

Abstract
The vacuolar H+-ATPase (V-ATPase) mediates ATP-driven H+ transport across membranes. This pump is present at the apical membrane of kidney proximal tubule cells and intercalated cells. Defects in the V-ATPase and in proximal tubule function can cause renal tubular acidosis. We examined the role of protein kinase A (PKA) and AMP-activated protein kinase (AMPK) in the regulation of the V-ATPase in the proximal tubule as these two kinases coregulate the V-ATPase in the collecting duct. As the proximal tubule V-ATPases have different subunit compositions from other nephron segments, we postulated that V-ATPase regulation in the proximal tubule could differ from other kidney tubule segments. Immunofluorescence labeling of rat ex vivo kidney slices revealed that the V-ATPase was present in the proximal tubule both at the apical pole, colocalizing with the brush-border marker wheat germ agglutinin, and in the cytosol when slices were incubated in buffer alone. When slices were incubated with a cAMP analog and a phosphodiesterase inhibitor, the V-ATPase accumulated at the apical pole of S3 segment cells. These PKA activators also increased V-ATPase apical membrane expression as well as the rate of V-ATPase-dependent extracellular acidification in S3 cell monolayers relative to untreated cells. However, the AMPK activator AICAR decreased PKA-induced V-ATPase apical accumulation in proximal tubules of kidney slices and decreased V-ATPase activity in S3 cell monolayers. Our results suggest that in proximal tubule the V-ATPase subcellular localization and activity are acutely coregulated via PKA downstream of hormonal signals and via AMPK downstream of metabolic stress.
Keywords: AMPK, PKA, acid-base homeostasis, metabolic stress
the kidney plays a vital role in acid-base homeostasis, by H+ secretion and/or reabsorption of filtered HCO3−, in a variety of nephron segments, including the proximal tubule (26, 34). The proximal tubule epithelium reabsorbs ∼80% of the filtered HCO3− (21). Proximal tubular mechanisms to reabsorb luminal HCO3− require H+ secretion into the lumen. Both apical Na+/H+ exchangers (NHEs) and the vacuolar H+-ATPase (V-ATPase) contribute to these processes (13, 21, 51, 54, 67). Apical membrane NHE3 contributes approximately two-thirds of the total H+ secretion, while approximately one-third is mediated by the V-ATPase (21). The V-ATPase is a protein complex that transports H+ across membranes by hydrolyzing ATP, and in the proximal tubule it has been identified by microscopy in endocytic vesicles, at the base of the brush border and also sometimes in microvilli (9). H+ transport by the V-ATPase is also important in apical endocytosis (19, 42, 63). Defects in V-ATPase and in proximal tubule function can cause proximal renal tubular acidosis (RTA) (29).
Changes in V-ATPase subcellular localization are associated with the regulation of H+ secretion (8, 39, 56). However, the potential role of kinases in V-ATPase regulation in the proximal tubule has not yet been elucidated. This pump exhibits similar regulation in kidney A-type intercalated cells and epididymal proton-secreting clear cells, which share a common developmental origin (29a). For example, previous work in our laboratory has shown that carbonic anhydrase (CA), luminal [HCO3−], HCO3−-activated soluble adenylyl cyclase (sAC), and cAMP/PKA are part of a signaling cascade that is likely downstream of acid-base changes regulating the activity and apical accumulation of the V-ATPase in kidney collecting duct and epididymis (2, 22, 46). Furthermore, other studies in our laboratory have shown that PKA directly phosphorylates the V-ATPase A subunit at Ser-175, a residue necessary for apical V-ATPase accumulation and activity in kidney cells (2). Other kinases are also known to be involved in V-ATPase regulation (11, 14). For example, glucose and PI-3-kinase in proximal tubule-derived cells regulate V-ATPase assembly and cell surface abundance (38). In addition, the V0 sector “a” subunit (ATP6V0A) senses endosomal acidification by recruiting ARNO in the proximal tubule (30). Our group also showed that the metabolic sensor AMP-activated protein kinase (AMPK) inhibits V-ATPase apical accumulation in kidney intercalated cells and in epididymal clear cells (22, 23) via phosphorylation at Ser-384 in the V-ATPase A subunit (1).
AMPK is a Ser/Thr protein kinase that regulates cellular processes, including membrane transport (27, 28, 47). During metabolic stress (e.g., hypoxia or ischemia), cellular ATP levels may drop slightly with a more substantial rise in [AMP] and thus the [AMP]:[ATP] ratio, leading to activation of AMPK (27) and the subsequent decrease of cellular ATP consumption and stimulation of ATP generation, thereby maintaining cellular ATP levels in the face of energy depletion. Furthermore, AMPK prevents PKA-mediated V-ATPase apical accumulation in A-type intercalated cells (22), an antagonism that also exists in the regulation of other membrane transport proteins (24). Although AMPK is abundantly expressed in kidney (20), an understanding of its role in kidney physiology and disease is less developed than in other organs (24, 47). We have recently identified a key AMPK phosphorylation site in the V-ATPase A subunit that when mutated abrogates the inhibitory effects of AMPK on the V-ATPase in a cell line of intercalated cell origin (1). Of clinical significance, AMPK activation in the proximal tubule during ischemia protects the epithelial cells against apoptosis and loss of polarity (37, 58). However, patients develop metabolic acidosis after AKI, and therefore AMPK-mediated acute V-ATPase downregulation in this segment may contribute to bicarbonate loss in the urine (55).
