

Abstract
Our previous studies support the protective effect of cGMP and cGMP-dependent protein kinase I (PKG-I) pathway on the development of renal fibrosis. Therefore, in the present studies, we determined whether pharmacologically or genetically increased PKG activity attenuates renal fibrosis in a unilateral ureteral obstruction (UUO) model and also examined the mechanisms involved. To increase PKG activity, we used the phosphodiesterase 5 inhibitor sildenafil and PKG transgenic mice. UUO model was induced in wild-type or PKG-I transgenic mice by ligating the left lateral ureteral and the renal fibrosis was observed after 14 days of ligation. Sildenafil was administered into wild-type UUO mice for 14 days. In vitro, macrophage and proximal tubular cell function was also analyzed. We found that sildenafil treatment or PKG transgenic mice had significantly reduced UUO-induced renal fibrosis, which was associated with reduced TGF-β signaling and reduced macrophage infiltration into kidney interstitial. In vitro data further demonstrated that both macrophages and proximal tubular cells were important sources of UUO-induced renal TGF-β levels. The interaction between macrophages and tubular cells contributes to TGF-β-induced renal fibrosis. Taken together, these data suggest that increasing PKG activity ameliorates renal fibrosis in part through regulation of macrophage and tubular cell function, leading to reduced TGF-β-induced fibrosis.
Keywords: PKG, UUO, renal fibrosis, TGF-β1
progressive renal fibrosis is a final common feature of all chronic kidney diseases. It affects 8–16% of the general population (8) and is ranked 18th of causes of total number of global deaths worldwide (16). Renal fibrosis is characterized by massive inflammatory cell infiltration, increased synthesis and deposition of extracellular matrix proteins (ECM), and increased number of myofibroblasts with the expression of α-smooth muscle actin (SMA) (7). Despite the many therapeutic interventions that have been suggested, the effective therapy to treat renal fibrosis is still lacking.
Accumulating evidence suggests that nitric oxide (NO)/cyclic guanosine monophosphate (cGMP)/cGMP-dependent protein kinase-I (PKG) signaling pathway plays an important role in renal physiology and pathology (20, 27). Our previous studies demonstrated that increasing PKG activity protected mice from acute kidney injury by downregulation of inflammation in an ischemia-reperfusion-induced kidney injury model (13) or by protection of mitochondria function in a cisplatin-induced kidney injury model (17). In addition to protecting against acute kidney injury, we also found that increasing PKG activity inhibited high glucose-induced thrombospondin 1-dependent transforming growth factor (TGF)-β activity and prevented ECM accumulation in kidney mesangial cells, suggesting an anti-fibrotic effect of PKG-I in chronic kidney disease, e.g., diabetic renal complications (29). Importantly, this concept is supported by a recent study showing that treatment with the guanylyl cyclase activator or NO donor (isosorbide dinitrate) suppresses renal fibrosis via PKG-I (23). Taken together, these studies strongly suggest the therapeutic potential of increasing PKG activity in treating renal fibrosis.
PKG activity can be increased by several ways. In the present study, both pharmacological and genetic approaches were utilized to increase PKG activity. A phosphodiesterase 5 (PDE5) inhibitor (sildenafil) was used in this study to increase renal PKG activity for a translational purpose. Sildenafil specifically inhibits cGMP hydrolyzation and is widely used in clinics to treat erectile dysfunction and pulmonary hypertension (1, 18). Our previous studies demonstrated that sildenafil administration increased renal PKG activity (17). In addition to the pharmacological approach, PKG transgenic mice (Tg) with increased renal PKG activity were also utilized. The therapeutic effects of increasing PKG activity on renal interstitial fibrosis were determined in a well-established unilateral ureteral obstruction (UUO) mouse model (2, 11) in this study.
MATERIALS AND METHODS
Experimental animals and protocols.
All experiments involving mice conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Kentucky Institutional Animal Care and Use Committee. Male wild-type (WT) littermates or PKG Tg mice at 8 wk of age were used. All of these mice were on a B6C3H background and were generated by our laboratory previously (13, 17). UUO was achieved by ligating the left lateral ureteral and sham-operated animals were used as a control. Mice were killed after 14 days of UUO. The obstructed kidneys were harvested for histology or other molecular biology analysis. Each group contained six mice. For sildenafil treatment group, 10 male WT littermates at 8 wk age were used. After UUO operation, sildenafil (12 mg·kg−1·day−1, Sigma) in saline was subcutaneously injected into mice twice daily for 14 days. At the same time, for control group, the same amount of saline was administered. Each group contained five mice.
