

Abstract
Chronic allergic asthma leads to airway remodeling and subepithelial fibrosis via mechanisms not fully understood. Airway remodeling is amplified by profibrotic mediators, such as transforming growth factor-β1 (TGF-β1), which plays a cardinal role in various models of fibrosis. We recently have identified a critical role for c-Jun-NH2-terminal-kinase (JNK) 1 in augmenting the profibrotic effects of TGF-β1, linked to epithelial-to-mesenchymal transition of airway epithelial cells. To examine the role of JNK1 in house dust mite (HDM)-induced airway remodeling, we induced allergic airway inflammation in wild-type (WT) and JNK1−/− mice by intranasal administration of HDM extract. WT and JNK1−/− mice were sensitized with intranasal aspirations of HDM extract for 15 days over 3 wk. HDM caused similar increases in airway hyperresponsiveness, mucus metaplasia, and airway inflammation in WT and JNK1−/− mice. In addition, the profibrotic cytokine TGF-β1 and phosphorylation of Smad3 were equally increased in WT and JNK1−/− mice. In contrast, increases in collagen content in lung tissue induced by HDM were significantly attenuated in JNK1−/− mice compared with WT controls. Furthermore HDM-induced increases of α-smooth muscle actin (α-SMA) protein and mRNA expression as well as the mesenchymal markers high-mobility group AT-hook 2 and collagen1A1 in WT mice were attenuated in JNK1−/− mice. The let-7 family of microRNAs has previously been linked to fibrosis. HDM exposure in WT mice and primary lung epithelial cells resulted in striking decreases in let-7g miRNA that were not observed in mice or primary lung epithelial cells lacking JNK1−/− mice. Overexpression of let-7g in lung epithelial cells reversed the HDM-induced increases in α-SMA. Collectively, these findings demonstrate an important requirement for JNK1 in promoting HDM-induced fibrotic airway remodeling.
Keywords: lung, epithelium, fibrosis, airway, epithelial-to-mesenchymal transition
allergic asthma is a chronic airway inflammatory-driven disease that is characterized by airway remodeling. The remodeling of the airways includes goblet cell hyperplasia, increased epithelial cell turnover, mucus metaplasia, extracellular matrix deposition, and subepithelial fibrosis (8, 10, 24). Airway remodeling has been shown in all degrees of asthma severities, and the remodeling process has been associated with an increase in the duration of asthma, reduced lung function, increased hyperresponsiveness, and greater use of medication (6). Airway remodeling is amplified by profibrotic mediators, such as transforming growth factor-β1 (TGF-β1) as well as interleukins-1, -13, and -17 (38, 43, 46). TGF-β1 expression in airways is sufficient to drive pulmonary fibrosis and is suggested as a key regulator of collagen deposition in asthma (31).
c-Jun-N-terminal kinases (JNK) are members of the family of mitogen-activated protein kinases (MAPK), which have been linked to diverse cellular processes that include apoptosis (41), proliferation (5), and survival (17). We and others have recently identified a critical role for JNK1 in various models of fibrosis. JNK1-deficient mice had decreased hepatic fibrosis (29), and JNK1−/− mice showed a significant attenuation of ovalbumin-induced subepithelial lung fibrosis as well as bleomycin- and TGF-β1-induced fibrosis (3). We have previously shown that JNK1 plays an important role in transducing TGF-β signals linked to epithelial-to-mesenchymal transition (EMT) of airway epithelial cells (1, 44), in part by controlling phosphorylation of Smad3 in the linker domain (44). EMT is an important process during embryonic development, tumor progression, and fibrotic tissue repair after injury (42). During EMT, epithelial cells enhance their migratory capacity by downregulation of epithelial markers, such as tight junction proteins and cytokeratins, and upregulation of mesenchymal proteins, such as α-smooth muscle actin (α-SMA) and vimentin (27, 42).
House dust mite (HDM; Dermatophagoides pteronyssinus) is one of the most common aeroallergens worldwide, and up to 85% of asthmatics are typically allergic to HDM (7). Exposure of HDM in mice has been shown to induce sustained airway inflammation, increased mucus production, and airway hyperresponsiveness (26). Increased collagen deposition and α-SMA expression indicative of airway remodeling have been observed in a mouse model of chronic HDM exposure (15). Furthermore, HDM exposure in vivo (25) and in vitro (22) has been shown to promote EMT, and primary epithelial cells from asthmatics are more susceptible to TGF-β1-induced EMT (20). These reports, together with the previous published data from our laboratory, led us to hypothesize that HDM-induced JNK1 activation is important in the induction of a mesenchymal expression profile in lung epithelial cells and subsequent remodeling of the airways. Therefore, the goal of this study was to investigate the role of the JNK1 in HDM-induced allergic airway remodeling.
MATERIALS AND METHODS
Animals and reagents.
JNK1−/− mice on a C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME) and were maintained as a breeding colony under pathogen-free conditions. Mice were housed with a 12-h:12-h light/dark cycle and allowed free access to standard laboratory chow and water. All experiments were conducted with age-matched JNK1−/− and wild-type (WT) littermate controls. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Vermont. All chemicals utilized were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Smad3, phospho-Smad3, and phospho-JNK antibodies were obtained from Cell Signaling Technology (Danvers, MA); antibodies to JNK1 and actin were from Santa Cruz Biotechnology (Santa Cruz, CA).
HDM administration.
HDM Dermatophagoides pteronyssinus extract (Greer Laboratories, Lenoir, NC) was resuspended in sterile PBS at a concentration of 1 μg protein/ml. The experimental group was sensitized with intranasal instillation of 50 μl of the HDM suspension for 15 days over three consecutive weeks, according to the schematic in Fig. 1A. The control group was sham sensitized with PBS.
Fig. 1.

