb is to remove the blocks to elongation imposed by NELF and DSIF and to stimulate efficient elongation and co-transcriptional processing of proviral transcripts. However, because promoter proximal RNAP II is continuously replaced, there is no loss of RNAP II from the promoter proximal region (Jadlowsky et al., 2014).
Tat/P-TEFb is known to remain associated with the elongating RNAPII machinery throughout transcription (Bourgeois et al., 2002; Keen et al., 1997; Sobhian et al., 2010) raising the possibility that it exerts an influence throughout the transcription cycle. The temporal and spatial recruitment of transcriptional and RNA processing factors to actively transcribed genes is guided by the dynamic changes in the phosphorylation marks on the CTD of RNAPII that are observed to occur over the course of a transcriptional cycle (Drogat and Hermand, 2012; Perales and Bentley, 2009). Since CDK9 kinase activity is crucial for controlling regular and alternative RNA splicing and for the recruitment of polyadenylation factors at the 3′ ends of RNAPII-transcribed genes (Ahn et al., 2004; Lenasi et al., 2011) it seems likely that Tat can also influence these processes. For example, the splicing-associated c-Ski-interacting protein (SKIP) which had previously been shown to be a stable component of activated mammalian spliceosome complexes that are essential for global RNA splicing is recruited by Tat/P-TEFb to the HIV LTR (Bres et al., 2005). SKIP recruitment dramatically enhances HIV transcription elongation, alternative splicing of viral RNA, and Tat-dependent LTR-driven proviral gene expression. How splicing and transcription termination is modified by Tat remains one of the unsolved problems in HIV transcription control.
The Tat/P-TEFb super elongation complex
Although P-TEFb was regarded for many years to be a discrete elongation factor, recent work has shown that P-TEFb is an integral component of larger “super elongation complex” (SEC) (Chou et al., 2013; He et al., 2010; Sobhian et al., 2010) (Fig. 3A). siRNA knockdowns of individual SEC components strongly inhibits Tat-dependent LTR-driven reporter gene expression, with knockdown of the elongation factor ELL2 producing the greatest effect (He et al., 2010; Sobhian et al., 2010). In actively replicating cells ELL2 is present at limiting levels due to rapid protein degradation by the polyubiquitination-proteasomal pathway (He et al., 2010; Liu et al., 2012). Both Tat and the SEC scaffold protein AFF4 prevent the degradation of ELL2 leading to higher levels of SEC associated with P-TEFb (He et al., 2010; Liu et al., 2012). Thus Tat is not only able to recruit the SEC to the HIV LTR but it is also able to significantly enhance the intracellular fraction of P-TEFb that becomes associated with the SEC (He et al., 2010; Sobhian et al., 2010), providing a complementary mechanism leading to enhanced proviral reactivation.
Fig. 3.
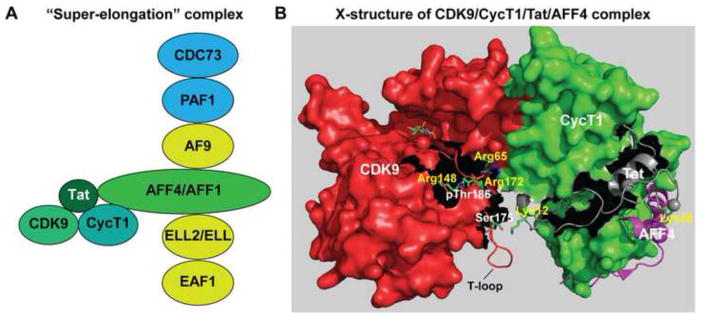
The super elongation complex (SEC). (A) Schematic diagram of the SEC. AFF4 (or AFF1) provides a molecular scaffold that organizes the assembly of the SEC. By preventing the rapid degradation of ELL2, Tat also facilitates SEC assembly at the HIV LTR. The SEC is linked to the paused polymerase complex via an interaction between ENL or AF9 and the PAF1 subunit. (B) Cartoon and surface renderings of the Tat/P-TEFb 3D structure (Tahirov et al., 2010) superposed onto AFF4/P-TEFb (Schulze-Gahmen et al., 2013) showing CDK9 T-loop contacts with Tat and the close proximity of Tat to AFF4. The T-loop of CDK9 can potentially make contacts with the N-terminal region of Tat as reported by Tahirov et al. (2010) and these interactions would be strengthened by the signal-dependent phosphorylation of Ser175. Hydrogen bonding coordination of the CDK9 Thr186 phosphate moiety by three highly conserved arginine residues reduces the flexibility of the upper T-loop region and may be essential for stabilizing the CDK9/hCycT1 heterodimer complex.
AFF4 and its structural homolog AFF1 are large flexible proteins (1218 and 1163 residues, respectively) that are believed to act as scaffolds to organize the assembly of the SEC (Chou et al., 2013; He et al., 2011). Recently a 2.9 Å resolution 3D structure of the N-terminal CycT1 interacting region of AFF4 (AFF42-73) in complex with P-TEFb was solved (Schulze-Gahmen et al., 2013), which we have modeled on the earlier P-TEFb structure (Tahirov et al., 2010), as shown in Fig. 3B. Tat and AFF4 bind to adjacent sites on hCycT1 and AFF4 can potentially form direct contacts with three Tat residues (Met1, Lys28, and Phe32). These structural observations help to explain why acetylation of Tat Lys28 by P/CAF (Bres et al., 2002; Kiernan et al., 1999) is able to stabilize the Tat/P-TEFb/TAR ternary complex while mutation of Lys28 (K28A) significantly diminish the ability of Tat and P-TEFb to trans-activate HIV in vivo (D’Orso and Frankel, 2009; Kiernan et al., 1999). Thus, Tat association with AFF4 is highly regulated by post-translational modifications and these biochemical pathways help orchestrate the recruitment of the SEC to the proviral transcription unit.
Establishment of HIV latency
Epigenetic silencing of HIV transcription
HIV preferentially integrates within intragenic regions of actively transcribed regions of the host genome (Lewinski et al., 2006). Thus, somewhat surprisingly, the majority of repressed, but inducible, proviruses are found within the introns of expressed cellular genes (Lewinski et al., 2005; Shan et al., 2011; Sherrill-Mix et al., 2013). The implication is that establishment of HIV latency is regulated independently of the control of the expression of the host gene into which the provirus has integrated.