In addition, PKA is involved in many regulatory cascades in the proximal tubule that affect epithelial transport. This kinase has been studied downstream of parathyroid hormone (PTH) and dopamine (31, 45, 49, 66), yet dopamine does not affect flow-mediated V-ATPase proton secretion in the proximal tubule (17). Another important regulator of proton secretion in the proximal tubule is angiotensin II (ANG II), as this hormone stimulates NHE3 (40). ANG II can induce decreases in cAMP levels in the proximal tubule (10, 52), yet it can increase V-ATPase activity via PKC (12). While long-term regulation of the V-ATPase by ANG II in proximal tubule cells appears to be dependent on mechanisms involving tyrosine kinases and PI-3-kinase (11), others have found that ANG II stimulates V-ATPase-dependent proton extrusion in the proximal tubule by insertion of vesicles into the brush-border membrane (64). Therefore, the role of PKA in proximal tubule regulation of the V-ATPase downstream from hormones and in the absence of ANG II requires further investigation.
In this study, we examined whether the subcellular localization and activity of the V-ATPase are regulated by PKA and AMPK in the S3 segment of the kidney proximal tubule. We posed this question because the S3 segment of the proximal tubule or pars recta is more suitable for measurements of V-ATPase subcellular localization in kidney slices. Moreover, the V-ATPase isoform composition is different from that in distal tubule intercalated cells (7, 63). Despite the differences in isoform composition in the proximal tubule, we found that regulation of the V-ATPase in this segment is analogous to the regulation observed in A-type intercalated cells.
MATERIALS AND METHODS
Reagents and chemicals.
All chemical compounds used in the studies presented here were purchased from Sigma-Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Pittsburgh, PA) unless otherwise stated. 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) was purchased from Toronto Research Chemicals (Toronto, Canada). The cell-permeant cAMP analog and PKA-specific activator N6-monobutyryl-cAMP (6-MB-cAMP) was obtained from BIOLOG Life Science Institute (Bremen, Germany). The phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) was obtained from Acros Organics (Fair Lawn, NJ). The specific PKA inhibitor mPKI was purchased from Biomol (Plymouth Meeting, PA).
Antibodies.
The phospho-Thr172-AMPK-α (pThr172-AMPKα catalog no. 3531), and anti-phospho-(Ser/Thr) PKA (anti-phospho-PKA) substrate antibodies were obtained from Cell Signaling Technology (Boston, MA). Chicken anti-V-ATPase E subunit and mouse anti-β-actin antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Rabbit anti-zonula occludens-1 (ZO-1) antibody, and secondary goat anti-chicken antibody conjugated with Alexa 488 were obtained from Life Technologies (Grand Island, NY). Secondary goat anti-rabbit antibody conjugated with Cy3 was obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Rabbit anti-V-ATPase A subunit antibody was obtained from GenScript USA (Piscataway, NJ). Secondary horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG antibodies were obtained from GE Healthcare Biosciences (Pittsburgh, PA). Rabbit anti-V-ATPase a4 subunit antibody was obtained from Abcam (Cambridge, MA).
Kidney tissue preparation and confocal immunofluorescence microscopy.
All animal experiments were approved by the Institutional Animal Care and Utilization Committee (IACUC) of the University of Pittsburgh, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Kidney slices (0.5 mm) were prepared from Sprague-Dawley rat kidneys removed after anesthesia with pentobarbital sodium (65 mg/kg ip). The anesthetized animals were perfused via the left ventricle with a buffer containing sodium (Ringer buffer), at pH 7.4 containing (in mM) 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, and 10 d-glucose, in 5% CO2 and air, at 37°C as described previously (2, 4–6, 46, 62). The kidneys were quickly sectioned into 1- to 2-mm-thick sections and submerged into additional Ringer buffer. The thinner slices were then prepared using a Stadie-Riggs microtome. The slices were then rapidly transferred to fresh vials containing Ringer buffer with or without various pharmacological agents, including a PKA-activating cell-permeant drug cocktail 6-MB-cAMP (1 mM) and IBMX (0.5 mM), the specific PKA inhibitor mPKI (10 μM), and/or AICAR. The timing of the kidney slice incubations was based on our previous experience with V-ATPase trafficking in intercalated cells in kidney slices (22). For example, the 30-min incubation time point was used when testing activators of V-ATPase apical accumulation in intercalated cells (such as cAMP analogs) because in tissues incubated without any agonist (Ringer buffer alone) the V-ATPase at 30 min remains cytosolic. On the other hand, at the 75-min time point in Ringer buffer alone, the V-ATPase accumulated at the apical membrane in intercalated cells (22). Therefore, this time point was used when testing potential inhibitors of V-ATPase apical accumulation. Treated slices were paired with control slices incubated in Ringer buffer alone for the same treatment time periods. After the treatments, kidney slices were fixed in 4% paraformaldehyde in PBS for 4 h at room temperature or 16 h at 4°C, as previously described (4, 5, 22). After cryoprotecting the slices by immersion in 30% sucrose in PBS-0.02% azide, we obtained 4-μm-thick cryosections. Cryosections were immunolabeled with an antibody against V-ATPase E subunit raised in chicken (Sigma-Aldrich) at 1:2,000 dilution in Dako low background diluent (Dako North America, Carpinteria, CA), followed by a secondary antibody raised in goat (Life Technologies, Grand Island, NY) coupled to Alexa 488. Slices were colabeled with CY3-labeled wheat germ agglutinin (WGA) lectin (Vector Laboratories, Burlingame, CA) to specifically mark the proximal tubule S3 segment (41). Immunolabeled tissues were mounted in VECTASHIELD mounting medium (Vector Laboratories) and imaged in a confocal laser scanning microscope (Leica TCS SP5, model upright DM 6000S, Leica Microsystems, Buffalo Grove, IL) using a 63× objective with identical laser settings for all samples.
Quantification of V-ATPase E subunit apical membrane accumulation in kidney slices.