Renal histopathological and immunohistochemical staining.
Kidneys were collected and immersion-fixed in 10% neutral formalin, embedded in paraffin, and sectioned into 4-μm-thick sections onto glass slides. After deparaffinization, kidney tissue sections were rehydrated and stained by Masson's trichrome (Sigma) for renal fibrosis, which was analyzed by calculating relative collagen-positive areas.
For immunohistochemical staining, kidney tissue sections (4-μm-thick) were deparaffinized in xylene and were rehydrated in graded mixtures of ethanol/water. Endogenous peroxidase activity was blocked with 3% H2O2 for 10 min at room temperature. The slides were placed in PBS buffer containing 5% bovine serum albumin for 30 min. Anti-F4/80 (AbD serotec), anti-α-SMA (Abcam), anti-E-cadherin (Cell Signaling), or anti-TGF-β1 (R&D Systems) antibodies were applied for 1 h at room temperature. A negative control was included by substituting control IgG for the primary antibody. After being washed with PBS, biotinylated secondary antibody was applied for 30 min. After another 15-min washing, an avidin-biotin-peroxidase complex was applied to the slides for 30 min. The slides were washed once again with PBS before color development with DAB using Vectastain ABC system (Vector Lab).
Cell experiments.
Bone marrow-derived cells were isolated from femurs and tibias of WT littermates or PKG Tg mice (male, 8–10 wk old) using the method as previously described (14). Bone marrow-derived cells were cultured in RPM1640 medium containing 20% FBS, 2% penicillin/streptomycin, and 25 ng/ml M-CSF for 7 days to allow proliferation and differentiation into mature macrophages. Then, macrophages were plated and treated with 1 μM human angiotension II (Sigma) for 24 h. After treatment, conditioned media were collected and used for measurement of active TGF-β levels using ELISA kit (R&D Systems).
Primary proximal tubular cells were isolated from both WT littermates and PKG Tg mice (male, 8–10 wk old) using the method as previously described (17). Cells were plated in collagen-coated dishes with DMEM/F12 culture medium supplemented with 25 ng/ml EGF, 1 ng/ml PGE1, 0.05 nM triiodothyronine, 0.05 nM hydrocortisone, 1% penicillin/streptomycin, 5% FBS and insulin-transferrin-sodium selenite medium. After overnight culture, the unattached cells were removed and fresh media were added. Then quiescent cells went through the following treatment: 1) cells were treated with human TGF-β1 (at 5 ng/ml, R&D Systems) for 24 h. After treatment, cells were harvested and the protein levels of α-SMA and collagen I in cell lysates were determined by Western blotting. 2) Cells were treated with 1 μM human angiotension II for 24–48 h. The active TGF-β levels in the conditioned media were analyzed by ELISA. The protein levels of α-SMA and collagen I in cell lysates were determined by Western blotting.
Western blotting analysis.
Mouse kidney cortex, proximal tubular cells, or macrophages were homogenized in lysis buffer. After concentration being measured, homogenates were subjected to SDS-PAGE gel under reducing conditions and transferred onto a nitrocellulose membrane. After being blocked with 5% milk, the membranes were incubated with anti-GAPDH (Millipore), anti-α-SMA (Sigma), anti-E-cadherin (Cell Signaling), anti-collagen type I (Abcam), anti-collagen type III (Abcam), anti-TGF-β1 (Santa Cruz Biotechnology), anti-Smad2 (Cell Signaling), anti-pSmad2 (Cell Signaling), anti-angiotension II (generously provided by Dr. Alana Daugherty at the University of Kentucky), anti-phospho-VASP (Ser 239), and anti-VASP (Calbiochem) antibodies at 4°C overnight. After being washed, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson Labs). The reaction was visualized by using an enhanced chemiluminescence system (Pierce). Immunoblots were analyzed by scanning densitometry and quantified by Quantity One gel analysis software (Bio-Rad Laboratories).
Real-time PCR.