Activation of c-Jun-NH2-terminal-kinase (JNK) and transforming growth factor (TGF)-β1 signaling pathways in house dust mite (HDM)-challenged wild-type (WT) and JNK1−/− mice. A: schematic depicting the time course of HDM exposure and euthanasia. For instillation, 50 μg HDM or PBS as the vehicle control was administered intranasally once at the days indicated via the arrows. Mice were euthanized 72 h after the last challenge. B: lung tissue was homogenized from PBS- or HDM-challenged mice for assessment of phosphorylated (p)-JNK1 and 2 and p-Smad3. Equal amounts of protein (20 μg) were separated by SDS-PAGE and subjected to Western blot analysis for p-JNK1/2 and p-Smad3. Total content of JNK1/2 and Smad3 was used as loading controls. Lanes are from individual mice in each treatment group.
Assessment of airway mechanics and airway hyperresponsiveness.
Mice were anesthetized with intraperitoneal pentobarbital sodium (90 mg/kg), tracheotomized, and mechanically ventilated at 200 breaths/min with a tidal volume of 0.25 ml and positive end-expiratory pressure of 3 cmH2O (FlexiVent; SCIREQ, Montreal, Quebec, Canada). Airway mechanics were assessed by the forced oscillation technique in which respiratory impedance was partitioned using the constant-phase model into measures of Newtonian resistance (Rn), a measure of conducting airway resistance, tissue dampening (G), a measure of tissue resistance, and tissue elastance (H), a measure of the stiffness of the lung (43). Airway responsiveness was assessed during airway challenge with increasing doses of aerosolized methacholine (saline control, 12.5, 25, and 50 mg/ml), as previously described (38). The response to methacholine at each dose was quantified as the average of the three peak measurements for each parameter of airway mechanics.
Collection of bronchoalveolar lavage.
Lungs were lavaged with 1.0 ml of 1× Dulbecco's PBS. Total and differential cell counts were performed as previously described (36). Briefly, cells were isolated by centrifugation, and total cell counts were performed using the Advia 120 (Siemens, Malvern, PA) automated hematology analyzer. Cytospins were performed and stained using the Hema3 kit (Fisher Scientific, Kalamazoo, MI). Differential cell counts were performed on a minimum of 300 cells.
Bronchoalveolar lavage fluid analyses.
Bronchoalveolar lavage fluid (BALF) was centrifuged at 1,200 g for 5 min to remove cells and debris and was then snap-frozen in liquid N2. BALF cytokine levels were determined by a 13-plex cytokine array (Bio-Rad, Hercules, CA), as directed by the manufacturer. Total TGF-β1 protein levels were determined by a Quantikine ELISA kit (R&D Systems, Minneapolis, MN).
Homogenization of lung tissue and Western blotting.
Protein lysates were prepared by mincing lung tissue in cold lysis buffer immediately followed by homogenization as previously described (3). Lysates were incubated on ice for 30 min, followed by 30 min of centrifugation at 16,000 g. A portion of the supernatant was saved for protein determination, before the addition of Laemmli sample buffer. Total protein was assessed by the Bio-Rad DC Protein Assay kit (Bio-Rad). Total JNK1/2, phospho JNK1/2, total Smad3, phospho Smad3, α-SMA, and β-actin protein abundance were evaluated by Western blotting.
Assessment of inflammation and mucus metaplasia.
Lungs were inflated to 25 cm H2O and fixed with 4% paraformaldehyde in PBS followed by paraffin imbedding. Paraffin blocks were cut into 5-μm sections and mounted to slides. Tissue histopathology and inflammation were assessed by hematoxylin and eosin staining. In mice challenged with PBS and HDM, mucus metaplasia was assessed by periodic acid Schiff staining (PAS) and quantified by scoring airway PAS reactivity using a scale of 0 to 3 (0 representing no positive staining and 3 being the highest intensity) by two independent, blinded observers. The cumulative score from each mouse was then averaged according to treatment group as described previously (3). MUC5AC and Gob5 mRNA expression was assessed via real-time PCR analysis.
α-SMA immunohistochemistry.
α-SMA staining was performed on lung sections after antigen retrieval by incubation of slides for 20 min in 0.01 M sodium citrate pH 6.0 at 95°C. Slides were then blocked with 2% normal goat serum for 30 min, followed by incubation with monoclonal mouse antibody against α-SMA (1:5,000 dilution; Sigma) overnight at 4°C. Biotinylated anti-mouse IgG was then applied for 30 min at room temperature, followed by addition of the avidin-biotin complex-alkaline phosphatase (Vectastain ABC-AP, Vector Laboratories) for another 30 min at room temperature. After the sections were rinsed in PBS, the substrate, Vector Red (Vector Laboratories), was added for 20 min. The Vector Red reacts with the bound alkaline phosphatase, producing an intense red color. Slides were counterstained with Mayer’s hematoxylin.
Assessment of HDM-induced collagen deposition.
Lung sections were stained with Masson's trichrome reagent to stain collagen. Slides were scored using a scale of 0 to 3 (0 being the least stain intensity, 3 the highest intensity) for airway-associated collagen deposition by two independent, blinded investigators. The cumulative score from each mouse was then averaged according to treatment group. Total lung collagen was measured in the upper right lobe of the lung after overnight digestion with 10 mg/ml pepsin in 0.5 M acetic acid using the Sircol Assay (Biocolor, Belfast, UK) as directed by the manufacturer.
RNA isolation and q-PCR.
RNA was extracted using miRNeasy columns (Qiagen, Valencia, CA) as directed by the manufacturer. Gene expression analysis (miRNA and mRNA) was performed by reverse transcriptase-qPCR using the miScript Reverse Transcription kit and SYBR Green PCR kit (Bio-Rad). PCR data were analyzed by using the ΔΔCt method of relative quantification. For microRNA expression, snoRNA202 was used as endogenous control. Primer sequences were taken from GeneBank or miRBase. All accession numbers are listed in Table 1.
Table 1.