V-ATPase E subunit accumulation was quantified at the apical membrane in proximal tubule cells using confocal microscopy images and Metamorph software (Molecular Devices, Sunnyvale, CA), adapting methods that our group has previously described for intercalated cells (22). Only cells in S3 segment tubules colabeled with WGA-coupled to CY3 and with open lumens were selected for analysis. At least three nuclei had to be visualized in an epithelial cell ribbon, and the cells used for analysis were not at the edges of such “ribbon.” The mean pixel intensity (MPI) of V-ATPase-associated fluorescence was measured for a region of interest (ROI) within each selected cell at the apical border of the cell where it colocalized with WGA-associated fluorescence. We then measured the V-ATPase-associated fluorescence in an ROI of the same shape and size in the cytoplasmic area next to the nucleus of the same cell, not colocalizing with WGA labeling, using similar methods validated in previous studies by the authors and others (4, 5, 22). The ROI measurements were performed in a blinded manner to ensure the accuracy of the evaluation and validated by at least two independent users. At least 10 cells per treatment condition were evaluated each from at least three separate rat kidney slice experiments. For each treatment, the apical-to-cytoplasmic ratio from MPI of V-ATPase E subunit-associated fluorescence was used to measure V-ATPase apical accumulation. This value was calculated for each cell and then a mean was obtained for each kidney. The V-ATPase apical membrane accumulation for each condition was expressed as the mean ± standard error of the mean (SE).
Cell culture.
We used a cell line of mouse S3 segment proximal tubule origin (a gift from Dr. Robert Bacallao, Indiana University). These “S3 cells” were originally derived from individual dissection of mouse S3 segments (33, 36). Our group has used these previously to study the regulation of proximal tubule S3 segment creatine transporter (36). Cells were used at passage 82–83 and cultured under conditions similar to those described previously (36). Briefly, stock cultures were maintained in a humidified 5% CO2-95% air incubator in a medium similar to that used to culture the mpkCCDc14 cell line (3, 36). This “CCD media” is composed of equal volumes of DMEM and Ham's F-12 plus 60 nM sodium selenate, 5 mg/ml transferrin, 2 mM glutamine, 50 nM dexamethasone, 1 nM triiodothyronine, 10 ng/ml epidermal growth factor, 5 mg/ml insulin, 20 mM d-glucose, 2% (vol/vol) FBS, and 20 mM HEPES, pH 7.4 (reagents from Life Technologies and Sigma-Aldrich). Cells were grown to ∼90% confluency in 75-cm2 plastic culture flasks and then seeded onto Transwell filters (Corning Life Sciences, Tewksbury, MA) prior to use in experiments.
Immunofluorescence labeling and confocal microscopy of polarized S3 cells.
Cells were seeded onto permeable polyester Transwell inserts (0.4-μm pore size; 0.33-cm2 surface area) at a density of 0.3 × 106/well. Cells were maintained in culture on these permeable supports for 4–5 days to form a polarized monolayer. In the set of experiments testing the effects of PKA activators, the medium was then replaced with 1 × PBS containing Ca2+ and Mg2+ (Thermo Fisher Scientific; with pH adjusted to = 7.2) in the absence or presence a PKA activating drug cocktail (6-MB-cAMP/IBMX; 1 mM/0.5 mM) for 30 min, using the same conditions that we previously published in cultured intercalated cells (2). As opposed to kidney slices, the S3 cell monolayers required a 4-h incubation time to achieve activation of AMPK by AICAR. After treating cells with either vehicle or pharmacological agents (see results and/or figure legends for specific conditions), the polarized monolayers were fixed in 2% paraformaldehyde in PBS buffer for 30 min and permeabilized by the addition of a buffer containing 1 × PBS, 1% BSA, and 0.1% Triton X-100 for 10 min at 37°C as described previously (36). After an additional wash with PBS, the filters were immunolabeled with a primary antibody raised in chicken against the V-ATPase anti-E subunit antibody (1:2,000 dilution) in Dako antibody diluent with background reducing components along with rabbit anti-ZO-1 (tight junction protein antibody; 1:100 dilution, Life Technologies), for 75 min and then with secondary goat anti-chicken antibody conjugated with Alexa 488 (1:800 dilution, Life Technologies) and secondary goat anti-rabbit antibody conjugated with Cy3 (1:800 dilution). Filters were mounted in ProLong Gold antifade reagent (Invitrogen). The filters were imaged in a Leica confocal microscope using a 100X 1.4-numerical aperture oil objective. Photomultipliers were set to 600–900 V (2 × Zoom). Images were collected every 0.20 μm, and frame average was conducted three times. Identical laser, stack acquisition, and X–Z reconstruction settings were used for all samples. The images were saved in a TIF format, and E subunit accumulation at the apical membrane was quantified on X–Z reconstructions using Metamorph software and methods validated in previous studies (2, 4, 5).
V-ATPase apical accumulation was determined by measuring the MPI of V-ATPase-associated fluorescence for a ROI within each selected cell at the apical border of the cell, identified by the tight junction marker ZO-1, and compared with an ROI of the same shape and size in the cytoplasmic area immediately subjacent to the chosen apical ROI, and above the nucleus of the same cell. The ROI measurements were performed in a blinded manner to ensure the accuracy of the evaluation. At least 30 cells per treatment condition were evaluated each from at least three separate experiments. The degree of apical accumulation of the V-ATPase E subunit was determined by calculating the MPI ratio of the apical-to-cytoplasmic ROI of V-ATPase E-subunit-associated fluorescence for each cell, and then a mean was obtained for each independent experiment (±SE).
Tissue lysate preparation from kidney slices and immunoblotting.