Total RNA was isolated from kidney cortex or cells using TRIzol reagent (Invitrogen) and treated with DNaseI (Roche). The treated RNA was cleaned up using an RNeasy kit (Qiagen). Two micrograms of total RNA were used for cDNA synthesis with a high-capacity cDNA reverse transcription kit (Invitrogen). Real-time PCR analyses were performed using a SYBR Green PCR Master Mix kit with a MyiQ real-time PCR Thermal Cycler (Bio-Rad Laboratories). All reactions were performed in triplicate in a final volume of 25 μl. Dissociation curves were run to detect nonspecific amplification, and we confirmed that single products were amplified in each reaction. The quantities of each test gene and internal control 18S RNA were then determined from the standard curve using MyiQ system software, and mRNA expression levels of test genes were normalized to 18S RNA levels as described previously (3). The genes that were tested include collagen type I, collagen type III, α-SMA, F4/80, TGF-β1, TNF-α, ICAM1, and E-cadherin. The sequence of these primers was listed in Table 1.
Table 1.
Primer sequences used for the study
| Genes | |
|---|---|
| Type I collagen | |
| Forward | 5′-TTCTCCTGGCAAAGACGGACTCAA-3′ |
| Reverse | 5′-AGGAAGCTGAAGTCATAACCGCCA-3′ |
| Type III collagen | |
| Forward | 5′-TCCTAACCAAGGCTGCAAGATGGA-3′ |
| Reverse | 5′-TCCTAACCAAGGCTGCAAGATGGA-3′ |
| α-SMA | |
| Forward | 5′-ATTGTGCTGGACTCTGGAGATGGT-3′ |
| Reverse | 5′-TGATGTCACGGACAATCTCACGCT-3′ |
| F-4/80 | |
| Forward | 5′-CTTTGGCTATGGGCTTCCAGTC-3′ |
| Reverse | 5′-GCAAGGAGGACAGAGTTTATCGTG-3′ |
| TGF-β1 | |
| Forward | 5′-ACTGCTTCCCGAATGTCTGACGTA-3′ |
| Reverse | 5′-TAAAGAGGTCACCCGCGTGCTAAT-3′ |
| TNF-α | |
| Forward | 5′-AGC CGA TGG GTT GTA CCT-3′ |
| Reverse | 5′-TGA GTT GGT CCC CCT TCT-3′ |
| ICAM1 | |
| Forward | 5′-ACACTATGTGGACTG GCAGTGGTT-3′ |
| Reverse | 5′-TGAGGCTCG ATTGTTCAGCTGCTA-3′ |
| E-cadherin | |
| Forward | 5′-CTGCTGCTCCTACTGTTTCTAC-3′ |
| Reverse | 5′-TCTTGGGAACACACACACTATC-3′ |
α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; ICAM1, intracellular adhesion molecule-1.
Statistical analysis.
Data are means ± SE. Differences between groups were determined by one-way ANOVA followed by Tukey's post hoc tests or Student's t-test as appropriate. The significance level was P <0.05.
RESULTS
Increasing PKG activity attenuates UUO-induced tubulointerstitial fibrosis.
PKG Tg mice and sildenafil administration (at the dose of 12 mg·kg−1·day−1, twice daily) were used to increase renal PKG activity genetically and pharmacologically, respectively. PKG activity in the kidney was analyzed by phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at serine 239. VASP is a ubiquitously expressed endogenous substrate for PKG, and phosphorylation of VASP at serine 239 has been used as a biomarker for PKG activation (25). As shown in Fig. 1, A–B, kidney PKG activity was increased in PKG Tg mice or in sildenafil-treated mice. UUO was achieved by ligating the left lateral ureteral of mice. Sham-operated animals were used as controls. After 14 days of UUO, the renal interstitial fibrosis was examined. UUO mice had decreased renal PKG activity (Fig. 1C). Masson staining results showed that increased collagen disposition was observed in the renal interstitial area from WT UUO mice, which was significantly reduced in either PKG Tg mice or sildenafil-treated UUO mice (Fig. 2, A–D). In addition, collagen type I (Fig. 1, E–H) and collagen type III (Fig. 2, I–L) mRNA as well as protein levels were significantly increased in kidney cortex from UUO mice, which were reduced in PKG Tg mice or in sildenafil-treated UUO mice (Fig. 2, I–L).
Fig. 1.