Primer sequences
| Gene or miRNA | Accession | Sequences (5′ → 3′) | |
|---|---|---|---|
| Gene | |||
| Hmga2 | NM_010441 | Forward | aaggcagcaaaaacaagagc |
| Reverse | gcaggcttcttctgaacgac | ||
| TGF-β1 | NM_011577 | Forward | tgcttcagctccacagagaa |
| Reverse | tggttgtagagggcaaggac | ||
| Acta2 (α-SMA) | NM_007392.2 | Forward | ctgacagaggcaccactgaa |
| Reverse | catctccagagtccagcaca | ||
| Col1A1 | NM_007742.3 | Forward | gagcggagagtactggatcg |
| Reverse | gttcgggctgatgtaccagt | ||
| Muc5ac | NM_010844.1 | Forward | gctacacccaggttgagaagtg |
| Reverse | tcctcactttccttggacttga | ||
| Clca3 (GOB5) | NM_017474 | Forward | actaaggtggcctacctccaa |
| Reverse | ggaggtgagtcaaggtgaga | ||
| Actin | NM_007393.3 | Forward | ctgaatggcccaggtctga |
| Reverse | ccctcccagggagaccaa | ||
| miRNA | |||
| let-7 g | MIMAT0000121 | Forward | tgaggtagtagtttgtacagtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7a | MIMAT0000521 | Forward | tgaggtagtaggttgtatagtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7b | MIMAT0000522 | Forward | tgaggtagtagttgtgtggtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7c | MIMAT0000523 | Forward | tgaggtagtaggttgtatggtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7d | MIMAT0000383 | Forward | agaggtagtagttgcatagtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7f | MIMAT0000525 | Forward | tgaggtagtagattgtatagtt |
| Reverse | Qiagen miScript universal primer | ||
| let-7i | MIMAT0000122 | Forward | tgaggtagtagtttgtgctgtt |
| Reverse | Qiagen miScript universal primer | ||
| snoRNA202 | SO:0000593 | Forward | cttttgaacccttttccatctg |
| Reverse | Qiagen miScript universal primer | ||
Primer sequences for quantitative PCR cycling conditions. Sequences were taken from GeneBank, and all accession numbers are denoted.
TGF, transforming growth factor; SMA, smooth muscle actin.
Cell culture and transfection.
Primary mouse tracheal epithelial (MTE) cell cultures were isolated as previously published (44). MTE cells were stimulated with 50 μg of HDM for 72 h. A line of spontaneously transformed murine alveolar type II epithelial cells (C10) (32) was cultured as described elsewhere (44). Transient transfections were performed using Nanofectin (PAA, Pasching, Austria) according to the manufacturer's instructions. Twenty-four hours before HDM (50 μg) stimulation for 48 h, 1 μg of precursor (pre)-let-7g expression vector (Cell Biolaboratories, San Diego, CA) was transfected into C10 cells. Both MTE and C10 cells were lysed for Western blotting and gene expression analysis.
Statistical analysis.
Data were evaluated using SPSS (version 21) by one-way ANOVA using the Bonferroni test to adjust for multiple comparisons. Histological scoring was analyzed using the Kruskal-Wallis test and Dunn's multiple-comparison post hoc tests. Results with P < 0.05 or smaller were considered statistically significant.
RESULTS
Activation of JNK and TGF-β1 signaling pathways in HDM-challenged WT and JNK1−/− mice.
Recent work in our laboratory identified JNK1 as a crucial mediator of lung fibrosis in different models (3). To identify a role of JNK in HDM-induced allergic airway disease, WT and JNK1−/− C57BL/6 mice were challenged with HDM (Fig. 1A), and phosphorylated JNK (phospho-JNK), which is reflective of JNK activation, was assessed by Western blot using lung homogenates. Results in Fig. 1B demonstrate an increase in phospho-JNK 1 and 2 in HDM-challenged WT mice compared with the PBS control group, suggesting activation of the JNK signaling pathway. A comparable increase in phospho-JNK 2 in HDM-challenged JNK1−/− mice was observed, suggesting that JNK2 does not compensate for the loss of JNK1. Note that two isoforms of JNK1 and JNK2 are expressed in WT mice. Hereafter only the major bands of JNK1 and 2 will be indicated. TGF-β1 has been shown to be critical in the pathogenesis of lung fibrosis (12), including subepithelial fibrosis (40). To examine TGF-β1 pathway activation, we evaluated phosphorylation of the receptor Smad, Smad3. Assessment of WT and JNK1−/− mice following HDM challenge demonstrated similar increases in phosphorylation of Smad3 compared with PBS-challenged mice (Fig. 1B), indicating activation of proximal TGF-β1 signaling in response to HDM.
Because JNK and TGF-β1 signaling pathways are activated following HDM exposure, we also examined the expression of TGF-β1 protein and mRNA in the lungs of WT and JNK1−/− mice exposed to daily administrations of HDM or PBS (5 days/wk) for 3 wk. Similar increases in total TGF-β1 protein in the BALF and mRNA in lung homogenates were observed in WT and JNK1−/− mice following HDM exposure compared with PBS-treated mice (data not shown), demonstrating that HDM-induced TGF-β1 expression is similar in WT and JNK1−/− mice.
HDM-induced allergic airway inflammation and airway hyperresponsiveness in WT and JNK1−/− mice.