These experiments were performed according to published methods (36) and adapted as follows. The snap-frozen kidney slices were briefly thawed on ice and placed in ice-cold lysis buffer containing 150 mM NaCl, 10 mM Tris (pH 7.4), 1% Triton X-100, 0.5% Igepal, 1 mM EDTA, 1 mM EGTA (pH 7.4), 200 mM PMSF, 50 mM NaF, 5 mM Na pyrophosphate, 0.1 mM Na orthovanadate, and one tablet each of complete protease inhibitor cocktail (Roche) and complete phosphatase inhibitor cocktail (Roche) per 10 ml of buffer solution. The samples were homogenized on ice using VWR Pestle Microtub 1.5 ml (VWR Laboratory Products, Westbury, NY) and then centrifuged at 13,000 rpm for 30 min at 4°C. The cleared supernatant was transferred to a new tube, and the protein concentration in each tissue lysate was determined by the Bradford micromethod using a BioPhotometer Plus (Eppendorf North America, Westbury, NY). Kidney tissue lysates (50 μg protein/sample) were subjected to SDS-PAGE on a 4–20% gradient gel (Mini-PROTEAN precast gels; Bio-Rad, Hercules, CA), and proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon transfer membranes; Millipore, Billerica, MA). Then, the blots were probed with an anti-V-ATPase A subunit antibody raised in rabbit (GenScript USA, Piscataway, NJ) followed by immunolabeling using an anti-β-actin antibody raised in mouse (Sigma-Aldrich) to confirm equal loading of protein. After washing in Tris-buffered saline with Tween (0.05%), blots were incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG; GE Healthcare Biosciences, Pittsburgh, PA), or anti-mouse IgG (GE Healthcare Biosciences). The probed protein was then visualized and quantified using a VersaDoc Imager with Quantity One software (Bio-Rad).
Cell monolayer lysates and immunoblotting.
S3 cells were grown on Transwell filters in CCD media for 5 days at 37°C and 5% CO2/95% air. After the various treatments, the cells were harvested using ice-cold lysis buffer similar to the one that was used with kidney slices. The 4-h incubation time point with AICAR was only used for S3 cell monolayers. At earlier time points we did not see the effects of AICAR on either V-ATPase apical accumulation or activation of AMPK (not shown). After the different treatments samples were then centrifuged at 13,000 rpm for 15 min at 4°C. The cleared supernatant was transferred to a new tube and the protein concentration was determined by the Bradford micromethod. Immunoblot analysis was performed as described previously (36) using 20 μg cellular protein/sample. Following SDS-PAGE on a 4–12% gradient gel (Nu-PAGE, Life Technologies), proteins were transferred to PVDF membranes. Then, the blots were probed with an anti-pThr172-AMPKα (Cell Signaling Technology), anti-V-ATPase A subunit, and anti-β-actin antibodies. After washing in Tris-buffered saline with Tween (0.05%), blots were incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (GE Healthcare Biosciences). The probed protein was then visualized and quantified using a VersaDoc Imager with Quantity One software.
For the slot immunoblot assay (slot-blot), the Bio-Dot SF Microfiltration Apparatus (Bio-Rad) was used, and the protocol was followed as recommended in the instruction manual by the manufacturer. Briefly, 15 μg cellular protein diluted in 200 μl lysis buffer were used. Vacuum was applied only during the washing step, while sample solutions or blocking solutions (5% bovine serum albumin; Sigma-Aldrich) were left to pass freely (gravity) through a nitrocellulose membrane. Then, the blot was probed with an anti-phospho-(Ser/Thr) PKA substrate (anti-phosphoPKA substrate; Cell Signaling Technology), and anti-β-actin (Sigma-Aldrich) antibodies. After washing in Tris-buffered saline with Tween (0.05%), blots were incubated for 1 h at room temperature with horseradish peroxidase conjugated anti-rabbit or anti-mouse IgG. The probed protein was then visualized and quantified as above.
Measurements of changes in V-ATPase-dependent extracellular acidification rates.
S3 cells were seeded onto 24-well plates at an initial concentration of 0.3 × 106 cells/well. The cells were grown in CCD medium (3, 36) for 5 days at 37°C and 5% CO2/95% air. The medium of these S3 cells was then replaced with a Na+-free, low buffering capacity solution (“0 Na/0 Bicarbonate pH 7.4”) containing (in mM) 135 N-methyl-d-glucamine, 5 KCl, 2 CaCl2, 1.2 MgSO4, 5.5 d-glucose, 6 l-alanine, 4 lactic acid, 1 HEPES, titrated to pH 7.43 using 1 HCl (modified from Ref. 15) for 20 min in a protocol based on Ref. 2. Then, extracellular pH (pHo) measurements were performed after each of two additional 20-min incubation periods in low buffering capacity solution. In the first incubation period, the cells were treated with vehicle alone in the absence/presence of a PKA-activating drug cocktail (6-MB-cAMP/IBMX; 1 mM/0.5 mM) and/or AMPK activator AICAR. During the second incubation period, bafilomycin (1 μM; Sigma-Aldrich), a specific V-ATPase inhibitor, was added to all wells in the presence/absence of agonists or antagonists. The samples were maintained at 37°C during the whole experiment in a water bath. The V-ATPase-dependent rate of pHo acidification was defined as the difference in the measured acidification rates in the presence/absence of bafilomycin. Following the pHo measurements, the number of viable cells at the end of the experiment was assessed by Trypan blue exclusion assay (57).
Measurement of the average rate of change in extracellular proton concentration using SNARF-5F.