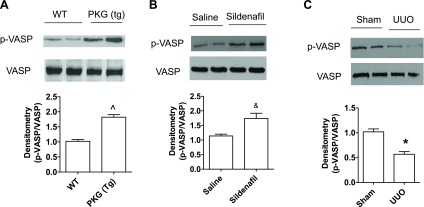
Changes of cGMP-dependent protein kinase I (PKG) activity in the kidneys from unilateral ureteral obstruction (UUO) mouse model. Representative immunoblotting of p-vasodilator-stimulated phosphoprotein (VASP) and total VASP in kidney cortex from wild-type (WT) or PKG transgenic (Tg) mice (A), from saline- or sildenafil-treated mice (B), or from WT mice after 14 days of UUO or sham surgery (C). Data are presented as means ± SE (n = 4 mice/group). *P < 0.05 vs. WT/sham group. ^P < 0.05 vs. WT mice. &P < 0.05 from saline-treated mice.
Fig. 2.

Increasing PKG activity attenuates renal fibrosis in a UUO mouse model. Representative light photomicrograph of the Masson staining of the kidney sections from 4 groups of WT and PKG-I Tg mice after 14 days of UUO or sham surgery (A) or from 2 groups of saline- and sildenafil-treated UUO mice (C). The positive collagen staining was shown as blue color. The scale bar represents 100 μm. Semiquantitative analysis was performed by calculating the positive area (B and D). Collagen type I (E–H) and collagen type III (I–L) mRNA and protein levels in kidney cortex from UUO or sham mice were determined by real-time PCR and Western blotting, respectively. Data are presented as means ± SE (n = 5–6 mice/group). *P < 0.05 vs. WT/sham group or saline group. #P < 0.05 vs. WT/UUO group. &P < 0.05 vs. Tg/sham group.
Effect of increasing PKG activity on α-SMA and E-cadherin expression in UUO model.
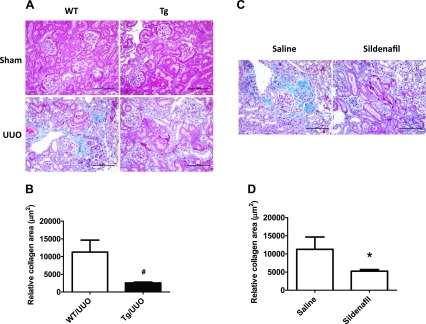
We determined whether increasing PKG activity affects the renal levels of α-SMA and E-cadherin. As shown in Fig. 3, α-SMA (Fig. 3, A–D) and E-cadherin (Fig. 3, E–H) mRNA as well as protein levels were significantly increased in kidney cortex from WT UUO mice. However, these changes were inhibited in either PKG Tg UUO mice or in sildenafil-treated UUO mice. The immunohistochemical staining of the kidney section showed similar findings (Fig. 3, I–J).
Fig. 3.

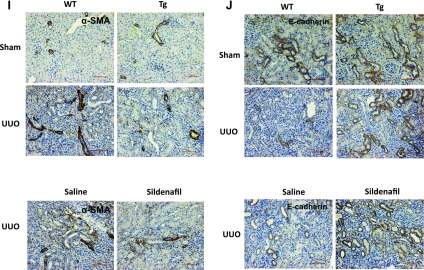
Increasing PKG activity affects kidney α-smooth muscle actin (SMA) and E-cadherin levels in a UUO mouse model. α-SMA (A–D) and E-cadherin (E–H) mRNA and protein levels in the kidney cortex from UUO or sham mice were determined by real-time PCR and Western blotting, respectively. Data are presented as means ± SE (n = 5–6 mice/group). *P < 0.05 vs. WT/sham group or saline group. #P < 0.05 vs. WT/UUO group. &P < 0.05 vs. Tg/sham group. I–J: kidney sections were stained with anti-α-SMA or E-cadherin antibody. The positive staining is shown as brown. Representative light micrographs are shown.
Increasing PKG activity inhibits TGF-β/Smad pathway in a UUO mouse model.
It is well-known that TGF-β/Smad pathway plays an important role in the development of renal fibrosis. Therefore, we determined how this pathway changed in our mouse model. We found that kidney TGF-β1 levels (mRNA and protein and immunohistochemical staining; Fig. 4, A–E) and pSmad2 protein levels (Fig. 4, F, G) were markedly increased in kidney cortex from WT UUO mice, which were reduced in either PKG Tg UUO mice or in sildenafil-treated UUO mice.