Inflammation is a significant hallmark of allergic airway disease. We next addressed the impact of genetic ablation of JNK1 on HDM-induced allergic airway inflammation. Evaluation of lung histopathology revealed infiltration of inflammatory cells in both WT and JNK1−/− mice following HDM exposure (Fig. 2A). Assessment of HDM-induced airway inflammation via enumeration of total and differential cell counts in the BALF revealed no clear differences between WT and JNK1−/− mice (Fig. 2B). Comparable increases in eosinophils (Fig. 2C) and lymphocytes (data not shown) were observed in WT and JNK1−/− mice following HDM exposure, whereas neutrophils and macrophages were not significantly changed in response to HDM at this time point (data not shown). BAL inflammatory cytokines and mRNA expression were also comparably elevated in WT and JNK1−/− mice in response to HDM (data not shown) with the exception granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-6 mRNA. Increases in GM-CSF and IL-6 were observed in lung tissue from JNK1−/− mice exposed to HDM compared with WT mice (data not shown). Despite these differences in mRNA expression, which were modest, our data overall show that HDM-induced allergic airway inflammation occurred to a similar extent in WT and JNK1−/− mice. To address the role of JNK1−/− in the manifestation of airway hyperresponsiveness following HDM exposure, we measured the changes in airway mechanics in response to methacholine. HDM exposure did not significantly affect conducting airway resistance (Rn) during methacholine challenge in WT or JNK1−/− mice (Fig. 2D). In contrast, HDM exposure led to increases in tissue dampening (G) and tissue elastance (H) at the higher doses of the methacholine challenge in WT and JNK1−/− mice (Fig. 2, E and F, respectively). However, there was no significant difference in the increase in tissue dampening or tissue elastance between HDM-exposed WT and JNK1−/− mice, with the exception that increases in tissue elastance were apparent at a lower dose of methacholine in JNK1−/− mice compared with WT animals.
Fig. 2.

Assessment of HDM-induced inflammation and airway hyperresponsiveness in WT and JNK1−/− mice. A: lung histopathology in WT or JNK1−/− mice exposed to HDM or PBS as the vehicle control. Lung sections were stained with hematoxylin and eosin (magnification: ×200). Inflammatory cell profile in bronchoalveolar lavage fluid. Total cells (B) and eosinophils (C) were enumerated. Changes in respiratory mechanics were analyzed in PBS- or HDM-challenged WT or JNK1−/− mice 72 h following the last HDM exposure. Ascending doses of methacholine were administered to determine airway resistance (Rn) (D), tissue dampening (G) (E), and elastance/stiffness (H) (F) parameters. The response to methacholine at each dose was quantified as the average of the 3 peak measurements for each parameter of airway mechanics. Data shown represent means ± SE from 2 independent experiments (n = 9 WT PBS, 10 WT HDM, 7 JNK1−/− PBS, 9 JNK1−/− HDM). *P < 0.05 (ANOVA) compared with respective controls.
HDM-induced mucus metaplasia in WT and JNK1−/− mice.
Another clinical manifestation of allergic airway disease is the development of mucus metaplasia. Assessment of mucus metaplasia in WT or JNK1−/− mice exposed to HDM revealed comparable reactivity with the PAS stain (Fig. 3, A and B). In addition, evaluation of the airway mucin MUC5AC and Gob5 (Clca3), a calcium-activated chloride channel involved in the regulation of mucus production and secretion, showed similar increases in WT and JNK1−/− mice (Fig. 3C), suggesting that, in these experimental settings, JNK1 does not play a dominant role in mucus metaplasia.
Fig. 3.

Evaluation of HDM-induced mucus metaplasia in WT and JNK1−/− mice. A: periodic Acid-Schiff (PAS) staining of airway mucus in WT or JNK1−/− mice exposed to HDM or PBS as the vehicle control (magnification: ×200). B: quantification of airway mucus staining intensity was determined by scoring of slides by 2 blinded investigators. *P < 0.05 (Kruskal-Wallis) compared with respective controls. C: quantification of mRNA levels for MUC5AC and CLCA3 (GOB5) in lung tissue homogenates by q-PCR. Results are presented as fold change compared with PBS controls. Data shown represent means ± SE from 2 independent experiments (n = 9 WT PBS, 10 WT HDM, 7 JNK1−/− PBS, 9 JNK1−/− HDM). *P < 0.05 (ANOVA) compared with respective controls.
HDM-induced airway remodeling and fibrotic gene expression in WT and JNK1−/− mice.
We next evaluated the role of JNK1 in HDM-induced fibrotic airway remodeling via analysis of lung histopathology using the Masson's trichrome collagen stain. HDM exposure in WT mice resulted in a significantly enhanced collagen content around the bronchioles. In contrast, the HDM-induced peribronchiolar deposition of collagen was significantly attenuated in JNK1−/− mice (Fig. 4, A and B). Quantitative assessment of total lung collagen content also demonstrated decreases in JNK1−/− mice compared with WT animals exposed to HDM (Fig. 4C), demonstrating that the presence of JNK1 contributes to HDM-induced increases in collagen deposition. Recent work in our laboratory identified JNK1 as a crucial mediator of TGF-β1-induced EMT (1) and lung fibrosis (3). Histological evaluation of the mesenchymal protein, α-SMA, demonstrated a significant increase in WT mice exposed to HDM, which was almost completely abrogated in JNK1−/− mice (Fig. 5A), consistent with decreases in overall α-SMA protein content and mRNA expression in JNK1−/− mice compared with WT animals (Fig. 5, B and C). In addition, increases in mRNA expression of high-mobility group AT-hook 2 (HMGA2), a critical regulator of EMT, observed in HDM-exposed WT mice were not observed in mice lacking JNK1−/− (Fig. 5C). Consistent with the attenuation of HDM-mediated increases in collagen content in JNK1−/− mice (Fig. 4), HDM-mediated increases in collagen1A1 (Col1a1) mRNA were also attenuated in JNK1−/− mice compared with WT littermate controls (Fig. 5C). The family of let-7 microRNAs (miRNA) are effective repressors of HMGA2 (23) and have been previously shown to play a role on fibrosis (35). Analysis of the Let-7 miRNA family revealed a significant decrease of let-7g miRNA in WT mice following HDM exposure, which was not observed in JNK1−/− mice (Fig. 5D). Overall, these data demonstrate that JNK1 is required for HDM-induced peribronchiolar fibrotic remodeling and mesenchymal gene expression in association with increases in the EMT regulator, HMGA2, and decreases in let-7g miRNA.
Fig. 4.