Indeed, control of the chromatin structure formed specifically on the proviral LTR appears to be a key mechanism leading to proviral silencing. Independent of the proviral integration site, the viral 5′ LTR is invariably occupied by two precisely positioned nucleosomes namely, Nuc-0 and Nuc-1 (Verdin et al., 1993). Nuc-1 is situated around the transcription start site and therefore provides a potential block to the release of promoter proximal transcription complexes.
A wide array of epigenetic modifications contribute to the silencing of latent proviruses. In particular nucleosomes on the 5′ LTR of latent proviruses typically display deacteylated histones and trimethylated histones, a pattern of histone modifications that is associated with formation of repressive heterochromatic structures (Friedman et al., 2011; Keedy et al., 2009). In addition the histone methyltransferases (HMTs) themselves (SUV39H1, G9a, and EZH2) have been found to be associated with the latent proviral LTR (du Chene et al., 2007; Friedman et al., 2011; Imai et al., 2010; Marban et al., 2007). The dominant histone methyltransferase controlling HIV latency in T-cells is EZH2 (Friedman et al., 2011), which in addition to its H3K27 methyltransferase activity, is an important structural component of polycomb repressive complex 2 (PRC2). During heterochromatin formation PRC2 offers a binding platform for multiple chromatin-modifying enzymes including HDACs and DNA methyltransferase-1 (DNMT1) (Cheng et al., 2011; Tae et al., 2011; Vire et al., 2006). Methylation of histone H3K9 due to SUV39H1 and G9a activity additionally stablizes heterochromatin structures by tethering the chromodomain-containing adaptor proteins HP1α, -β, and -γ (du Chene et al., 2007).
Numerous trans-acting factors have been found that facilitate the recruitment of HDACs, PRC2 and other repressor complexes to the core promoter, suggesting that repression mechanisms may be both redundant and utilized in different cell types. For example, the cooperative binding of LSF and YY1 to the LTR allows YY1 to exert its repressive effect on proviral transcription by interacting with and recruiting HDAC1 to Nuc-1 (He and Margolis, 2002). Occupancy of the NF-κB element at the LTR enhancer by p50 homodimers has also been shown to mediate the recruitment of HDAC1 (Williams et al., 2006). Similarly CBF-1, a key effector of the Notch signaling pathway, recruits HDACs to LTR of latent proviruses in both infected primary CD4+ T cells and a Jurkat T-cell model of HIV latency (Tyagi and Karn, 2007; Tyagi et al., 2010). Finally, the bromodomain protein BRD2 has recently been reported to mediate repression of the HIV LTR (Boehm et al., 2013). It is possible that potential BRD2 recruitment to the proviral LTR, mediated by an interaction with the transcriptional repressor E2F1, may in turn promote the recruitment of repressor complexes carrying acetylated lysine residues (Karn, 2013).
DNA methylation at two CpG islands flanking the transcription start site has also been associated with silencing of the HIV promoter in latently infected Jurkat T cells and primary CD4+ T cells (Blazkova et al., 2009; Kauder et al., 2009). However, two recent studies analyzing the latent viral reservoir of HAART-treated aviremic patients found very low or no methylation of the LTR of their HIV-1 DNA suggesting that extensive proviral DNA methylation is unlikely to be a marker of latent proviruses (Blazkova et al., 2012; Palacios et al., 2012).
Control of HIV latency by transcriptional interference
An alternative explanation for proviral silencing is that transcriptional interference, characterized by occlusion or dislodgement of transcription initiation or elongation complexes from the provirus by readthrough transcription of the viral-containing host gene or termination of host-initiated transcripts at the polyadenylation site in the 5′ LTR inactivates the provirus (Han et al., 2008; Lenasi et al., 2008). While there are specific cases where transcriptional interference has been documented, there is only a slight preference for proviral integration in the same orientation as the host gene (63.8% vs 36.2%) in latently infected primary CD4+ T cells (Shan et al., 2011). Therefore, it seems likely that epigenetic repression of the provirus always makes a contribution to maintenance and establishment of latency.
Host and viral control of P-TEFb activation
Control of Cyclin T1 expression and P-TEFb assembly
Activation of P-TEFb in latently infected T cells is a sequential process that involves de-repression of hCycT1 expression, phosphorylation of CDK9 at Thr186, assembly of P-TEFb into 7SK snRNP, signal-dependent and Tat-dependent dissociation of 7SK snRNP, and eventually, Tat-dependent recruitment of P-TEFb and the SEC to the HIV provirus (Fig. 1). Expression of hCycT1 in resting CD4+ T cells is highly restricted at the post-transcriptional level, and this is likely to be mediated by several micro-RNAs (Budhiraja et al., 2012a; Chiang et al., 2012) and direct translational control mechanisms (Hoque et al., 2011). Levels of hCycT1 protein are elevated very rapidly (within 1 h) upon stimulation of resting T cells with cytokines, protein kinase C agonists, or ligation of the T cell receptor complex (Mbonye et al., 2013; Ramakrishnan et al., 2009; Sung and Rice, 2006). Undifferentiated monocytes which, unlike monocyte-derived macrophages and dendritic cells, are known to be non-permissive to HIV replication also show micro-RNA dependent CycT1 restriction (Chiang et al., 2012; Sung and Rice, 2009).
In contrast to hCycT1, CDK9 is constitutively expressed in resting T cells and its levels are only modestly increased after prolonged periods (>24 h) of T-cell activation (Ghose et al., 2001; Mbonye et al., 2013; Ramakrishnan et al., 2009). However, in resting cells the CDK9 kinase is inactive as a consequence of dephosphorylation of its regulatory activation loop (T-loop) at the highly conserved CDK residue Thr186 (Budhiraja et al., 2012a). Activation of T cells rapidly induces Thr186 phosphorylation of CDK9 concomitant with the elevation in hCycT1 expression (Mbonye et al., 2013; Ramakrishnan et al., 2009). Using an elegant chemical genetic approach Larochelle et al. (2012) have recently identified the CDK7 subunit of TFIIH to be the nuclear CDK9-activating kinase. Similarly, siRNA knockdown studies have identified PPM1A as the inactivating phosphatase responsible for maintaining Thr186 dephosphorylation of CDK9 in resting CD4+ T cells (Budhiraja et al., 2012b; Wang et al., 2008).
