

Abstract
This report describes the development of a novel dual color Southern protocol to visualize two distinct genomes or genic regions simultaneously on a single Southern blot. The blot is developed with IRDye-conjugated antibody (Ab) and streptavidin that recognize Digoxigenin- (Dig) or biotin-labeled probes, respectively and visualized on an infrared imager. This protocol was validated by visualizing viral and host genomes of human cytomegalovirus (HCMV)-infected human fibroblasts. This technique utilizes extremely sensitive fluorescent imaging, allowing the detection of nanogram quantities of DNA, as opposed to microgram quantities needed in Southerns using radioactively labeled probes, and eliminates the inherent loss in signal after stripping and reprobing a Southern blot. The probes are labeled with non-radioactive Dig and biotin and can be stored for extended periods of time. This protocol will aid in studies of any system with two genomes, such as cells infected with numerous types of microorganisms (virus/parasites/bacteria), or studies of mitochondrial and nuclear DNA within the same cells.
Keywords: Dual-color Southern, DNA analysis, simultaneous two-genome detection in infected cells, IRDyes, Li-Cor Odyssey, non-radioactive Southern
Type of research
The study of the effects of pathogenic infection on DNA is hampered by the current techniques available to visualize the separate host and parasite genomes. Visualization of these genomes on Southern or slot blots typically involves hybridizing the blot with a radioactively-labeled probe specific for one of the two genomes followed by exposure to x-ray film. Subsequently the blot must be stripped and hybridized with a second labeled probe recognizing the alternate genome before a second exposure to film. Probing, stripping, and re-probing the blot takes a minimum of four days and often requires microgram amounts of DNA to ensure a strong signal. Stripping the blot between successive hybridizations diminishes the signal acquired from the second genome. Therefore, development of a protocol allowing visualization of both the genomes simultaneously using stable, non-radioactive labels would be beneficial.
This report describes a sensitive dual color Southern blot protocol that allows discrete visualization of nanogram quantities of both host and pathogen DNA simultaneously in less than two days. Host and virus genome probes were labeled with Dig- and biotin-modified nucleotides and detected using IRDye-conjugated Ab and streptavidin, eliminating the need for radioactive label. Developed blots were scanned at the two corresponding wavelengths using the Li-Cor Odyssey infrared imager. The signals from the IRDye-conjugated Ab and streptavidin were extremely stable and did not decay, allowing for reanalysis weeks after initial development as well as extended storage of probes.
This novel dual color Southern will aid in studies of any system with two genomes, such as cells infected with numerous types of microorganisms (virus/parasites/bacteria), or even studies of mitochondrial and nuclear DNA within the same cells. In addition, it will allow Southern detection of two genes that cannot be separated readily on a gel without the unavoidable signal loss associated with stripping and successive probing.
This dual color protocol could be modified for Northern analysis to examine changes in expression of two different genes following exposure to a compound of interest. Slot blot analysis would provide a relatively rapid and efficient screening tool to determine what conditions and at what times during a timecourse would benefit from more detailed qPCR analysis.
Combining the dual color Southern technique with different methods of electrophoretic separation (e.g. CHEF, alkaline agarose) will only increase the strength of this new method. We have combined this protocol with T4 endonuclease alkaline agarose electrophoresis to study nucleotide excision repair in HCMV-infected human cells (O'Dowd et al., 2012).
Time required
This protocol takes approximately five days: one day to run the gel, two days for capillary transfer of the gel to the membrane, one day for prehybridization and overnight hybridization of probes, and one day for blocking and development of the blot.
Materials
Special equipment
Odyssey Infrared Imaging System (Li-Cor).
Chemicals and reagents
Note: All solutions are prepared in distilled deionized H2O (ddH2O) unless otherwise specified.
Buffer A - 0.3M Sucrose, 60 mM KCl, 60 mM Tris pH 8.0, 15 mM NaCl, 2 mM EDTA-Na2, 0.5 mM spermidine, 0.15 mM spermine. Filter and store at 4oC.
Buffer A + Ipegal - Buffer A with 0.5% Ipegal CA-630.
Buffer B - 0.15 M NaCl, 5mM EDTA-Na2.
Buffer C - 20 mM Tris pH 8.0, 20 mM NaCl, 20 mM EDTA-Na2, 0.3 mg/ml proteinase K.
PCI - phenol:chloroform:isoamyl alcohol at a ratio of 25:24:1.
CI - chloroform: isoamyl alcohol at a ratio of 24:1.
3M Sodium Acetate.
TE Buffer - 10 mM Tris-HCl pH 8.0, 1mM EDTA-Na2.