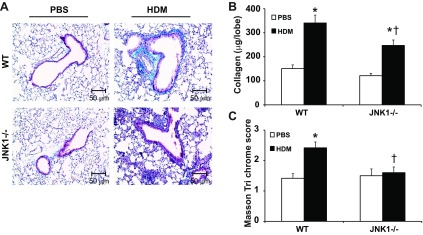
Impact of JNK1 gene ablation for HDM-induced fibrotic airway remodeling. A: histopathological analysis of Masson's trichrome-stained airway sections from WT or JNK1−/− mice exposed to PBS or HDM (magnification: ×200). B: semiquantitative analysis of peribronchiolar collagen deposition. Peribronchiolar Masson's trichrome reactivity in airways of similar dimension/mouse was scored by 2 blinded investigators, and cumulative scores were averaged. *P < 0.05 (Kruskal-Wallis) compared with respective control and †P < 0.05 (Kruskal-Wallis) compared with WT HDM. C: assessment of total collagen content in the upper right lobe of mice after HDM exposure. *P < 0.05 (ANOVA) compared with respective control and †P < 0.05 (ANOVA) vs. WT HDM. Data shown represent means ± SE from 2 independent experiments (n = 9 WT PBS, 10 WT HDM, 7 JNK1−/− PBS, 9 JNK1−/− HDM).
Fig. 5.
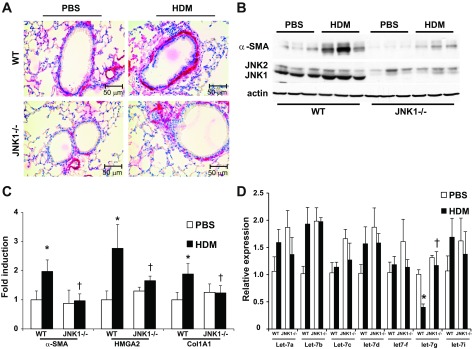
Impact of JNK1 gene ablation on HDM-induced expression of profibrotic mediators. A: assessment of α-smooth muscle actin (α-SMA) immunolocalization in lung sections from WT and JNK1−/− mice exposed to PBS or HDM (magnification: ×200). B: evaluation of α-SMA content in lung tissue from WT and JNK1−/− mice exposed to HDM. Equal amounts of protein (10 μg) were separated by SDS-PAGE and subjected to Western blot analysis for α-SMA and JNK1/2. Actin was used as a loading control. Lanes represent individual mice in each group. Assessment of mRNA abundance of α-SMA, HMGA2, Coll1A1 (C) and miRNA let-7a,b,c,d,f,g and i by q-PCR in lung homogenates (D) from WT and JNK1−/− mice exposed to HDM. mRNA abundance was normalized to β-actin, whereas let-7 miRNAs were normalized to SNO202 (miRNA). Results are expressed as fold change compared with WT PBS and reflect means ± SE from 2 independent experiments (n = 9 WT PBS, 10 WT HDM, 7 JNK1−/− PBS, 9 JNK1−/− HDM). *P < 0.05 (ANOVA) compared with respective controls. †P < 0.05 compared with the WT HDM group.
HDM-induced mesenchymal protein and gene expression in WT and JNK1−/− MTE cells.
We previously demonstrated that TGF-β1-induced mesenchymal increases in lung epithelial cells are JNK1 dependent (1). To further investigate the mechanism by which HDM exerts its effects, we extended our observations by using WT and JNK1−/− primary MTE cells. WT and JNK1−/− primary mouse tracheal epithelial cells were exposed to either PBS or HDM for 3 days. HDM stimulation resulted in an increase in α-SMA protein (Fig. 6A) and a significant increase in α-SMA, HMGA2, and collagen1A1 mRNA compared with PBS-treated cells (Fig. 6B). In contrast, HDM-mediated increases in these mesenchymal mediators were almost completely absent in JNK1−/− MTE cells (Fig. 6). Analysis of the Let-7 miRNA family revealed a similar decrease of let-7g miRNA in WT MTE cells following HDM exposure (Fig. 6C), similar to our observations in HDM-exposed WT mice (Fig. 5D). Similar to results in JNK1−/− mice, let-7g was not attenuated in JNK1−/− MTE cells exposed to HDM. These data suggest a functional link between JNK1 and repression of let-7g miRNA.
Fig. 6.

HDM-induced mesenchymal protein and gene expression in WT and JNK1−/− mouse tracheal epithelial (MTE) cells. A: evaluation of α-SMA content in WT and JNK−/− MTE cells stimulated with HDM (50 μg) for 3 days. Equal amounts of protein (20 μg) were separated by SDS-PAGE and subjected to Western blot analysis for α-SMA and JNK1/2. Actin was used as a loading control. Assessment of mRNA abundance of α-SMA, HMGA2, Coll1A1 (B) and miRNA let-7a,b,c,d,f,g and i (C) isolated from WT and JNK1−/− MTE cells exposed to HDM. mRNA abundance was normalized to β-actin, whereas let-7 miRNAs were normalized to SNO202 (miRNA). Results are expressed as fold change compared with WT PBS and reflect means ± SE from 3 independent experiments *P < 0.05 (ANOVA) compared with respective controls. †P < 0.05 compared with the WT HDM group.
Let-7g expression in lung epithelial cell prevents HDM-induced expression of α-SMA.
We next sought to further explore the functional role of let-7g in HDM-induced increases in α-SMA in lung epithelial cells. We therefore overexpressed precursor (pre)-let-7g miRNA in C10 mouse lung epithelial cells before exposure of cells to PBS or HDM. Exposure of C10 cells to HDM led to an increase in α-SMA protein, α-SMA mRNA, and a decrease in let-7g miRNA expression (Fig. 7). The observed increases in α-SMA protein and α-SMA mRNA after stimulation with HDM were no longer apparent after the overexpression of let-7g miRNA (Fig. 7). These findings demonstrate the ability of let-7g miRNA to prevent HDM-induced increase in α-SMA.