Fig. 4.

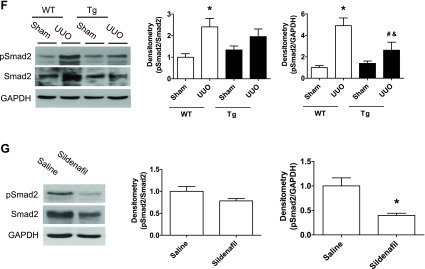
Increasing PKG activity reduced transforming growth factor (TGF)-β/Smad pathway in a UUO mouse model. A–D: kidney TGF-β1 mRNA and protein levels from UUO or sham group of mice were determined by real-time PCR and Western blotting, respectively. E: kidney sections from mice were stained with anti-TGF-β1 antibody. The positive staining is shown as brown. Representative light micrographs are shown. F and G: pSmad2 protein levels in kidney cortex from UUO or sham group of mice were determined by Western blotting. Data are presented as means ± SE (n = 5–6 mice/group). *P < 0.05 vs. WT/sham group or saline group. #P < 0.05 vs. WT/UUO group. &P < 0.05 vs. Tg/sham group.
Increasing PKG activity reduces UUO-induced renal macrophage infiltration and proinflammatory cytokine levels.
Our previous studies demonstrated that increasing PKG activity reduced ischemia-reperfusion-induced macrophage infiltration into kidney (13). Similarly, in this obstructive renal fibrosis mouse model, we found that UUO-induced kidney macrophage infiltration was significantly reduced in PKG Tg mice or in sildenafil-treated mice (Fig. 5). In addition, kidney proinflammtory cytokine levels, e.g., TNF-α (Fig. 6, A, B) and ICAM-1 (Fig. 6, C, D) were increased in WT UUO mice, which were reduced by increasing PKG activity.
Fig. 5.
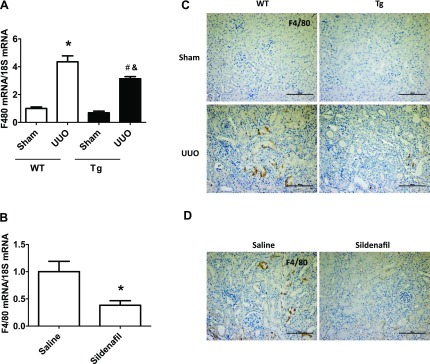
Increasing PKG activity reduced renal macrophage infiltration in a UUO mouse model. A and B: F4/80 mRNA levels in kidney cortex from UUO or sham groups of mice were determined by real-time PCR. Data are presented as means ± SE (n = 5–6 mice/group). *P < 0.05 vs. WT/sham group or saline group. #P < 0.05 vs. WT/UUO group. &P < 0.05 vs. Tg/sham group. C and D: kidney sections from mice were stained with anti-F4/80 antibody. The positive staining is shown as brown. Representative light micrographs are shown. Scale bars = 100 μm.
Fig. 6.

Increasing PKG activity reduced kidney inflammation in a UUO mouse model. TNF-α (A, B) and ICAM-1 (C, D) mRNA levels in the kidney cortex from different groups of mice were determined by real-time PCR. Data are presented as means ± SE (n = 5–6 mice/group). *P < 0.05 vs. WT/sham group or saline group. #P < 0.05 vs. WT/UUO group. &P < 0.05 vs. Tg/sham group.
Increasing PKG activity reduced TGF-β1 production in the conditioned media from angiotensin II-treated macrophages or proximal tubular cells.
We further determined the possible cellular sources of UUO-induced TGF-β1 levels in the kidney by in vitro studies. As an upstream mediator to promote the process of fibrosis in UUO kidney (2), angiotension II protein levels were increased in UUO mouse kidney cortex in our present study. Moreover, no significant difference was found between WT UUO mice and Tg UUO mice (data not shown). Therefore, angiotension II was used as a stimulator of TGF-β1 production in vitro to mimic UUO model in vivo. Both bone marrow-derived macrophages and proximal tubular cells were isolated from WT and PKG Tg mice. Macrophages or proximal tubular cells isolated from PKG Tg mice had increased PKG transgene expression and PKG activity (Fig. 7A) (17). Angiotensin II-stimulated TGF-β1 production in macrophages as well as in proximal tubular cells was determined. We found that angiotensin II stimulated TGF-β1 production in both macrophages and proximal tubular cells from WT mice, but not the cells from the PKG Tg mice (Fig. 7, A, B). In addition, TGF-β1 or angiotensin II treatment stimulated α-SMA or collagen I production in proximal tubular cells from WT mice, but not the cells from the PKG Tg mice (Fig. 8). Taken together, all of these data suggest that both macrophages and proximal tubular cells are two important cellular sources of TGF-β production in the kidney from a UUO mouse model. The interaction between macrophage and proximal tubular cell contributes to TGF-β-induced renal fibrosis.