S3 cells were seeded onto Transwell filters at an initial concentration of 0.3 × 105 cells/well. The cells were grown in CCD medium (3, 36) for 5 days at 37°C and 5% CO2/95% air. After incubating the cells in the absence or presence of the AMPK activator AICAR (2 mM) for 4–5 h at 37°C and 5% CO2/95% air, the media of these S3 cells were then replaced with 10 μM SNARF-5F (Invitrogen) in a “0 Na/0 Bicarbonate pH 7.4” solution (2), in the absence/presence of a PKA-activating drug cocktail (6-MB-cAMP/IBMX; 1 mM/0.5 mM) and in the continuous absence or presence of the AMPK activator AICAR (2 mM). Then, the filters were maintained at room temperature on the stage of a Nikon Ti microscope (Nikon). The SNARF dye was excited by a metal arc lamp filtered with a 485/25 nm bandpass filter, and the ratio of fluorescence emission at 640 and 580 nm (F640/F590) was captured every minute with a Retiga EVi Blue camera (QImaging) for 25 min. A standard curve of F640/F590 vs. pHo was created with HEPES-buffered solutions between pH 6 and 8, and sample pH was calculated with linear regression. The change in extracellular proton concentration [H+] was then calculated using the following formula: [H+] = 10−pH. The rate of [H+] change was defined as the difference in the calculated [H+] at two time points divided by the time of incubation period (20 min). For all samples the first time point was measured after 5 min of equilibration time of the cells with the 0 Na/0 Bicarbonate pH 7.4 SNARF-5F-containing solution. To evaluate the contribution of the V-ATPase in the extracellular acidification in the presence of the different agonists, pHo measurements were repeated in filters treated with the V-ATPase inhibitor bafilomycin (50 nM). The bafilomycin was added to the apical surface of the filters diluted in the 0 Na/0 Bicarbonate pH 7.4 SNARF-5F-containing solution in the presence/absence of the various agonists and/or inhibitors being tested.
Cell surface biotinylation assays.
S3 cells were grown on Transwell filters in CCD medium for 5 days at 37°C and 5% CO2/95% air. The medium was then replaced with 1 × PBS containing Ca2+ and Mg2+ (Thermo Fisher Scientific and with pH adjusted to 7.2) in the absence or presence a PKA activating drug cocktail (6-MB-cAMP/IBMX; 1 mM/0.5 mM) for 30 min, using the same conditions that we previously published in cultured intercalated cells (2). Apical surface biotinylation assays were then conducted as previously described (36), in which cells were first washed with ice cold PBS containing Mg2+ and Ca2+ three times for 5 min. Then, the apical membrane was biotinylated using 1 mg/ml EZ link sulfo-NHS-SS-biotin (Thermo Fisher Scientific) in PBS for two consecutive incubation periods of 10 min. Free NHS-biotin was then quenched by washing cells with ice-cold 10% FBS in PBS. Before harvesting cells, monolayers were washed with ice-cold PBS containing Ca2+ and Mg2+ three times, using PBS containing 1% Triton X-100, 50 mM EGTA, 10 mM Tris-Cl (pH 7.4), 0.4% deoxycholate, and one tablet of complete protease inhibitor cocktail (Roche), and complete phosphatase inhibitor cocktail (Roche). Protein concentration was determined, and 500 μg of protein was incubated with 30 μl of streptavidin-agarose beads (50%, Thermo Fisher Scientific) in a total volume of 1 ml overnight at 4°C, while kept in constant rotation. The beads were washed three times in lysis buffer, and the biotinylated proteins were eluted from the beads by boiling samples in 2× Laemmli sample buffer containing 20% DTT. The protein samples were then separated by SDS-PAGE on a 4–12% gradient gel (Nu-PAGE) and subjected to immunoblot analysis using anti-V-ATPase a4 and E subunits and anti-β-actin antibodies.
Statistical analysis.
All statistical analyses were performed using commercially available software (SIGMASTAT, Systat Software). Single comparisons were performed using the Student's t-test. Group comparisons were performed using one-way analysis of variance (ANOVA). When ANOVA showed significant differences among treatments, Student's t-tests (vs. control) were performed to determine whether the specific treatment was significantly different from the control. In all cases, P values <0.05 were considered significant.
RESULTS
PKA agonists induce apical membrane accumulation of the V-ATPase in S3 segment proximal tubule cells in kidney slices.
To investigate the role of PKA in the regulation of V-ATPase subcellular localization in proximal tubule cells, we performed ex vivo experiments using kidney tissue slices. We incubated kidney slices in Ringer buffer for 30 min in the presence or absence of the PKA activators 6-MB-cAMP (1 mM) and IBMX (0.5 mM), a method that is used routinely in our laboratory (22). The slices were fixed after the different treatments, and cryosections were then labeled with CY3-coupled WGA, a marker of the proximal tubule S3 segment brush-border membrane (41, 60), and with an antibody against the V-ATPase E subunit, a marker for active V-ATPase at the membrane (8, 39, 56). We used the 30-min time point as a control for the slice incubation because in previous studies the V-ATPase was mostly localized to cytoplasmic domains in intercalated cells in the presence of Ringer buffer (which contains bicarbonate) at this time point. Thus the effects of PKA activators in inducing apical V-ATPase accumulation can be readily observed at the 30-min time point (22).
Indeed, confocal images of immunofluorescently stained kidney slices incubated in Ringer buffer alone (30 min) revealed that in the proximal tubule S3 segment the V-ATPase was present both at the apical pole in microvilli-like structures and also in a diffuse cytosolic distribution (Fig. 1A). The apical structures colocalized with WGA immunolabeling (Fig. 1, B and C). This fluorescently labeled lectin also allowed us to better localize the S3 segments in the outer stripe of the outer medulla, as WGA immunolabeling is more intense in S3 segments compared with other proximal tubule segments (41). In the presence of PKA activators (6-MB-cAMP and IBMX in Ringer buffer) for the same time period, more apical accumulation of the V-ATPase E subunit was observed in the proximal tubule and less diffuse cytosolic staining in slices (Fig. 1, D–F). Accumulation of the V-ATPase subunits at the apical pole of cells has been used as a surrogate for V-ATPase activity at the membrane (8, 22, 39, 56). Quantification revealed that PKA promotes apical membrane accumulation of the V-ATPase in the proximal tubule S3 segment, as measured by the apical-to-cytoplasmic ratio of mean pixel intensity (MPI) of V-ATPase E subunit-associated fluorescence (Fig. 1G). This previously published method involves the comparison of MPI of V-ATPase-associated fluorescence in an apical ROI (colocalizing with WGA) with that in a subjacent cytoplasmic ROI (Fig. 1, A and D, insets) (22).