500 μg/ml RNase A.
10X TBE Buffer - 0.9 M Tris base, 0.9 M boric acid, 20 mM EDTA.
0.25N HCl.
Magnacharge nylon transfer membrane, 0.45 μm (GE Water and Process Technologies).
20X SSC- 3M NaCl, 300mM sodium citrate-2H2O, pH 7.0.
SYBR Gold (Invitrogen).
100X Denhardt’s Solution −2% (w/v) BSA, 2% (w/v) Ficoll 400, 2% (w/v) polyvinylpyrrolidone. Filter and store at −20°C.
20X SSPE - 3M NaCl, 0.2M NaH2PO4-H2O, 0.02M EDTA, pH to 7.4 with NaOH.
Hybridization Buffer - 5X SSPE, 2% (w/v) SDS, 10% (w/v) dextran sulfate, 1x Denhardt’s solution, 10μg/ml sheared sonicated salmon sperm DNA. Store at −20°C.
Wash Buffer 1 - 2x SSPE.
Wash Buffer 2 - 2xSSPE, 1% SDS.
Wash Buffer 3 - 0.1X SSPE.
Blocking Buffer - 0.6% (w/v) gelatin from cold water fish skin (Sigma) in TBST.
TBST- 50mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.05% Tween 20.
Digoxigenin-11-dUTP, alkali stable (Roche).
Biotin-16-dUTP (Biotium).
Mouse anti-Dig IgG antibody (Jackson Labs)
IRDye 800-conjugated Streptavidin (Rockland).
IRDye 700-conjugated goat anti-mouse IgG (Rockland).
Detailed Procedure
Isolating DNA ~6h
-
i
Harvest approximately 106 uninfected and infected cells as well as isolated pathogens per established protocols (Luo et al., 2007; O'Dowd et al., 2012).
-
ii
Resuspend cell pellets in 5 ml cold Buffer A + NP40. Note: Steps ii. through vi. isolate nuclei from cells. To isolate DNA from pelleted pathogens without nuclei skip to step vii.
-
iii
Incubate 10 min on ice.
-
iv
Centrifuge at 1,500 rpm for 5 min at 4°C to pellet nuclei. Aspirate supernatant without disturbing pellet.
-
v
Wash with 10 ml Buffer A.
-
vi
Centrifuge at 1,500 rpm for 5 min at 4°C to pellet nuclei. Aspirate supernatant without disturbing pellet.
-
vii
Resuspend pellet in 1.5 ml Buffer B.
-
viii
Add 1.5 ml Buffer C and mix thoroughly. Incubate for 2 h at 63°C.
-
ix
PCI/CI extract and isopropanol precipitate DNA at −20°C for 1 h. Centrifuge at 10,000 rpm for 30 min at 4°C. Aspirate the supernatant, being careful not to disturb the pellet, and wash with 70% ethanol. Repeat the spin. Remove the supernatant and air dry the pellet for 10 min.
-
x
Resuspend the pellet in 400 μl TE buffer.
-
xi
Add 4 μl RNase A and incubate for 1 hr at 37°C.
-
xii
PCI/CI extract the DNA. Isopropanol precipitate the DNA and centrifuge in a microcentrifuge at maximum speed for 15 min at 4°C to pellet. Wash the pellet in 70% ethanol and centrifuge again.
-
xiii
Air dry the pellet for 10 min and dissolve in an appropriate amount of TE. The solution can be incubated for 1 hr at 65°C to aid in dissolving the pellet.
Labeling probes ~6h
-
xiv
Digest isolated host and pathogen DNA with a preferred restriction endonuclease, such as HindIII.
-
xv
PCI/CI extract and isopropanol precipitate DNA at −20°C for 1 h. Aspirate the supernatant, being careful not to disturb the pellet, and wash with 70% ethanol. Repeat the spin. Remove the supernatant and air dry the pellet for 10 min before resuspending in TE buffer.
-
xvi
Label digested host DNA with Dig-11-dUTP and digested viral DNA with biotin-16-dUTP by random priming per kit instructions (BioPrime Array CGH genomic labeling module, Invitrogen).
Optional: While labeling genomic probes also random prime label Lambda HindIII size markers to allow DNA fragment size determination.
Gel electrophoresis and capillary transfer ~4 h + overnight (O/N) and up to 72 h
-
xvii
Quantitate DNA extracted from experimental samples and electrophorese on a 0.8 - 1% agarose gel dissolved in 0.5X TBE.
-
xviii
Depurinate gel in 0.25N HCl for 15 min with shaking at room temperature (RT).
-
xix
Rinse gel in ddH2O.