Fig. 7.

let-7g expression in lung epithelial cell prevents HDM-induced expression of α-SMA. A: evaluation of α-SMA content in C10 cells transfected with pre-let-7g before stimulation with HDM (50 μg) for 48 h. Equal amounts of protein (20 μg) were separated by SDS-PAGE and subjected to Western blot analysis for α-SMA. Actin was used as a loading control. Assessment of miRNA let-7g (B) and mRNA abundance of α-SMA (C) in C10 cells transfected with control (con) or pre-let-7g miRNA, before stimulation with PBS or HDM. let-7 miRNAs were normalized to SNO202 (miRNA) and mRNA abundance was normalized to β-actin. Results are expressed as fold change compared with the PBS group and reflect means ± SE from 2 independent experiments *P < 0.05 (ANOVA) compared with respective controls. †P < 0.05 compared with the control HDM group.
DISCUSSION
The molecular events that contribute to airway remodeling in patients with allergic airway disease are not completely unraveled. In this study, we set out to investigate the role of JNK1 for the development of airway inflammation, hyperresponsiveness, and remodeling in an HDM model of allergic airway disease. We demonstrate herein that genetic ablation of JNK1 does not affect HDM-induced inflammation, airway hyperresponsiveness, or mucus metaplasia. However, we demonstrate the importance of the JNK1 signaling pathway in augmenting peribronchiolar fibrotic remodeling and enhancing the expression of profibrotic mediators. Notably JNK1−/− mice were considerably protected against HDM-induced increased α-SMA content and profibrotic mediator expression compared with WT animals, potentially by preserving miRNA let-7g expression.
Hallmarks of the pathophysiology associated with exposure to allergen are increased inflammatory cells, mucus metaplasia, and airway hyperresponsiveness, in association with increases in Th2 cytokines. JNK has been shown to regulate innate and adaptive immune responses (11), including Th2 polarization (13). JNK controls the expression of proinflammatory cytokines, such as IL-2, IL-6, and TNF-α in inflammatory bowel disease (39). However, in the present study, HDM-induced airway inflammation, cytokine mRNA expression, and content in the BALF were not different in WT or JNK1−/− mice following HDM exposure, with the exception of slight increases in IL-6 and GM-CSF mRNA expression in JNK1−/− mice compared with WT animals. Similarly, mucus metaplasia, a Th2-dependent process, was not affected following ablation of JNK1. The lack of an impact of ablation of JNK1 on HDM-triggered airway inflammation observed herein is puzzling. Because JNK1 and JNK2 have often been considered to have overlapping or redundant functions, it is conceivable that compensatory activation of JNK2 could replace the function of JNK1. However, phospho-JNK2 was increased comparably in response to HDM in both WT and JNK1−/− mice (Fig. 1B), suggesting that observed differences herein are not due to JNK2. Recent studies have shown opposing roles of JNK1 and 2, based on the observation that JNKs can activate a large number of different substrates dependent on the specific stimulus and cell type (7).
Previous studies also demonstrated activation of the JNK signaling pathway in models of allergic inflammation, confirming our results (14, 34). However, these two studies revealed conflicting data about the role of JNK in allergic inflammation and airway hyperresponsiveness based upon the use of a nonspecific JNK inhibitor (SP600125) (4). In a rat model of ovalbumin-induced allergic inflammation, inhibition of JNK with SP600125 had no effect on mucosal inflammation and airway hyperresponsiveness (14), whereas, in a mouse model of ovalbumin-induced allergic asthma, SP600125 attenuated allergen-induced airway hyperresponsiveness and significantly inhibited eosinophil and lymphocyte inflammation (34). The lack of an impact on proinflammatory responses and airway hyperresponsiveness in JNK1−/− mice compared with WT animals observed in the present study suggests that, in the setting of an asthma-relevant allergen and sensitization via the airways, JNK1 does not play a dominant role in allergic inflammation or airway hyperresponsiveness. However, additional studies will be required to address the contribution of JNK1 in specific cell types and the timing of its activation during the pathogenesis of allergic airways disease, which could dictate its functional outcome.
An important feature of the pathophysiology of asthma is airway remodeling, which includes changes in the composition and organization of the airway cellular and structural components. In agreement with previous studies (15, 19, 26), we observed an increase in fibrotic lung remodeling in HDM-induced allergic airway disease. The TGF-β1 signaling pathway has been highlighted as a cardinal mediator in the lung-remodeling process in asthma (21). The present study shows that TGF-β1 cytokine expression and mRNA were upregulated significantly in the lung following HDM exposure, similar to what has been shown in previous studies involving both humans and mice with allergic airway disease (15, 26, 28). Conflicting data about the role of TGF-β1 in antigen-induced lung remodeling exists. TGF-β1 has been shown to be a mediator of remodeling in the Ova model of allergic airway disease, yet it attenuated airway hyperresponsiveness (2, 33). However, TGF-β1 was found to be not critically required for the development of HDM-induced remodeling (15). Similar to previously described results in the Ova model of allergic airway disease, no differences in TGF-β1 production were observed in JNK1 mice compared with WT control following HDM exposure (3). Despite the lack of differences in TGF-β1, we demonstrate herein a functional contribution of JNK1 in HDM-induced lung remodeling, consistent with a previous study in which a JNK inhibitor inhibited allergen-induced fibrotic remodeling (34) and our previous studies demonstrating a critical role for JNK1 in various models of lung fibrosis (3). The full details of the mechanisms whereby JNK1 elicits profibrotic signals require additional investigation.