Fig. 1.

Protein kinase A (PKA) agonists induce apical membrane accumulation of the vacuolar H+-ATPase (V-ATPase) in the proximal tubule S3 segment in kidney slices. A: the V-ATPase displays a cytoplasmic distribution in confocal images of kidney slices treated with Ringer buffer alone for 30 min and immunolabeled with an antibody against the E subunit (green) with partial colocalization with the brush-border marker Cy3-coupled WGA (B, red). C: WGA labeling is partially colocalizing with E subunit labeling. D: addition of the cell-permeant N6-monobutyryl-cAMP (6-MB-cAMP) (1 mM) plus IBMX (0.5 mM) induced an apical/luminal accumulation of the V-ATPase. E: these kidney slices were also colabeled with CY3-coupled WGA, which did not show a significant change in distribution in the presence of the PKA agonist. F: in the merged image the E subunit immunolabeling becomes dominant to the WGA labeling at the apical pole of the S3 segment epithelial cells in the presence of the PKA activating compounds. Regions of interest (ROIs) were outlined for each cell at apical and cytoplasmic regions for quantification using previously described method (2, 4, 5) as illustrated in A and D, insets. G: quantification of the mean (±SE) of V-ATPase E subunit-associated mean pixel intensity (MPI) apical-to-cytoplasmic ratio [arbitrary units (AU)] was used as a measure of apical V-ATPase accumulation, which was greater in the presence of PKA agonists. Data were obtained from at least 3 separate experiments measuring a total of at least 30 cells per condition (*P < 0.05 vs. Ringer, pH 7.4, 30 min). Scale bar = 20 μm.
The PKA inhibitor myristoylated PKI blocks V-ATPase apical accumulation in the S3 segment proximal tubule cells in kidney slices.
Incubation of kidney slices in Ringer buffer (which contains bicarbonate) for 75 min induced apical accumulation of the V-ATPase (Fig. 2A). This result in the proximal tubule is parallel to our findings in collecting duct intercalated cells (22). Compared with slices incubated in Ringer buffer for 75 min, slices incubated with the PKA inhibitor mPKI (10 μM) displayed more diffuse cytosolic V-ATPase labeling in S3 segments (Fig. 2, B). Quantification of the apical-to-cytoplasmic MPI of V-ATPase E subunit-associated fluorescence confirmed that inhibition of PKA with mPKI prevents V-ATPase apical accumulation in S3 proximal tubule epithelial cells (Fig. 2C). These findings also support the concept that the 75-min time point in kidney slices incubated in Ringer buffer represents a time point where the effects of inhibitors of V-ATPase apical accumulation can be tested.
Fig. 2.

The PKA inhibitor mPKI blocks V-ATPase apical accumulation in S3 segment cells in kidney slices. Confocal images of V-ATPase E subunit immunofluorescence labeling in kidney slices incubated either in Ringer buffer alone for 75 min or in the presence of the specific PKA inhibitor mPKI (10 μM). A: incubation of kidney slices in the Ringer buffer (which contains bicarbonate) for 75 min induced apical accumulation of the V-ATPase. B: addition of the membrane-permeant PKA inhibitor mPKI (10 μM) induced a more diffuse cytosolic V-ATPase labeling in S3 segments. C: quantification of apical-to-cytoplasmic MPI of V-ATPase E subunit-associated fluorescence from three separate experiments demonstrated that mPKI prevented V-ATPase apical accumulation that is induced by buffer alone in S3 proximal tubule epithelial cells (*P < 0.05 vs. Ringer buffer control, 75 min). Scale bar = 20 μm.
AMPK activation prevents V-ATPase apical accumulation in S3 segment epithelial cells in kidney slices.
For these experiments we also used the 75-min time point of incubation in Ringer buffer. As demonstrated above (Fig. 2), kidney slices incubated in Ringer buffer at pH 7.4 and 37°C for 75 min show apical V-ATPase distribution in proximal tubule epithelial cells, a process that is mediated by PKA. Therefore, the 75-min time point in kidney slices incubated in Ringer buffer can be used to study the effects of inhibitors of PKA-mediated V-ATPase apical accumulation. Our results revealed that a 75-min incubation in Ringer buffer alone caused apical V-ATPase accumulation in S3 proximal tubules (Fig. 3A), whereas addition of the AMPK activator AICAR induced more diffuse cytosolic V-ATPase labeling compared with Ringer buffer alone (Fig. 3B). Quantification of apical-to-cytoplasmic MPI of V-ATPase E subunit-associated fluorescence confirmed that AICAR induced a cytoplasmic redistribution of the V-ATPase in S3 proximal tubule epithelial cells (Fig. 3C).
Fig. 3.