-
xx
Denature gel for 30–60 min at RT in 0.5M NaOH, 1.5M NaCl.
-
xxi
Capillary transfer the DNA out of the gel onto a charged nylon membrane in 20X SSC pH 7.0 for at least 12 h up to 72 h, depending on the size of the DNA being transferred. Do not prewet the membrane before laying it on the gel surface to begin transfer.
-
xxii
Wash the membrane in 2X SSC 5 min at RT.
-
xxiii
Irradiate the membrane at 254 nm to UV cross-link the DNA to the surface (approximately 2 min).
-
xxiv
Stain the gel with a 1:10,000 dilution of SYBR gold in 0.5x TBE for at least 1 h to check that the transfer was efficient.
Hybridization 4 h + O/N
-
xxv
Prewet the membrane in Wash Buffer 1.
-
xxvi
Pre-hybridize the blot in Hybridization Buffer for 2–4 h at 65°C in a hybridization oven.
-
xxvii
Denature the probes by boiling for 5–10 min, then immediately place on ice.
-
xxviii
Discard the Hybridization Buffer and replace with fresh Hybridization Buffer containing the denatured probes. Hybridize O/N at 65°C.
-
xxix
Wash the membrane twice in Wash Buffer 1 for 5 min at RT.
-
xxx
Wash the membrane twice in Wash Buffer 2 for 15 min at 60°C.
-
xxxi
Wash the membrane twice in Wash Buffer 3 for 15 min at 60°C.
Blocking and developing ~4 h
-
xxxii
Incubate the blot in Blocking Buffer with 1% SDS for 1 h at RT with shaking.
-
xxxiii
Wash the blot twice in TBST for 5 min with shaking.
-
xxxiv
Incubate the blot with the α-Dig Ab diluted 1:6,000 in Blocking Buffer for 1 h at RT with shaking.
-
xxxv
Wash the blot in TBST for 5 min with shaking.
-
xxxvi
Wash the blot in TBST + 1% SDS for 10 min with shaking.
-
xxxvii
Wash the blot three times in TBST for 5 min with shaking.
-
xxxviii
Dilute IRDye-conjugated α-Mouse secondary Ab 1:4,000 and IRDye-conjugated streptavidin 1:20,000 in Blocking Buffer with 0.02% SDS (secondary solution).
-
xxxix
Incubate the blot with the secondary solution for 30–60 min at RT with shaking and protected from light.
-
xl
Wash in TBST for 5 min with shaking.
-
xli
Wash the blot in TBST + 1% SDS for 15 min with shaking.
-
xlii
Wash the blot three times in TBST for 5 min with shaking.
-
xliii
Wash the blot twice for 5 min in TBS without Tween.
-
xliv
Scan the blot on the Li-Cor Odyssey Infrared Imaging System.
Results
This dual color Southern blot protocol yielded a blot that, when scanned on the Li-Cor Odyssey infrared imager, gave specific signals at 700 and 800 nm wavelengths. This protocol was used to visualize the host and HCMV virus genomes in infected cells (O'Dowd et al., 2012). Figure 1 shows that the host genome could be detected in isolated uninfected host cellular DNA as well as in DNA extracted from infected cells, while the virus genome was specifically detected in DNA extracted from both isolated virions and infected cells. No significant cross hybridization was seen between the probed genomes.
Figure 1. Dual color Southern blot.