Like other CDKs, CDK9 is activated by binding of the cyclin subunit which relieves steric blockade at the catalytic cleft by inducing large conformational changes to the T-loop (Jeffrey et al., 1995; Russo et al., 1996). The 3D structures of the T-loop phosphorylated CDK9/hCycT1 and CDK2/cyclin A reveal that a hydrogen bonding coordination of the phosphate moiety by three conserved arginine residues appear to be essential for stabilizing intermolecular hydrogen bonding interactions at the CDK/cyclin dimer interface (Fig. 3) (Russo et al., 1996; Tahirov et al., 2010). Since the phosphorylation of CDK9 at Thr186 is required to stabilize its heterodimer interactions with hCycT1 (Mbonye et al., 2013; Russo et al., 1996), the majority of CDK9 in resting T cells is likely to be present within Hsp90/Cdc37 kinase-specific chaperone complexes (O’Keeffe et al., 2000).
Regulation of P-TEFb pools by the 7SK RNP complex
The majority of P-TEFb in activated T-cells is found in the 7SK small nuclear ribonucleoprotein (snRNP) complex (Kim et al., 2011; Nguyen et al., 2001; Yang et al., 2001; Yik et al., 2003). The 7SK snRNP provides an exchangeable pool from which the catalytically competent and transcriptionally active form of P-TEFb can be readily extracted and directed to genes. The sequestration of P-TEFb by 7SK snRNP in T-cells occurs shortly after its signal-dependent biogenesis and requires Thr186 phosphorylation of CDK9 (Budhiraja et al., 2012a; Chen et al., 2004; Mbonye et al., 2013).
7SK snRNA provides a scaffold for binding two molecules of P-TEFb in complex with homo- or heterodimers of the inhibitory proteins HEXIM1 or HEXIM2 (Li et al., 2005; Peterlin et al., 2012) (Fig. 1). Within the 7SK snRNP the RNA is protected from nuclease degradation by the 5′ methylphosphate capping enzyme MEPCE which remains bound at the 5′ end, and the La-related protein LARP7 which binds the polyuridine sequence at the 3′ end (Jeronimo et al., 2007; Krueger et al., 2008; Markert et al., 2008).
Recruitment of P-TEFb by Tat to the provirus and competition with BRD4
As described above, Tat is able to induce release of P-TEFb from 7SK snRNP (Fig. 1). The Tat-dependent release mechanism is thought to involve RNA binding by Tat and induction of an RNA conformational change that poises the viral factor to competitively displace HEXIM1 from association with P-TEFb (Krueger et al., 2010; Muniz et al., 2010; Sedore et al., 2007). Therefore, in addition its classical role as a trans-activator of HIV transcription these studies have established an important role for Tat as a viral activator of P-TEFb.
Formation of the Tat/P-TEFb complex is in competition with P-TEFb binding by the chromatin-binding bromodomain protein BRD4 (Jang et al., 2005; Yang et al., 2005). Like Tat, BRD4 has a well-defined C-terminal P-TEFb interacting domain (Bisgrove et al., 2007) and can induce dissociation of P-TEFb from 7SK snRNP (Krueger et al., 2010; Schroder et al., 2012). Knockdown of BRD4 in the background of a latent HIV infection, or treatment of latently infected cell lines and primary T cells with the bromodomain inhibitor JQ1, results in a robust reactivation of Tat-dependent proviral gene expression because in the absence of competing BRD4 Tat binding to P-TEFb is strongly enhanced (Bartholomeeusen et al., 2012; Boehm et al., 2013; Li et al., 2013; Mbonye et al., 2013; Zhu et al., 2012).
In addition to BRD4, other factors may help recruit P-TEFb to transcription complexes. Both the DNA-binding transcriptional repressor CTIP2 and the SR-splicing factor SRSF2 have been identified as additional regulatory components within 7SK snRNP. CTIP2 mediates the recruitment of 7SK snRNP to promoters (Cherrier et al., 2013) while SRSF2 has been proposed to aid transfer of P-TEFb to promoter-proximally paused RNAPII on cellular genes by binding nascent RNA transcripts, employing a mechanism analogous to the recruitment of P-TEFb to TAR by Tat (Ji et al., 2013).
D’Orso and colleagues (D’Orso and Frankel, 2010; D’Orso et al., 2012) have recently developed the provocative hypothesis that Tat is recruited to the DNA template before TAR is synthesized. As RNAP II transcribes through the TAR region there is an exchange that leads to displacement of P-TEFb from the promoter-associated 7SK snRNP to TAR RNA. Key experiments supporting this idea come from ChIP and ChIP-seq data showing HEXIM and other 7SK associated proteins bound to upstream sites on both HIV and cellular promoters (D’Orso and Frankel, 2010; D’Orso et al., 2012; Ji et al., 2013). However, other groups, including ourselves, have been unable to detect P-TEFb and 7SK snRNP components upstream of the transcription start site. One possible explanation for these discrepancies is that D’Orso and Frankel (D’Orso and Frankel, 2010) performed their experiments in HeLa cells which typically display a much higher level of basal HIV transcription and less restriction by heterochromatic structures compared to the latently infected Jurkat T-cell lines that we have used in our studies. It therefore possible that the P-TEFb recruitment they observed in the absence of Tat is due to residual transcription in the HeLa cell system, perhaps mediated by the high levels of BRD4 that they have found to be associated with the HIV provirus. It is also possible that the 7SK RNP complex is only loosely associated with promoters and can only be detected by ChIP after extensive crosslinking (Ji et al., 2013).
Emergence from Latency
Activation of P-TEFb in resting memory T-cells
Activation of latently infected T cells through engagement of the T-cell receptor (TCR) or stimulation with PKC-agonists results in the immediate stimulation of processive proviral transcription (Brooks et al., 2003; Kim et al., 2011; Natarajan et al., 2010). The early rounds of proviral transcription coincide with the recruitment of P-TEFb to the proviral LTR and requires functional Tat, even though the short duration of T-cell stimulation is not sufficient to build-up Tat expression above its undetectable sub-threshold basal levels (Mbonye et al., 2013).
Recently, we identified pSer175 as an additional major phospho-modification on the CDK9 T-loop that is induced by TCR and PKC-agonist stimulation (Mbonye et al., 2013). Signal-dependent Ser175 phosphorylation of CDK9, although not required for Tat binding to P-TEFb, appears to favor binding of Tat over BRD4 thereby shifting the equilibrium in favor of Tat. The pSer175 modification also provides an extremely useful marker to identify the transcriptionally active forms of P-TEFb since it is absent from P-TEFb in the 7SK RNP complex (Mbonye et al., 2013). In a related study, Fujinaga et al. (2012) demonstrated that the TCR- or PMA-mediated disassembly of 7SK sRNP to release P-TEFb requires activation of the protein kinase C isoform PKC-θ which in turn phosphorylates HEXIM1 protein at Ser158, which is situated right in the middle of the protein’s bipartite RNA binding sequence. Consequently, the Ser158 phosphorylated HEXIM1 is unable to bind to 7SK snRNA and is therefore unable to bind and inhibit P-TEFb. Thus, multiple PTMs on 7SK RNP components appear to weaken protein-protein and protein-RNA interactions and shift the equilibrium to favor P-TEFb/Tat complex formation instead of P-TEFb/BRD4 complex formation. With increasing cycles of HIV transcription and a progressive elevation in Tat expression, Tat-dependent dissociation of P-TEFb from 7SK snRNP is expected to amplify the signal-dependent release of P-TEFb.