We recently demonstrated that JNK1 facilitates mesenchymal expression in epithelial cells stimulated with TGF-β1 via phosphorylation of Smad3 in the linker domain (44), raising the possibility that JNK1 controls fibrotic airway remodeling downstream of TGF-β1 via control of Smads. Interestingly, epithelial-specific overexpression of Smad2 results in an increased susceptibility of fibrotic remodeling features following HDM exposure (18), in an IL-25-dependent manner (19). Smad3 has been shown to play a pivotal role in signal transduction pathways associated with TGF-β1-mediated wound healing and fibrosis (16). Indeed we observe an increase in Smad3 phosphorylation following HDM exposure. Furthermore, Smad2/3 was found to be activated after allergen challenge in epithelial and subepithelial cells within bronchial biopsies. However, mice lacking Smad3 were not protected against HDM-induced fibrotic lung remodeling (15), suggesting that Smad signaling pathway may not be the only pathway by which the fibrogenic effects of TGF-β1 are mediated. Alternatively, differences in the strains of mouse or protocols used to elicit allergic airway disease in the aforementioned studies could account for the apparent discrepant findings. Numerous studies have shown that the phenotype and model severity of antigen-induced airway disease in mice are strain dependent (9).
Previous work from our laboratory demonstrated the functional importance of JNK1 in transducing TGF-β signals linked to EMT of airway epithelial cells (1). In vitro studies have demonstrated that HDM proteins can collaborate with TGF-β1 to promote EMT in bronchial epithelial cells (22) and cleavage of junction proteins (45). Furthermore, primary airway epithelial cells from asthmatics reveal an increased susceptibility to TGF-β1-induced EMT via a Smad3-dependent process, supporting the notion that the asthmatic airway epithelium displays an altered epithelial repair phenotype. In vivo, EMT has recently been identified as a significant contributor in HDM- induced lung remodeling (25). In the present study, we observed increases in mesenchymal gene expression following HDM exposure in WT mice, which were attenuated in JNK1−/− mice. In addition, our previous studies demonstrated that TGF-β1-induced mRNA expression of HMGA2 requires JNK1 and depends on the phosphorylation status of the Smad3 linker region (44). HMGA2 is a nonhistone chromosome protein that is a proximal regulator of EMT. In the present study, HDM-induced upregulation of HMGA2 was almost completely inhibited in JNK1−/− mice. Altogether, these findings suggest the requirement of JNK1 and EMT in HDM-induced lung remodeling.
Members of the let-7 miRNA family have been shown to be the most abundantly expressed miRNAs in lung tissue (37). Significantly, let-7 miRNAs are effective repressors of HMGA2. Our findings that let-7g miRNA is suppressed following HDM exposure of mice and primary tracheal epithelial cells in vitro and requires JNK1 are novel. let-7 miRNA was previously shown to posttranscriptionally inhibit IL-13 in an allergic asthma model in mice. Consequently, intranasal delivery of a let-7 mimic alleviated asthma features such as inflammation and subepithelial fibrosis (30). Although significant decreases in let-7g miRNA were observed following HDM exposure, which did not occur in JNK1−/− mice, no differences in IL-13 mRNA were detected between the different groups. These results suggest that JNK1-dependent modulation of let-7g miRNA specifically does not control expression of IL-13, or that the current study design was not optimal to determine associations between let-7g miRNA and expression of IL-13. In contrast, in a different study using a similar murine model of asthma, let-7 miRNAs have been suggested to play a proinflammatory role (37). We have recently demonstrated a TGF-β1-induced interaction between Smad3 and JNK1 and that TGF-β1-induced Smad3 phosphorylation in the linker region and Smad transcriptional activity are controlled by JNK1 (44). Our findings, together with the observation that in idiopathic pulmonary fibrosis miRNA let-7d expression is repressed via Smad3 binding to the let-7d promoter (35), suggest a putative role of JNK1 in regulating let-7 miRNAs through a Smad3-dependent mechanism. However, additional studies will be required to unravel the exact interplay between JNK1 and Smad3 in the regulation of let-7 miRNAs, which also will require differentiation between let-7d and let-7g.
In conclusion, findings of the present study demonstrate an important role for JNK1 in promoting HDM-induced fibrotic airway remodeling, independent of the recruitment of inflammatory cells, airway hyperresponsiveness, and mucus metaplasia. Therefore, new strategies aimed at attenuation of fibrotic airway remodeling in asthmatics, which is not targeted with current therapeutics, could involve targeting facets of the JNK1 pathway.
GRANTS
This work was supported by grants R01 HL085464 and T32 HLO76122 from the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.L.J.v.d.V. and Y.M.W.J.-H. conception and design of research; J.L.J.v.d.V., S.M.H., K.G.L., A.S.G., M.A., and N.D. performed experiments; J.L.J.v.d.V., J.E.T., and N.D. analyzed data; J.L.J.v.d.V., D.G.C., L.K.L., C.G.I., and Y.M.W.J.-H. interpreted results of experiments; J.L.J.v.d.V. prepared figures; J.L.J.v.d.V. drafted manuscript; J.L.J.v.d.V., S.M.H., J.F.A., J.E.T., D.G.C., K.G.L., L.K.L., C.G.I., and Y.M.W.J.-H. edited and revised manuscript; J.L.J.v.d.V., S.M.H., J.F.A., J.E.T., D.G.C., K.G.L., A.S.G., L.K.L., M.A., N.D., C.G.I., and Y.M.W.J.-H. approved final version of manuscript.