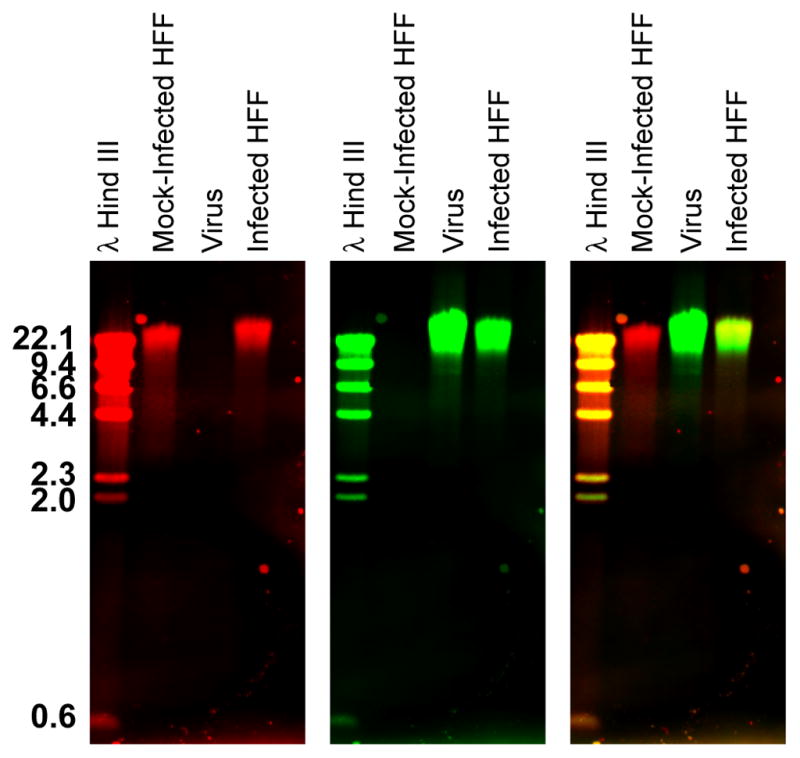
DNA was extracted from uninfected or HCMV-infected human fibroblasts (HFF) as well as isolated HCMV viral particles. DNAs were separated on a 0.8% agarose gel before transferring to a charged nylon membrane. Subsequently the blot was hybridized with a mixture of Dig-labeled host genomic probes and biotin-labeled virus genomic probes. Probes were detected with IRDye 700-conjugated α-mouse secondary Ab and IRDye 800-conjugated streptavidin, respectively. The host and virus genomes were visualized simultaneously in the 700 (red- left panel) and 800 (green- middle panel) nm channels, respectively, on a Li-Cor Odyssey Infrared Imager. An overlay image of the red and green channels is shown at the right. The yellow color in the overlay image indicates lanes that contain both host and viral DNA. This figure has been modified from figure 7 in O’Dowd et. al. (O'Dowd et al., 2012).
The dual color Southern blot protocol was extremely sensitive, being able to detect as little as 10 ng of DNA (Figure 2). As will be discussed in detail below, care must be taken in optimizing DNA loading, probe labeling and Ab concentration to ensure quantifiable signals.
Figure 2. Sensitivity of the dual color Southern.
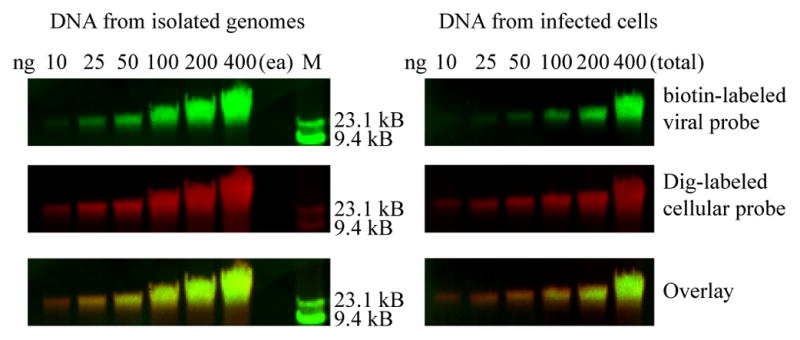
DNA extracted from uninfected HFF and pelleted HCMV viral particles were mixed in the amounts indicated and loaded on a 0.8% agarose gel (left panels) along with a lambda HindIII DNA size marker. DNA extracted from HCMV-infected HFFs was loaded in separate wells on the same gel in the amounts indicated (right panels). After capillary transfer the blot was hybridized with a mixture of Dig-labeled host genomic probes and biotin-labeled virus genomic probes. Probes were detected with IRDye 700-conjugated α-mouse secondary Ab and IRDye 800-conjugated streptavidin. The host and virus genomes were visualized simultaneously in the 800 (green- top panels) and 700 (red- middle panels) nm channels, respectively, on a Li-Cor Odyssey Infrared Imager. An overlay image of the red and green channels is shown in the bottom panels.
Discussion
Troubleshooting
Reducing background
As with any technique there are a number of factors that need to be considered and optimized. The greatest obstacle encountered was high background. The following are several factors that were addressed to obtain relatively clean, and, most importantly, quantifiable blots. Initial experiments with this technique produced some ugly results with significant background. Charged nylon membranes yielded lower background than uncharged nylon membranes. The membrane must be handled gently with forceps and gloves. Any creases in the membrane caused streaks in the final scan.
Certain blocking reagents also contributed to the background, including milk and BSA. Cold water fish skin gelatin produced the lowest background and cleanest blots. In addition, the final washes of the blot prior to scanning must exclude Tween because the detergent fluoresces and causes significant background in the green channel.
Transfer
Larger genomic DNA did not transfer efficiently using electroblotting. However, the very reliable and decidedly low-tech capillary transfer method, although slower, provided consistently good transfer and reproducible results. Efficient transfer of large DNA fragments required longer transfer times, up to 72 h.