Chromatin remodeling and activation of HIV transcription initiation
The absence of cellular transcription initiation factors in the nuclei of latently infected resting memory CD4+ T cells and non-activated macrophage lineage cells is another major contributor towards maintenance of HIV latency. In the Jurkat T-cell model, TCR stimulation induces an initial wave of HIV transcription initiation that is largely dependent on the nuclear localization of NF-κB p65/p50 heterodimer complexes and their binding to the duplicate NF-κB elements at the HIV LTR (Kim et al., 2011; Tyagi et al., 2010). Stimulation of latently infected T lymphocytes and macrophage lineage cells with PKC agonists or pro-inflammatory cytokines also activates HIV transcription initiation in a NF-κB p65-dependent manner (Kim et al., 2006; Nabel and Baltimore, 1987; Williams et al., 2006; Williams et al., 2007).
Nuclear NF-κB p65/p50 directs the recruitment of a co-activator complex of histone acetyltransferases (HATs) such as p300/CREB-binding protein (p300/CBP) and p300/CBP-associated factor (P/CAF) to the HIV LTR (Gerritsen et al., 1997; Perkins et al., 1997). Acetylation of Nuc-1 appears to a signal allowing recruitment of the chromatin remodeling SWI/SNF complexes. Rafati et al. (2011) have observed that two functionally distinct subclasses of SWI/SNF, BAF and PBAF are exchanged at the LTR. Maintenance of HIV latency requires BAF, which helps position the repressive Nuc-1. Upon activation, BAF is lost from the HIV promoter, while PBAF is selectively recruited to reposition Nuc-1 and facilitate LTR transcription (Rafati et al., 2011).
The NF-κB p65 subunit is itself subject to acetylation by p300/CBP which serves to maintain it in the nucleus by inhibiting interaction with IκBα (Chen et al., 2001; Furia et al., 2002). Deacetylation of p65 by HDAC3 switches off NF-κB-dependent transcription by allowing IkBα binding and subsequent nuclear export of p65/p50 (Chen et al., 2001). By contrast, lysine methylation of p65 stabilizes NF-κB binding to the LTR (Lu et al., 2010).
In primary T-cells NF-κB is typically replaced by NFAT (Bosque et al., 2011; Bosque and Planelles, 2009; Chan et al., 2013) which is activated by calcium/calcineurin signaling (Kim et al., 2011). Like NF-κB p65/p50, NFAT proteins can bind CBP/p300 and recruit its HAT activity to gene promoters (Garcia-Rodriguez and Rao, 1998). Surprisingly, NFAT is dispensable in most Jurkat T-cell models (Chan et al., 2013; Kim et al., 2011). Since NFAT and NF-κB bind in a mutually exclusive manner to overlapping sites on the proviral enhancer (Giffin et al., 2003) it seems likely they are used sequentially during T-cell activation, with NF-κB responsible for the initial responses and NFAT acting at later times.
Post-transcriptional modification of Tat during transcription
The complex interactions between Tat, histone modifying enzymes, and elongation factors are coordinated through a series of post-translational modifications of Tat. In addition to targeting Nuc-1, HATs are reported to directly modify Tat on lysine residues (Lys50 and Lys51) that are situated within its arginine-rich TAR RNA binding motif (ARM) (Col et al., 2001; Kiernan et al., 1999; Ott et al., 1999). Acetylation of the ARM region is thought to dissociate the binding of Tat to TAR, and simultaneously reinforce its interactions with chromatin modifying transcriptional co-activators by providing acetyl binding sites for their bromodomains (Lu et al., 2013; Ott et al., 2011). By contrast, acetylation of Tat by P/CAF at Lys28 situated within the cysteine-rich region of its activation domain has also been reported to enhance the association of Tat with P-TEFb and stabilize the Tat/P-TEFb/TAR ternary complex (Bres et al., 2002; D’Orso and Frankel, 2009; Kiernan et al., 1999). At the end of the transcription cycle SIRT1 binds and deacetylates Tat at Lys50 which mediates the recycling of unacetylated Tat thought to be necessary for subsequent rounds of viral transcription (Pagans et al., 2005). Following deacetylation of Tat at the ARM region, monomethylation at Lys51 by Set7/9 bound to TAR enhances Tat-dependent HIV transcription by facilitating Tat interactions with TAR and recruitment of the host elongation factor P-TEFb to the RNA (Pagans et al., 2010). Subsequent demethylation of Lys51 by LSD1/CoREST allows for the re-acetylation of the ARM of Tat which triggers entry into the TAR-independent phase of proviral transcription elongation (Sakane et al., 2011). Detailed studies monitoring the kinetics of each of these Tat modifications will be needed to provide a fuller understanding of their roles in regulating HIV transcription.
Stochastic events
Only a very small subset of latent proviruses (< 1%) in peripheral blood becomes reactivated following in vitro ‘maximum’ T-cell activation with the mitogen PHA and irradiated allogeneic PBMCs (Ho et al., 2013). Significant genomic sequence defects (eg. large internal deletions and APOBEC3G-mediated hypermutations) account for the lack of reactivation of most of the non-induced proviruses (~88 %) (Ho et al., 2013) but the remaining fraction of non-induced latent proviruses (~11%) have intact sequences, are integrated into actively transcribed regions, and have been demonstrated in viral reconstruction experiments to be replication-competent (Ho et al., 2013).
Why only a small fraction of proviruses can be reactivated is mysterious, and potentially creates a barrier to cure efforts. Within a clonal population of latently infected Jurkat T-cells, cell-to-cell stochastic fluctuations in the assembly of transcription factor and chromatin remodeling complexes at the HIV LTR produce occasional transient bursts of viral transcription in a sub-population of these cells (Pearson et al., 2008; Singh et al., 2012; Weinberger et al., 2005). Although the reactivation of latent HIV ex vivo is now deemed to be inherently stochastic (Ho et al., 2013; Weinberger and Weinberger, 2013), sustained cellular activation is clearly needed in order to maintain HIV transcription (Kim et al., 2011; Pearson et al., 2008; Williams et al., 2007). Thus the stochastic nature of proviral reactivation may contribute to inefficient proviral reactivation during a first round of T-cell activation (Ho et al., 2013).