Fig. 7.
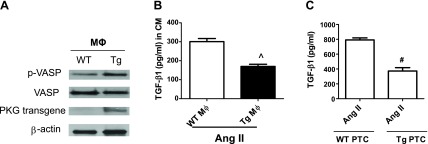
Increasing PKG activity reduced TGF-β1 production in the conditioned media (CM) from angiotensin II (ANG II)-treated macrophages or proximal tubular cells (PTC). A: macrophages were isolated from WT or PKG (Tg) mice. The expressions of PKG transgene and PKG activity (p-VASP) were determined by Western blotting. B–C: TGF-β1 protein levels in the CM from human ANG II (1 μM)-treated macrophages (MΦ) or PTC isolated from WT or PKG-I Tg mice were measured by ELISA. Data are presented as means ± SE (n = 3 individual experiments). ^P < 0.05 vs. WT MΦ. #P < 0.05 vs. WT/ANG II.
Fig. 8.

Increasing PKG activity reduced TGF-β1 or ANG II-induced α-SMA and collagen I production in PTC. PTC were isolated from WT or PKG (Tg) mice and treated by TGF-β1 (A–B; at 5 ng/ml) or human ANG II (C–D; 1 μM) for 24 h. α-SMA and collagen I protein levels were determined by Western blotting. Data are presented as means ± SE (n = 3 individual experiments). *P < 0.05 vs. WT/control. #P < 0.05 vs. WT/TGF-β1. ^P < 0.05 vs. WT/ANG II.
DISCUSSION
In the present study, we demonstrated that increasing PKG-I activity by both genetic (PKG-I Tg mice) and pharmacological (sildenafil treatment) approaches attenuated UUO-induced tubulointerstitial fibrosis, which was accompanied by decreased renal TGF-β/Smad signaling. In addition, UUO-induced renal macrophage infiltration was reduced by the increased PKG activity. In vitro data further suggested that PKG regulated both macrophage and proximal tubular cell function, leading to decreased TGF-β1 signaling and attenuated renal fibrosis. Taken together, these studies suggest that increasing PKG activity may provide a new therapeutic strategy for renal fibrosis.
PKG is a serine/threonine kinase. Two types of PKG (PKG-I and PKG-II) have been identified in mammalian cells. In the previous studies, we demonstrated that PKG-I has a protective effect on ischemia-reperfusion acute kidney injury partially through inhibiting inflammatory cell infiltration into the kidney, reducing kidney inflammation, and inhibiting tubular cell apoptosis (13). PKG-I also protected against cisplatin-induced acute kidney injury by preserving mitochondria function (17) and prevented the development of diabetic renal complications by inhibiting high-glucose-induced thrombospondin 1-dependent TGF-β activity (29). In the present study, we revealed another important protective role of PKG-I in renal fibrosis induced by UUO, which is in agreement with a recent study from Schinner et al. (23). By using both genetic and pharmacological approaches, the anti-fibrotic effect of PKG-I was further confirmed in this study. Moreover, increasing PKG activity by a PDE5 inhibitor sildenafil that was utilized in this study may have an important clinical relevance, since sildenafil is widely used clinically to treat erectile dysfunction or pulmonary hypertension. Therefore, the current study has translational importance.