REFERENCES
- 1.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YM. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J Cell Sci 121: 1036–1045, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcorn JF, Rinaldi LM, Jaffe EF, van Loon M, Bates JH, Janssen-Heininger YM, Irvin CG. Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med 176: 974–982, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alcorn JF, van der Velden J, Brown AL, McElhinney B, Irvin CG, Janssen-Heininger YM. c-Jun N-terminal kinase 1 is required for the development of pulmonary fibrosis. Am J Respir Cell Mol Biol 40: 422–432, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachleda P, Dvorak Z. Pharmacological inhibitors of JNK and ERK kinases SP600125 and U0126 are not appropriate tools for studies of drug metabolism because they activate aryl hydrocarbon receptor. Gen Physiol Biophys 27: 143–145, 2008 [PubMed] [Google Scholar]
- 5.Bantel H, Schmitz ML, Raible A, Gregor M, Schulze-Osthoff K. Critical role of NF-κB and stress-activated protein kinases in steroid unresponsiveness. FASEB J 16: 1832–1834, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Bergeron C, Al-Ramli W, Hamid Q. Remodeling in asthma. Proc Am Thorac Soc 6: 301–305, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Bode AM, Dong Z. The functional contrariety of JNK. Mol Carcinog 46: 591–598, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola Asthma AM. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 161: 1720–1745, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Boyce JA, Austen KF. No audible wheezing: nuggets and conundrums from mouse asthma models. J Exp Med 201: 1869–1873, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol 111: 215–225; quiz 226, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol 9: 1000–1004, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science 282: 2092–2095, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Eynott PR, Xu L, Bennett BL, Noble A, Leung SY, Nath P, Groneberg DA, Adcock IM, Chung KF. Effect of an inhibitor of Jun N-terminal protein kinase, SP600125, in single allergen challenge in sensitized rats. Immunology 112: 446–453, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fattouh R, Midence NG, Arias K, Johnson JR, Walker TD, Goncharova S, Souza KP, Gregory RC, Jr, Lonning S, Gauldie J, Jordana M. Transforming growth factor-beta regulates house dust mite-induced allergic airway inflammation but not airway remodeling. Am J Respir Crit Care Med 177: 593–603, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol 85: 47–64, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuichi S, Hashimoto S, Gon Y, Matsumoto K, Horie T. p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase regulate interleukin-8 and RANTES production in hyperosmolarity stimulated human bronchial epithelial cells. Respirology 7: 193–200, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, McKenzie AN, Lloyd CM. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 68: 82–90, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory LG, Mathie SA, Walker SA, Pegorier S, Jones CP, Lloyd CM. Overexpression of Smad2 drives house dust mite-mediated airway remodeling and airway hyperresponsiveness via activin and IL-25. Am J Respir Crit Care Med 182: 143–154, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med 180: 122–133, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Halwani R, Al-Muhsen S, Al-Jahdali H, Hamid Q. Role of transforming growth factor-beta in airway remodeling in asthma. Am J Respir Cell Mol Biol 44: 127–133, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol 42: 69–79, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Janssen-Heininger YM, Poynter ME, Aesif SW, Pantano C, Ather JL, Reynaert NL, Ckless K, Anathy V, van der Velden J, Irvin CG, van der Vliet A. Nuclear factor kappaB, airway epithelium, and asthma: avenues for redox control. Proc Am Thorac Soc 6: 249–255, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeffery PK. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 1: 176–183, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One 6: e16175, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD, Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med 169: 378–385, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112: 1776–1784, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kariyawasam HH, Pegorier S, Barkans J, Xanthou G, Aizen M, Ying S, Kay AB, Lloyd CM, Robinson DS. Activin and transforming growth factor-beta signaling pathways are activated after allergen challenge in mild asthma. J Allergy Clin Immunol 124: 454–462, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 138: 347–359, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA 105: 3903–3908, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer RJ, Elias JA. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med 200: 377–389, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malkinson AM, Dwyer-Nield LD, Rice PL, Dinsdale D. Mouse lung epithelial cell lines—tools for the study of differentiation and the neoplastic phenotype. Toxicology 123: 53–100, 1997 [DOI] [PubMed] [Google Scholar]
- 33.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol 174: 5774–5780, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Nath P, Eynott P, Leung SY, Adcock IM, Bennett BL, Chung KF. Potential role of c-Jun NH2-terminal kinase in allergic airway inflammation and remodelling: effects of SP600125. Eur J Pharmacol 506: 273–283, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, Richards T, Selman M, Watkins SC, Pardo A, Ben-Yehudah A, Bouros D, Eickelberg O, Ray P, Benos PV, Kaminski N. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 182: 200–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. Nuclear factor-kappaB activation in airway epithelium induces inflammation and hyperresponsiveness. Am J Respir Crit Care Med 177: 959–969, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polikepahad S, Knight JM, Naghavi AO, Oplt T, Creighton CJ, Shaw C, Benham AL, Kim J, Soibam B, Harris RA, Coarfa C, Zariff A, Milosavljevic A, Batts LM, Kheradmand F, Gunaratne PH, Corry DB. Proinflammatory role for let-7 microRNAS in experimental asthma. J Biol Chem 285: 30139–30149, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riesenfeld E, Allen GB, Bates JH, Poynter ME, Wu M, Aimiand S, Lundblad LK. The temporal evolution of airways hyperresponsiveness and inflammation. J Allergy Ther 1: 1–7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy PK, Rashid F, Bragg J, Ibdah JA. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol 14: 200–202, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc 3: 413–417, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sousa AR, Lane SJ, Soh C, Lee TH. In vivo resistance to corticosteroids in bronchial asthma is associated with enhanced phosyphorylation of JUN N-terminal kinase and failure of prednisolone to inhibit JUN N-terminal kinase phosphorylation. J Allergy Clin Immunol 104: 565–574, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 15: 740–746, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Tomioka S, Bates JH, Irvin CG. Airway and tissue mechanics in a murine model of asthma: alveolar capsule vs. forced oscillations. J Appl Physiol 93: 263–270, 2002 [DOI] [PubMed] [Google Scholar]
- 44.Velden JL, Alcorn JF, Guala AS, Badura EC, Janssen-Heininger YM. c-Jun N-terminal kinase 1 promotes transforming growth factor-beta1-induced epithelial-to-mesenchymal transition via control of linker phosphorylation and transcriptional activity of Smad3. Am J Respir Cell Mol Biol 44: 571–581, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan H, Winton HL, Soeller C, Gruenert DC, Thompson PJ, Cannell MB, Stewart GA, Garrod DR, Robinson C. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. J Exp Allergy 30: 685–698, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4: 583–594, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]