An alternative, but not exclusive explanation for why certain subsets of silenced proviruses fail to get reactivated when cells are stimulated is that epigenetic silencing of HIV-1 results in complex and heterogenous patterns of histone modifications and DNA methylation (Blazkova et al., 2009; Kauder et al., 2009; Pearson et al., 2008). Interestingly, latent proviruses that reactivate poorly in response to cellular activators are found to carry higher levels of H327me3 marks (Friedman et al., 2011).
Summary and perspectives
We believe that the most practical approach to HIV eradication therapy will be based on using drugs to induce the transcriptional activity of latent HIV-1 without inducing the polyclonal activation of non-infected cells (a “shock” phase) since this type of therapy can ultimately be delivered in normal clinical settings. Once the virus is reactivated a “kill” phase will be used to eliminate the induced cells through existing immune responses, viral cytopathogenicity or cytotoxic drugs.
The detailed molecular studies described above strongly imply that effective activation of the entire latent viral pool may ultimately require a cocktail of drugs that stimulate both transcription initiation and P-TEFb mobilization. Protein kinase C agonists are a major class of drugs that are able to induce transcriptional activity of latent HIV-1 (Beans et al., 2013; Sanchez-Duffhues et al., 2011). All of these drugs are expected to activate signaling pathways leading to both P-TEFb and NF-κB mobilization but they tend to lead to global T-cell activation and associated toxicity. Finding drugs that can selectively activate P-TEFb in resting T-cells is a major challenge for the field.
The second major class of activators that are under investigation are drugs that induce epigenetic modifications such as histone deacetylase (HDAC) inhibitors (e.g. SAHA, panobinostat, rombidepsin) (Archin et al., 2012; Friedman et al., 2011; Van Lint et al., 1996) and histone methyltransferase inhibitors (DZNep, BIX01294) (Friedman et al., 2011; Imai et al., 2010). SAHA has been shown in induce transient HIV RNA production in patients and is thus the first potential inducer of latent proviruses that has been evaluated as part of the HIV Cure agenda (Archin et al., 2012). As expected from the mechanisms described above, transcriptional activation of latent HIV by HDAC inhibitors occurs independently of NF-κB activity and is associated with significant remodeling of the proviral Nuc-1 region that includes displacement of Nuc-1, decreased HDAC occupancy and histone hyperacetylation (He and Margolis, 2002; Kim et al., 2011; Van Lint et al., 1996). Similarly, treatment of latently infected cells with selective HMT inhibitors can also reactivate silenced proviruses in various HIV latency cell line models (Bernhard et al., 2011; Friedman et al., 2011; Imai et al., 2010). Recently, Bouchat et al. (2012) reported that ex-vivo treatments of resting CD4+ T cells isolated from HIV-1-infected individuals with the histone methyltransferase inhibitors chaetocin or BIX01294 induced virus production to a comparable extent as treatment with either SAHA or the PKC agonist prostratin.
Although SAHA and other HDAC inhibitors are able to induce viral RNA transcription in clinical settings it does not typically lead to release of infectious virions. This may be because although it is able to allow release of the paused RNAP II it is unable to induce P-TEFb and ignite a full cycle of viral transcription. The big take home lesson from the molecular studies of HIV transcription is that efficient viral reactivation is only seen when there is a reversal of epigenetic blocks (which is normally accomplished by NFAT or NF-kB activation) and activation of P-TEFb (which is normally induced through TCR activation of PKC). It follows that specific combinations of drugs hitting these two pathways will be need. We are confident that new, highly potent, drug combinations will soon be identified. The hope for an HIV cure, only a pipedream a few years ago, remains high.
Acknowledgments
We thank current members of the Karn laboratory, Hongxia Mao, Kien Nyguen, Michael Greenberg, Curtis Dobrowolski, Biswajit Das, Mary Ann Checkley, David Alvarez, Yoelvis Garcia and Stephanie Milne for helpful discussions. Work in the Karn laboratory on HIV latency was supported by Public Health Service grants, R01-AI067093 and DP1-DA028869 to JK and the Martin Delaney CARE Collaboratory, U19-AI096113. UM was supported by amFAR postdoctoral award 108266-51-RFRL. We also thank the CWRU/UH Center for AIDS Research (P30-AI036219) for provision of services. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn SH, Kim M, Buratowski S. Phosphorylation of Serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell. 2004;13:67–76. doi: 10.1016/s1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and Extra-terminal (BET) Bromodomain Inhibition Activate Transcription via Transient Release of Positive Transcription Elongation Factor b (P-TEFb) from 7SK Small Nuclear Ribonucleoprotein. J Biol Chem. 2012;287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci U S A. 2013;110:11698–11703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011;585:3549–3554. doi: 10.1016/j.febslet.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17:7056–7065. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Murray D, Justement JS, Funk EK, Nelson A, Moir S, Chun TW, Fauci AS. Paucity of HIV DNA Methylation in Latently Infected, Resting CD4+ T Cells from Infected Individuals Receiving Antiretroviral Therapy. J Virol. 2012;86:5390–5392. doi: 10.1128/JVI.00040-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, Hirsch I. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, Zhoug MM, RFFS, Weinberger L, Verdin E, Ott M. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12:452–462. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011;7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, de Wit S, Clumeck N, Lambotte O, Rouzioux C, Rohr O, van Lint C. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1+ HAART-treated patients. AIDS. 2012 doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- Bourgeois CF, Kim YK, Churcher MJ, West MJ, Karn J. Spt5 cooperates with Tat by preventing premature RNA release at terminator sequences. Mol Cell Biol. 2002;22:1079–1093. doi: 10.1128/MCB.22.4.1079-1093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TP, Woods JO, Sedaghat AR, Siliciano JD, Siliciano RF, Wilke CO. Analysis of HIV-1 Viremia and Provirus in Resting CD4+ T Cells Reveals a Novel Source of Residual Viremia in Patients on Antiretroviral Therapy. J Virol. 2009;83:8470–8481. doi: 10.1128/JVI.02568-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bres V, Gomes N, Pickle L, Jones KA. A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev. 2005;19:1211–1226. doi: 10.1101/gad.1291705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bres V, Kiernan R, Emiliani S, Benkirane M. Tat acetyl-acceptor lysines are important for human immunodeficiency virus type-1 replication. J Biol Chem. 2002;277:22215–22221. doi: 10.1074/jbc.M201895200. [DOI] [PubMed] [Google Scholar]
- Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc Natl Acad Sci U S A. 2003;100:12955–12960. doi: 10.1073/pnas.2233345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhiraja S, Famiglietti M, Bosque A, Planelles V, Rice AP. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J Virol. 2012a;87:1211–1220. doi: 10.1128/JVI.02413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhiraja S, Ramakrishnan R, Rice AP. Phosphatase PPM1A negatively regulates P-TEFb function in resting CD4T+ T cells and inhibits HIV-1 gene expression. Retrovirology. 2012b;9:52. doi: 10.1186/1742-4690-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JK, Bhattacharyya D, Lassen KG, Ruelas D, Greene WC. Calcium/Calcineurin Synergizes with Prostratin to Promote NF-kappaB Dependent Activation of Latent HIV. PLoS One. 2013;8:e77749. doi: 10.1371/journal.pone.0077749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-κB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- Chen R, Yang Z, Zhou Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J Biol Chem. 2004;279:4153–4160. doi: 10.1074/jbc.M310044200. [DOI] [PubMed] [Google Scholar]
- Cheng AS, Lau SS, Chen Y, Kondo Y, Li MS, Feng H, Ching AK, Cheung KF, Wong HK, Tong JH, Jin H, Choy KW, Yu J, To KF, Wong N, Huang TH, Sung JJ. EZH2-Mediated Concordant Repression of Wnt Antagonists Promotes {beta}-Catenin-Dependent Hepatocarcinogenesis. Cancer Res. 2011;71:4028–4039. doi: 10.1158/0008-5472.CAN-10-3342. [DOI] [PubMed] [Google Scholar]
- Cheng B, Price DH. Analysis of factor interactions with RNA polymerase II elongation complexes using a new electrophoretic mobility shift assay. Nucleic Acids Res. 2008;36:e135. doi: 10.1093/nar/gkn630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrier T, Le Douce V, Eilebrecht S, Riclet R, Marban C, Dequiedt F, Goumon Y, Paillart JC, Mericskay M, Parlakian A, Bausero P, Abbas W, Herbein G, Kurdistani SK, Grana X, Van Driessche B, Schwartz C, Candolfi E, Benecke AG, Van Lint C, Rohr O. CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci U S A. 2013;110:12655–12660. doi: 10.1073/pnas.1220136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang K, Sung TL, Rice AP. Regulation of Cyclin T1 and HIV-1 Replication by MicroRNAs in Resting CD4+ T Lymphocytes. J Virol. 2012;86:3244–3252. doi: 10.1128/JVI.05065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Upton H, Bao K, Schulze-Gahmen U, Samelson AJ, He N, Nowak A, Lu H, Krogan NJ, Zhou Q, Alber T. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc Natl Acad Sci U S A. 2013;110:E123–131. doi: 10.1073/pnas.1216971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A. 1998;95:8869–8873. doi: 10.1073/pnas.95.15.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Davey RT, Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- Col E, Caron C, Seigneurin-Berny D, Gracia J, Favier A, Khochbin S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J Biol Chem. 2001;276:28179–28184. doi: 10.1074/jbc.M101385200. [DOI] [PubMed] [Google Scholar]
- Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory TJ, Schacker TW, Stevenson M, Fletcher CV. Overcoming pharmacologic sanctuaries. Curr Opin HIV AIDS. 2013;8:190–195. doi: 10.1097/COH.0b013e32835fc68a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR, Lomedico PT, Ju G. Transcriptional interference in avian retroviruses--implications for the promoter insertion model of leukaemogenesis. Nature. 1984;307:241–245. doi: 10.1038/307241a0. [DOI] [PubMed] [Google Scholar]
- Czudnochowski N, Bosken CA, Geyer M. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat Commun. 2012;3:842. doi: 10.1038/ncomms1846. [DOI] [PubMed] [Google Scholar]
- D’Orso I, Frankel AD. Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex. Proc Natl Acad Sci U S A. 2009;106:3101–3106. doi: 10.1073/pnas.0900012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orso I, Frankel AD. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol. 2010;17:815–821. doi: 10.1038/nsmb.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orso I, Jang GM, Pastuszak AW, Faust TB, Quezada E, Booth DS, Frankel AD. A transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol Cell Biol. 2012 doi: 10.1128/MCB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA. HIV-1 Tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990;9:4145–4153. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogat J, Hermand D. Gene-specific requirement of RNA polymerase II CTD phosphorylation. Mol Microbiol. 2012;84:995–1004. doi: 10.1111/j.1365-2958.2012.08071.x. [DOI] [PubMed] [Google Scholar]
- du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evering TH, Mehandru S, Racz P, Tenner-Racz K, Poles MA, Figueroa A, Mohri H, Markowitz M. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination antiviral therapy initiated during primary infection. PLoS Pathog. 2012;8:e1002506. doi: 10.1371/journal.ppat.1002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. Epigenetic silencing of HIV-1 by the Histone H3 lysine 27 Methyltransferase Enhancer of Zeste 2 (EZH2) J Virol. 2011;85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 2012;40:9160–9170. doi: 10.1093/nar/gks682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Cujec TP, Peng J, Garriga J, Price DH, Graña X, Peterlin BM. The ability of positive transcription elongation factor b to transactive human immodeficiency virus transcription depends on a functional kinase domain, cyclin T1 and Tat. J Virol. 1998;72:7154–7159. doi: 10.1128/jvi.72.9.7154-7159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furia B, Deng L, Wu K, Baylor S, Kehn K, Li H, Donnelly R, Coleman T, Kashanchi F. Enhancement of nuclear factor-κ B acetylation by coactivator p300 and HIV-1 Tat proteins. J Biol Chem. 2002;277:4973–4980. doi: 10.1074/jbc.M107848200. [DOI] [PubMed] [Google Scholar]
- Gallastegui E, Millan-Zambrano G, Terme JM, Chavez S, Jordan A. Chromatin reassembly factors are involved in transcriptional interference promoting HIV latency. J Virol. 2011;85:3187–3202. doi: 10.1128/JVI.01920-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewelRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez C, Rao A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP) J Exp Med. 1998;187:2031–2036. doi: 10.1084/jem.187.12.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose R, Liou LY, Herrmann CH, Rice AP. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J Virol. 2001;75:11336–11343. doi: 10.1128/JVI.75.23.11336-11343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR κB element. Nature Struct Biol. 2003;10:800–806. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- Han Y, Lin YB, An W, Xu J, Yang HC, O’Connell K, Dordai D, Boeke JD, Siliciano JD, Siliciano RF. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe. 2008;4:134–146. doi: 10.1016/j.chom.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann CH, Rice AP. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J Virol. 1995;69:1612–1620. doi: 10.1128/jvi.69.3.1612-1620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Shamanna RA, Guan D, Pe’ery T, Mathews MB. HIV-1 Replication and Latency Are Regulated by Translational Control of Cyclin T1. Journal of molecular biology. 2011;410:917–932. doi: 10.1016/j.jmb.2011.