The AMP-activated protein kinase (AMPK) activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) prevents V-ATPase apical accumulation in the proximal tubule S3 segment. Confocal images illustrate the cellular distribution changes of the V-ATPase E subunit in the S3 segment of kidney slices treated with Ringer buffer in the absence/presence of the AMPK activator AICAR (2 mM; 75 min). A: incubation of kidney slices in buffer alone for 75 min induced apical V-ATPase accumulation in S3 proximal tubules. B: addition of AICAR induced more diffuse cytosolic V-ATPase labeling in S3 segments compared with buffer alone. C: quantification of the mean (±SE) V-ATPase-associated MPI apical-to-cytoplasmic ratio from three separate experiments confirmed a significant cytoplasmic redistribution of the V-ATPase in the presence of AICAR (*P < 0.05 vs. Ringer, pH 7.4, 75 min. Scale bar = 10 μm). D: representative immunoblot of rat kidney lysates showing AMPK activity (pThr172-AMPKα; top) with β-actin as loading control (bottom) following treatment of kidney slices with Ringer buffer in the absence or presence of the AMPK activator AICAR (2 mM; 75 min). E: AICAR treatment induced a significant upregulation of phosphorylated pThr172-AMPKα compared with untreated cells. Values are relative means (±SE) of pThr172-AMPKα signal for 3 separate slices from 3 different animals normalized to β-actin (*P < 0.05 vs. Ringer, pH 7.4, 75 min).
We confirmed that AICAR activated AMPK in kidney slices by performing immunoblots of kidney slice lysates using the phosphospecific pThr172-AMPKα antibody (Fig. 3D, top). After normalization for loading, quantification revealed consistent AMPK activation with AICAR treatment in the kidney slices preparations (Fig. 3E).
Acute AMPK activation does not affect cellular expression of V-ATPase A subunit in kidney slices.
The diffuse intracellular localization induced by the AMPK activator AICAR was also associated with a qualitative decrease in V-ATPase-associated fluorescence in the S3 segment tubule cells, a finding that we had observed previously in collecting duct intercalated cells of rat kidney slices treated with AICAR (22). We then tested whether acute AMPK activation for 75 min affected the steady-state levels of the V-ATPase A subunit, a subunit that is directly phosphorylated by AMPK (1, 23). Quantification of the anti-V-ATPase A subunit immunoblot signal (Fig. 4A, top) normalized to β-actin (Fig. 4A, bottom) revealed that AICAR treatment for 75 min had no effect on the steady-state cellular expression of V-ATPase A subunit in rat kidney slice homogenates (Fig. 4B). Although these results reflect changes in overall kidney V-ATPase A-subunit expression, these studies confirm that acute AMPK activator treatments do not decrease total V-ATPase expression levels.
Fig. 4.

Acute AMPK activation does not affect cellular expression of V-ATPase A subunit in kidney slices. Rat kidney slices were treated for 75 min in either control Ringer buffer alone or Ringer buffer containing 2 mM AICAR followed by rapid freezing and homogenization. A: representative immunoblots of V-ATPase A subunit (top) or β-actin (bottom) expression in total kidney slices lysates. B: quantification of the anti-V-ATPase A subunit signal normalized to β-actin revealed that AICAR treatment for 75 min had no effect on the cellular expression of V-ATPase A subunit in these homogenates. Values are means (±SE) of 3 independent kidney slice preparations normalized to β-actin.
AMPK activation prevents PKA-mediated V-ATPase apical accumulation in kidney slice S3 segments.
We then asked whether AMPK activation prevents the V-ATPase apical accumulation induced by PKA activators at 30 min in the S3 segment (cf. Fig. 1). Indeed, incubation of slices with the AMPK activator AICAR for 30 min prevented any apical V-ATPase accumulation in the absence (Fig. 5A) or presence (Fig. 5B) of the PKA activators 6-MB-cAMP/IBMX. These effects were quantified using the same method as in Fig. 1 and are presented in Fig. 5C.
Fig. 5.
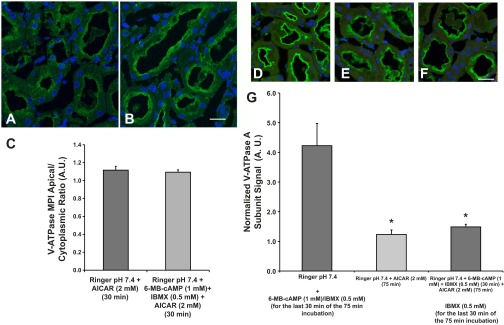
The AMPK activator AICAR prevents PKA-mediated V-ATPase apical accumulation in the S3 segment in kidney slices. Representative confocal images of immunofluorescence labeling of kidney slices using an antibody against the V-ATPase E subunit in S3 cells. A: incubation of kidney slices with 2 mM AICAR for 30 min. B: incubation of kidney slices with both AICAR and the PKA activators 6-MB-cAMP (1 mM) and IBMX (0.5 mM) for 30 min. C: quantification of the MPI of E subunit-associated fluorescence revealed that the apical-to-cytoplasmic V-ATPase ratio was unchanged in slices treated with AICAR vs. AICAR plus 6-MB-cAMP/IBMX for 30 min. Scale bar = 15 μm. D: incubation of kidney slices with Ringer buffer pH 7.4 for 75 min total, with the addition of 6-MB-cAMP/IBMX for the last 30 min of the 75 min incubation. E: incubation of kidney slices with Ringer buffer pH 7.4 with AICAR for 75 min. F: incubation of kidney slices with AICAR in Ringer buffer for 75 min with the addition of 6-MB-cAMP/IBMX for the last 30 min of the 75 min incubation. Scale bar for D–F = 25 μm. G: quantification of the MPI of E subunit-associated fluorescence revealed that the apical-to-cytoplasmic ratio of the V-ATPase was significantly decreased in proximal tubules of slices treated with AICAR whether these slices had been treated or not with PKA activators with respect to slices that were treated with PKA activators in the last 30 min of a 75 min incubation (*P < 0.05 vs. Ringer buffer pH 7.4 with PKA activators in the last 30 min).