Linear signal
The Odyssey scanner is extremely sensitive and the signal can be saturated by overloading the gel, using a probe that is too strongly labeled, or non-optimal Ab (or streptavidin) concentrations. For our experiments we found that 150 ng of DNA produced a strong signal in a linear range that could be accurately quantitated. The linear range was determined using the blot in Figure 2.
The signal from the biotin-labeled probes is extremely bright compared to the Dig-labeled probes. In order to balance the brightness of the probes a 1:20 dilution of biotin-dUTP was used in random primed reactions.
Slot blots with serial dilutions of DNA probed with different Ab (and streptavidin) concentrations were used to determine a linear signal range.
Quantitation
Quantitation should be performed on the original 16-bit Tagged Image File Format (TIFF) generated by the Odyssey imager. Any compression of the original image results in the loss of data and reduced accuracy in signal quantitation.
Alternative and Support Protocols
DNA was isolated using the protocol described in Adair et.al. (Adair et al., 2005). Alternate protocols may be substituted to isolate high quality DNA, including genomic DNA extraction kits from a plethora of sources.
Capillary transfer as well as the basic Southern blot protocol which uses radioactively labeled probes is described in detail in Molecular Cloning: A Laboratory Manual (Sambrook and Russell, 2001).
Many procedures can be used to fix DNA to the membrane for a Southern blot (step xxiii). Alternatives to UV irradiation include drying and baking, or microwaving. In addition, the alkaline transfer used in this protocol may be sufficient to fix the DNA to the membrane. Therefore, using existing laboratory protocols should be considered for this step.
This technique was based on the single channel Li-Cor Odyssey Southern blot (http://biosupport.licor.com/docs/09395_Southern.pdf).
Random priming of the probes can also be carried out as described by Feinberg and Vogelstein (Feinberg and Vogelstein, 1983).
When using probes for which wash conditions have been optimized previously try those optimized conditions first in steps xxix–xxxi.
Essential Literature References
Original papers
Book chapters
Equipment Support Website
Quick Procedure
Isolating DNA ~6 h
-
i
Harvest and pellet uninfected and infected cells as well as isolated pathogens.
-
ii
Isolate DNA.
Labeling probes ~ 6 h
-
iii
Digest extracted host and viral DNA, phenol chloroform extract, and isopropanol precipitate.
-
iv
Random prime label host and viral probes (one with Dig- and one with biotin-dUTP).
Gel electrophoresis and capillary transfer 1.5 h + O/N
-
v
Quantitate the DNA extracted from experimental samples and run on an agarose gel.
-
vi
Depurinate, denature and transfer the DNA from the gel to a charged nylon membrane.
Hybridization 4 h + O/N
-
vii
Pre-hybridize the blot for 2–4 h at 65°C.
-
viii
Denature the probe and hybridize to the blot O/N at 65°C.
-
ix
Wash the membrane and proceed with blocking.
Blocking and developing ~ 4 h
-
x
Block the blot for 1 h with shaking.
-
xi
Incubate the blot with α-Dig Ab for 1 h at RT with shaking.
-
xii
Wash the blot.
-
xiii
Incubate the blot with IRDye-conjugated α-mouse secondary Ab and streptavidin for 30–60 min at RT with shaking and protected from light.
-
xiv
Wash the blot.
-
xv
Wash the blot in TBS without Tween.
-
xvi
Scan the blot on a Li-Cor Odyssey Infrared Imaging System
Highlights.
A dual color Southern blot to visualize host and viral genomes simultaneously.
This protocol will aid studies of cells infected with any microorganism.
Very sensitive fluorescent imaging allows detection of nanogram quantities of DNA.
IRDye-based detection eliminates the need for radioactive labeling.
Eliminates the inherent loss in signal after stripping and reprobing of blots.
Acknowledgments
The authors thank John O’Dowd for critical reading and editing of the manuscript. This work was supported by NIH grants # RO1- AI51463, #P20 RR016454 (INBRE program) and #P20 RR015587 (COBRE program) to EAF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adair JE, Kwon Y, Dement GA, Smerdon MJ, Reeves R. Inhibition of nucleotide excision repair by high mobility group protein HMGA1. J Biol Chem. 2005;280:32184–92. doi: 10.1074/jbc.M505600200. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Analytical biochemistry. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol. 2007;81:1934–50. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dowd JM, Zavala AG, Brown CJ, Mori T, Fortunato EA. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012;8:e1003038. doi: 10.1371/journal.ppat.1003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW, editors. Molecular Cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York USA: 2001. [Google Scholar]