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation by BIX01294. J Biol Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, Fujinaga K, Karn J. The negative elongation factor (NELF) is required for the maintenance of proviral latency but does not induce promoter proximal pausing of RNAP II on the HIV LTR. Mol Cell Biol. 2014 doi: 10.1128/MCB.01013-13. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD. SR Proteins Collaborate with 7SK and Promoter-Associated Nascent RNA to Release Paused Polymerase. Cell. 2013;153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Gunthard HF. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A. 2008;105:16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by Tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Karn J. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr Opin HIV AIDS. 2011;6:4–11. doi: 10.1097/COH.0b013e328340ffbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J. A new BET on the control of HIV latency. Cell Cycle. 2013;12:545–546. doi: 10.4161/cc.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol. 2009;83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen NJ, Churcher MJ, Karn J. Transfer of Tat and release of TAR RNA during the activation of the human immunodeficiency virus type-1 transcription elongation complex. EMBO J. 1997;16:5260–5272. doi: 10.1093/emboj/16.17.5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, Benkirane M, Van Lint C. HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006;25:3596–3604. doi: 10.1038/sj.emboj.7601248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Mbonye U, Hokello J, Karn J. T-Cell Receptor Signaling Enhances Transcriptional Elongation from Latent HIV Proviruses by Activating P-TEFb through an ERK-Dependent Pathway. Journal of molecular biology. 2011;410:896–916. doi: 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6:235–244. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- Krueger BJ, Jeronimo C, Roy BB, Bouchard A, Barrandon C, Byers SA, Searcey CE, Cooper JJ, Bensaude O, Cohen EA, Coulombe B, Price DH. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008;36:2219–2229. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger BJ, Varzavand K, Cooper JJ, Price DH. The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One. 2010;5:e12335. doi: 10.1371/journal.pone.0012335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Guermah M, Roeder RG, Amini S, Khalili K. Interaction between cell cycle regulator, E2F-1, and NF-κB mediates repression of HIV-1 gene transcription. J Biol Chem. 1997;272:29468–29474. doi: 10.1074/jbc.272.47.29468. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012 doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laspia MF, Rice AP, Mathews MB. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell. 1989;59:283–292. doi: 10.1016/0092-8674(89)90290-0. [DOI] [PubMed] [Google Scholar]
- Lenasi T, Contreras X, Peterlin BM. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe. 2008;4:123–133. doi: 10.1016/j.chom.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenasi T, Peterlin BM, Barboric M. Cap-binding protein complex links pre-mRNA capping to transcription elongation and alternative splicing through positive transcription elongation factor b (P-TEFb) J Biol Chem. 2011;286:22758–22768. doi: 10.1074/jbc.M111.235077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol. 2005;79:6610–6619. doi: 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006;2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem. 2005;280:28819–28826. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hsu J, Chan C, Li Z, Zhou Q. The Ubiquitin Ligase Siah1 Controls ELL2 Stability and Formation of Super Elongation Complexes to Modulate Gene Transcription. Mol Cell. 2012;46:325–334. doi: 10.1016/j.molcel.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Li Z, Xue Y, Zhou Q. Viral-Host Interactions That Control HIV-1 Transcriptional Elongation. Chem Rev. 2013;113:8567–8582. doi: 10.1021/cr400120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, Gudkov AV, Stark GR. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci U S A. 2010;107:46–51. doi: 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert A, Grimm M, Martinez J, Wiesner J, Meyerhans A, Meyuhas O, Sickmann A, Fischer U. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 2008;9:569–575. doi: 10.1038/embor.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, Karn J. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013;9:e1003338. doi: 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missra A, Gilmour DS. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc Natl Acad Sci U S A. 2010;107:11301–11306. doi: 10.1073/pnas.1000681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz L, Egloff S, Ughy B, Jady BE, Kiss T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010;6:e1001152. doi: 10.1371/journal.ppat.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, Baltimore DA. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- Natarajan M, August A, Henderson AJ. Combinatorial signals from CD28 differentially regulate human immunodeficiency virus transcription in T cells. J Biol Chem. 2010;285:17338–17347. doi: 10.1074/jbc.M109.085324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan M, Schiralli-Lester GM, Lee C, Missra A, Wasserman GA, Steffen M, Gilmour DS, Henderson AJ. NELF coordinates RNA polymerase II pausing, premature termination and chromatin remodeling to regulate HIV transcription. J Biol Chem. 2013 doi: 10.1074/jbc.M113.496489. E print ahead of publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- O’Keeffe B, Fong Y, Chen D, Zhou S, Zhou Q. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem. 2000;275:279–287. doi: 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- Ott M, Geyer M, Zhou Q. The Control of HIV Transcription: Keeping RNA Polymerase II on Track. Cell Host Microbe. 2011;10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Schnölzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- Pagano JM, Kwak H, Waters CT, Sprouse RO, White BS, Ozer A, Szeto K, Shalloway D, Craighead HG, Lis JT. Defining NELF-E RNA Binding in HIV-1 and Promoter-Proximal Pause Regions. PLoS Genet. 2014;10:e1004090. doi: 10.1371/journal.pgen.1004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagans S, Kauder SE, Kaehlcke K, Sakane N, Schroeder S, Dormeyer W, Trievel RC, Verdin E, Schnolzer M, Ott M. The Cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription. Cell Host Microbe. 2010;7:234–244. doi: 10.1016/j.chom.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M. SIRT1 Regulates HIV Transcription via Tat Deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios JA, Perez-Pinar T, Toro C, Sanz-Minguela B, Moreno V, Valencia E, Gomez-Hernando C, Rodes B. Long-term nonprogressor and elite controller patients who control viremia have a higher percentage of methylation in their HIV-1 proviral promoters than aviremic patients receiving highly active antiretroviral therapy. J Virol. 2012;86:13081–13084. doi: 10.1128/JVI.01741-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Josefsson L, Coffin JM. HIV reservoirs and the possibility of a cure for HIV infection. J Intern Med. 2011;270:550–560. doi: 10.1111/j.1365-2796.2011.02457.x. [DOI] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell. 2009;36:178–191. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-κB and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993;12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Peterlin BM, Brogie JE, Price DH. 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip Rev RNA. 2012;3:92–103. doi: 10.1002/wrna.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R, Dow EC, Rice AP. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J Leukoc Biol. 2009;86:1345–1350. doi: 10.1189/jlb.0509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner DB, Yamaguchi Y, Wada T, Handa H, Price DH. A highly purified RNA polymerase II elongation control system. J Biol Chem. 2001;276:42601–42609. doi: 10.1074/jbc.M104967200. [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, Descours B, Guergnon J, Viard JP, Boufassa F, Lambotte O, Goujard C, Meyer L, Costagliola D, Venet A, Pancino G, Autran B, Rouzioux C AVS Group. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane N, Kwon HS, Pagans S, Kaehlcke K, Mizusawa Y, Kamada M, Lassen KG, Chan J, Greene WC, Schnoelzer M, Ott M. Activation of HIV Transcription by the Viral Tat Protein Requires a Demethylation Step Mediated by Lysine-specific Demethylase 1 (LSD1/KDM1) PLoS Pathog. 2011;7:e1002184. doi: 10.1371/journal.ppat.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Duffhues G, Vo MQ, Perez M, Calzado MA, Moreno S, Appendino G, Munoz E. Activation of Latent HIV-1 Expression by Protein Kinase C Agonists. A Novel Therapeutic Approach to Eradicate HIV-1 Reservoirs. Curr Drug Targets. 2011;12:348–356. doi: 10.2174/138945011794815266. [DOI] [PubMed] [Google Scholar]
- Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, Lau J, Bisgrove D, Schnolzer M, Verdin E, Zhou MM, Ott M. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem. 2012;287:1090–1099. doi: 10.1074/jbc.M111.282855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, Alber T. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife. 2013;2:e00327. doi: 10.7554/eLife.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35:4347–4358. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selliah N, Zhang M, DeSimone D, Kim H, Brunner M, Ittenbach RF, Rui H, Cron RQ, Finkel TH. The gamma-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology. 2006;344:283–291. doi: 10.1016/j.virol.2005.09.063. [DOI] [PubMed] [Google Scholar]
- Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir after Virus Reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Yang HC, Rabi SA, Bravo HC, Irizarry RA, Zhang H, Margolick JB, Siliciano JD, Siliciano RF. Influence of host gene transcription level and orientation on HIV-1 latency in a primary cell model. J Virol. 2011;85:5384–5393. doi: 10.1128/JVI.02536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrill-Mix S, Lewinski MK, Famiglietti M, Bosque A, Malani N, Ocwieja KE, Berry CC, Looney D, Shan L, Agosto LM, Pace MJ, Siliciano RF, O’Doherty U, Guatelli J, Planelles V, Bushman FD. HIV latency and integration site placement in five cell-based models. Retrovirology. 2013;10:90. doi: 10.1186/1742-4690-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Siliciano JD, Lai J, Callender M, Pitt E, Zhang H, Margolick JB, Gallant JE, Cofrancesco J, Jr, Moore RD, Gange SJ, Siliciano RF. Stability of the latent reservoir for HIV-1 in patients receiving valproic acid. J Infect Dis. 2007;195:833–836. doi: 10.1086/511823. [DOI] [PubMed] [Google Scholar]
- Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Razooky BS, Dar RD, Weinberger LS. Dynamics of protein noise can distinguish between alternate sources of gene-expression variability. Mol Syst Biol. 2012;8:607. doi: 10.1038/msb.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr W, Fidler S, McClure M, Weber J, Cooper D, Ramjee G, Kaleebu P, Tambussi G, Schechter M, Babiker A, Phillips RE, Porter K, Frater J. Duration of HIV-1 Viral Suppression on Cessation of Antiretroviral Therapy in Primary Infection Correlates with Time on Therapy. PLoS One. 2013;8:e78287. doi: 10.1371/journal.pone.0078287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TL, Rice AP. Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology. 2006;3:66. doi: 10.1186/1742-4690-3-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5:e1000263. doi: 10.1371/journal.ppat.1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tae S, Karkhanis V, Velasco K, Yaneva M, Erdjument-Bromage H, Tempst P, Sif S. Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 2011;39:5424–5438. doi: 10.1093/nar/gkr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–751. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras PJ, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, Boyer F, Parrinello H, Berkhout B, Terzian C, Benkirane M, Kiernan R. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell. 2012;150:1147–1157. doi: 10.1016/j.cell.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dow EC, Liang YY, Ramakrishnan R, Liu H, Sung TL, Lin X, Rice AP. Phosphatase PPM1A regulates phosphorylation of Thr-186 in the Cdk9 T-loop. J Biol Chem. 2008;283:33578–33584. doi: 10.1074/jbc.M807495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel cdk9-associated c-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Weinberger AD, Weinberger LS. Stochastic Fate Selection in HIV-Infected Patients. Cell. 2013;155:497–499. doi: 10.1016/j.cell.2013.09.039. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Wen Y, Shatkin AJ. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes Dev. 1999;13:1774–1779. doi: 10.1101/gad.13.14.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Kwon H, Chen LF, Greene WC. Sustained induction of NF-kappaB is required for efficient expression of latent human immunodeficiency virus type 1. J Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wires ES, Alvarez D, Dobrowolski C, Wang Y, Morales M, Karn J, Harvey BK. Methamphetamine activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaB) and induces human immunodeficiency virus (HIV) transcription in human microglial cells. J Neurovirol. 2012;18:400–410. doi: 10.1007/s13365-012-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Inukai N, Narita T, Wada T, Handa H. Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol Cell Biol. 2002;22:2918–2927. doi: 10.1128/MCB.22.9.2918-2927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chen Y, Gabuzda D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-κB. J Biol Chem. 1999;274:27981–27988. doi: 10.1074/jbc.274.39.27981. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- Yukl SA, Gianella S, Sinclair E, Epling L, Li Q, Duan L, Choi AL, Girling V, Ho T, Li P, Fujimoto K, Lampiris H, Hare CB, Pandori M, Haase AT, Gunthard HF, Fischer M, Shergill AK, McQuaid K, Havlir DV, Wong JK. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J Infect Dis. 2010;202:1553–1561. doi: 10.1086/656722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Klatt A, Gilmour DS, Henderson AJ. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J Biol Chem. 2007;282:16981–16988. doi: 10.1074/jbc.M610688200. [DOI] [PubMed] [Google Scholar]
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, Brass AL. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]