TGF-β/Smad pathway has been proved to play essential roles in renal fibrosis (15, 26). The TGF-β family is comprised of three isoforms (TGF-β1, TGF-β2, and TGF-β3). Among these isoforms, TGF-β1 is the most common inducer of renal fibrosis (10). By binding to specific receptors on the cell membrane surface and subsequently activating Smads, TGF-β regulates many fundamental cellular activities, including cell growth, differentiation, proliferation, as well as ECM remodeling (24). Mitogen-activated protein kinases and integrin-linked kinase were TGF-β downstream effectors that modulate the expression of profibrotic genes (12, 28). In addition, a series of microRNAs can be regulated by TGF-β and contributes to renal fibrosis (9, 31). Together, all of these studies suggest the important role of TGF-β/Smad signaling in renal fibrosis. In this study, we demonstrated that the protein levels of TGF-β1 and pSmad2 were increased in WT UUO mice, which were reduced in either Tg mice with UUO or in sildenafil-treated UUO mice. These data suggest that the antifibrotic effect mediated by increased PKG activity might be through downregulation of TGF-β/Smad signaling.
How does PKG-I regulate TGF-β/Smad signaling during renal fibrosis? Before answering this question, it is important to know the cellular sources of UUO-induced renal TGF-β1 levels. Many cell types including fibroblasts, myofibroblasts, endothelial cells, tubular epithelial cells, and macrophages involve renal interstitial fibrosis (6), and some of these cell types have been suggested to be important cellular sources of TGF-β1 (4, 5, 30). In this study, we found that increasing PKG activity significantly reduced macrophage infiltration into UUO kidney, which was associated with reduced kidney inflammation. These data are consistent with our previous report (13), suggesting that increasing PKG activity reduces macrophage migration and contributes to the decreased macrophage recruitment in the injured kidney, which is possibly through a Rho GTPase-mediated mechanism (19, 22). Another novel finding of the present study is that increasing PKG activity not only reduced the number of infiltrating macrophages but also downregulated the macrophage function, e.g., TGF-β1 secretion in macrophages. TGF-β production was significantly less in macrophage isolated from Tg mice compared with those from WT mice. In addition to the macrophage-derived TGF-β1, we also demonstrated that proximal tubular cells are another important source of kidney TGF-β levels in the UUO model. The interaction between these two cell types contributes to TGF-β1-induced renal fibrosis.
In this study, the exact mechanism by which PKG downregulates TGF-β levels remains to be determined. It has been reported that PKG interfered with TGF-β/Smad pathway through directing the proteasomal degradation of activated Smad in endothelial cells (21). In addition, our previous studies demonstrated that PKG inhibited high-glucose-induced active TGF-β levels in rat mesangial cells by downregulating the transcription of thrombospondin 1, a major physiological regulator of TGF-β activation (29). The additional molecular mechanisms involving PKG-mediated downregulation of TGF-β signaling will warrant further investigation.
In summary, the current study provides the first evidence that increasing PKG activity attenuates renal fibrosis in a UUO mouse model, partially through downregulation of TGF-β/Smad signaling. Importantly, pharmacologically increasing PKG activity by a PDE5 inhibitor sildenafil has clinical significance.
GRANTS
This study was supported in part by the Department of Veterans Affairs Merit Review Award (to S. Wang) and the National Institutes of Health Grant R01 DK081555 (to S. Wang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: W.C., H.M., X.Q., H.N., Q.Z., and X.W. performed experiments; W.C. analyzed data; W.C. and S.W. interpreted results of experiments; W.C. prepared figures; W.C. drafted manuscript; W.C., H.M., X.Q., H.N., Q.Z., X.W., J.F., and S.W. approved final version of manuscript; J.F. and S.W. edited and revised manuscript; S.W. conception and design of research.
REFERENCES
- 1.Buvat J, Buttner H, Hatzimouratidis K, Vendeira PA, Moncada I, Boehmer M, Henneges C, Boess FG. Adherence to initial PDE-5 inhibitor treatment: randomized open-label study comparing tadalafil once a day, tadalafil on demand, and sildenafil on demand in patients with erectile dysfunction. J Sex Med 10: 1592–1602, 2013 [DOI] [PubMed] [Google Scholar]
- 2.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Cui W, Maimaitiyiming H, Qi X, Norman H, Wang S. Thrombospondin 1 mediates renal dysfunction in a mouse model of high fat diet-induced obesity. Am J Physiol Renal Physiol 305: F871–F880, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diamond JR, Ricardo SD, Klahr S. Mechanisms of interstitial fibrosis in obstructive nephropathy. Semin Nephrol 18: 594–602, 1998 [PubMed] [Google Scholar]
- 5.Ding G, Pesek-Diamond I, Diamond JR. Cholesterol, macrophages, and gene expression of TGF-β1 and fibronectin during nephrosis. Am J Physiol Renal Fluid Electrolyte Physiol 264: F577–F584, 1993 [DOI] [PubMed] [Google Scholar]
- 6.Farris AB, Colvin RB. Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens 21: 289–300, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hewitson TD. Fibrosis in the kidney: is a problem shared a problem halved? Fibrogenesis Tissue Repair 5, Suppl 1: S14, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW. Chronic kidney disease: global dimension and perspectives. Lancet 382: 260–272, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Jiang L, Qiu W, Zhou Y, Wen P, Fang L, Cao H, Zen K, He W, Zhang C, Dai C, Yang J. A microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-beta1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int 84: 285–296, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein J, Kavvadas P, Prakoura N, Karagianni F, Schanstra JP, Bascands JL, Charonis A. Renal fibrosis: insight from proteomics in animal models and human disease. Proteomics 11: 805–815, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol 20: 1907–1918, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Tong X, Maimaitiyiming H, Clemons K, Cao JM, Wang S. Overexpression of cGMP-dependent protein kinase I (PKG-I) attenuates ischemia-reperfusion-induced kidney injury. Am J Physiol Renal Physiol 302: F561–F570, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLos One 6: e26656, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M, Liu YZ, Feng Y, Xu YF, Che JP, Wang GC, Zheng JH. Novel evidence demonstrates that epithelial-mesenchymal transition contributes to nephrolithiasis-induced renal fibrosis. J Surg Res 182: 146–152, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2095–2128, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maimaitiyiming H, Li Y, Cui W, Tong X, Norman H, Qi X, Wang S. Increasing cGMP-dependent protein kinase I activity attenuates cisplatin-induced kidney injury through protection of mitochondria function. Am J Physiol Renal Physiol 305: F881–F890, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, Trial R. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309: 1268–1277, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridley AJ. Regulation of macrophage adhesion and migration by Rho GTP-binding proteins. J Microsc 231: 518–523, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Sanches TR, Volpini RA, Massola Shimizu MH, Braganca AC, Oshiro-Monreal F, Seguro AC, Andrade L. Sildenafil reduces polyuria in rats with lithium-induced NDI. Am J Physiol Renal Physiol 302: F216–F225, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Saura M, Zaragoza C, Herranz B, Griera M, Diez-Marques L, Rodriguez-Puyol D, Rodriguez-Puyol M. Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ Res 97: 1115–1123, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Sawada N, Itoh H, Miyashita K, Tsujimoto H, Sone M, Yamahara K, Arany ZP, Hofmann F, Nakao K. Cyclic GMP kinase and RhoA Ser188 phosphorylation integrate pro- and antifibrotic signals in blood vessels. Mol Cell Biol 29: 6018–6032, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schinner E, Schramm A, Kees F, Hofmann F, Schlossmann J. The cyclic GMP-dependent protein kinase Ialpha suppresses kidney fibrosis. Kidney Int 84: 1198–1206, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Schnaper HW, Hayashida T, Poncelet AC. It's a Smad world: regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol 13: 1126–1128, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM, Walter U. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn Schmiedebergs Arch Pharmacol 358: 134–139, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLos One 7: e34026, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tapia E, Sanchez-Lozada LG, Soto V, Manrique AM, Ortiz-Vega KM, Santamaria J, Medina-Campos ON, Cristobal M, Avila-Casado C, Pedraza-Chaverri J, Rodriguez-Iturbe B, Franco M. Sildenafil treatment prevents glomerular hypertension and hyperfiltration in rats with renal ablation. Kidney Blood Press Res 35: 273–280, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Wang D, Warner GM, Yin P, Knudsen BE, Cheng J, Butters KA, Lien KR, Gray CE, Garovic VD, Lerman LO, Textor SC, Nath KA, Simari RD, Grande JP. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am J Physiol Renal Physiol 304: F938–F947, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Wu X, Lincoln TM, Murphy-Ullrich JE. Expression of constitutively active cGMP-dependent protein kinase prevents glucose stimulation of thrombospondin 1 expression and TGF-β activity. Diabetes 52: 2144–2150, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int 70: 1914–1919, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol 22: 1668–1681, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]