In addition, we performed experiments in kidney slices to test whether a 75-min treatment with AICAR would also prevent the cAMP-induced V-ATPase apical accumulation. We incubated kidney slices with either: 1) Ringer buffer alone for the first 45 min followed by addition of the PKA activators 6-MB-cAMP/IBMX for the last 30 min of the incubation (75 min total incubation; Fig. 5D); 2) Ringer buffer with AICAR alone for 75 min (Fig. 5E); or 3) Ringer buffer with AICAR for the entire 75 min with the addition of 6-MB-cAMP/IBMX during the last 30 min of the AICAR incubation (Fig. 5F). After immunofluorescence labeling of the fixed slices and quantification of V-ATPase-E subunit-associated fluorescence in confocal micrographs, we observed that AICAR treatment significantly decreased apical accumulation of the E subunit, whether or not the slices were treated with PKA agonists during the last 30 min of the 75-min incubation (Fig. 5G). Overall, these results suggest that AMPK activation prevents PKA-mediated V-ATPase apical accumulation in proximal tubule S3 segment cells in situ.
PKA agonists induce apical membrane accumulation of the V-ATPase in an S3 segment-derived cell line.
Another tool available for our studies is a mouse cell line originally derived by dissection of S3 segments (33, 36). These cells are a good model to perform functional and biochemical studies on the V-ATPase in a proximal tubule epithelial cell model. To characterize endogenous V-ATPase subcellular localization in polarized S3 cells in response to PKA modulation, cells grown on filters were placed in PBS pH 7.2 alone (Fig. 6A) or treated with PKA activators (6-MB-cAMP/IBMX; 1 mM/0.5 mM; Fig. 6B) for 30 min, using a previously published protocol from our laboratory that we had previously performed in a cell line of intercalated cell characteristics (2). The change in PBS pH from 7.4 to 7.2 was necessary to induce intracellular redistribution of the V-ATPase. When incubated in cell culture media at pH 7.4 (see below and Fig. 7A), S3 cells had mostly apical V-ATPase immunolabeling. We imaged the monolayers by confocal microcopy and obtained confocal Z stacks of these images. The X–Z reconstructions of the immunofluorescence labeling, using methods previously published by our laboratory (2), revealed that when S3 cells were incubated in Ringer buffer alone, the V-ATPase had a diffuse intracellular distribution (Fig. 6A, green). However, when the S3 cell monolayers were incubated in the presence of PKA activators, the V-ATPase E subunit significantly accumulated at the apical pole of these cells (Fig. 6B, green), with more intense signal on the apical side of the ZO-1 tight junction marker (Fig. 6B, red). Quantification confirmed that apical V-ATPase accumulation was greater in the presence of PKA agonists (Fig. 6C).
Fig. 6.

PKA agonists induce apical membrane accumulation of the V-ATPase in polarized S3 cells in culture. Confocal images of V-ATPase E subunit immunofluorescence labeling in polarized S3 cells grown on filters for 5 days. A: S3 monolayers incubated in PBS buffer pH 7.2 (with Ca2+ and Mg2+) alone for 30 min demonstrated apical and cytoplasmic distribution for the V-ATPase (A and A′: E subunit, green; A and A“: ZO-1, red; nucleus, blue). B: addition of PKA activators (6-MB-cAMP/IBMX; 1 mM/0.5 mM) induced an apical accumulation of the V-ATPase (B and B′: E subunit, green; B and B”: ZO-1, red; nucleus, blue). C: quantification of the mean (±SE) V-ATPase E subunit-associated MPI apical-to-cytoplasmic ratio under the different conditions. Data were obtained from three separate experiments measuring a total of at least 30 cells per condition (*P < 0.05 vs. PBS pH 7.2, 30 min). Scale bar = 7 μm. D: immunoblot of V-ATPase a4 subunit (ATP6V0A4 in the V-ATPase V0 integral membrane domain) in 5% of whole cell lysates or biotinylated protein samples from polarized S3 cells treated with PBS containing Ca2+ and Mg2+ (pH = 7.2) alone or in the presence of PKA activating drug cocktail (6-MBcAMP:IBMX; 1 mM:0.5 mM) for 30 min. E: quantification of the mean (±SE) V-ATPase band intensity measured in the biotinylated fraction and divided by that in the cell lysate and expressed relative to that in control cells. Addition of 1 mM 6-MB-cAMP plus 0.5 mM IBMX increased V-ATPase surface expression by 60–70% relative to vehicle control. (*P < 0.05; n = 3).
Fig. 7.

The AMPK activator AICAR induces V-ATPase cytoplasmic redistribution in a polarized S3 cells. Confocal images of V-ATPase E subunit immunofluorescence labeling in polarized S3 cells grown on filters and incubated in CCD media in the absence or presence of the AMPK activator AICAR (2 mM; 4 h). A: S3 monolayers grown in media containing HCO3− and hormones (potential PKA agonists) demonstrated an apical accumulation of the V-ATPase. B: addition of AICAR to the media caused a more diffuse cytosolic labeling of the V-ATPase E subunit. C: quantification of the mean (±SE) V-ATPase-associated MPI apical-to-cytoplasmic ratio under the different conditions confirmed a significant cytoplasmic redistribution of the V-ATPase in the presence of AICAR. Data were obtained from three separate experiments measuring a total of at least 30 cells per condition (*P < 0.05 vs. CCD media, 4 h). Scale bar = 7 μm. D: AMPK activity as measured by immunoblotting of pThr172-AMPKα (top) relative to β-actin as a loading control (bottom) in polarized S3 cell lysates incubated in CCD medium in the absence or presence of the AMPK activator AICAR (2 mM; 4 h). E: quantification of the mean (±SE) pThr172-AMPKα signal normalized to β-actin is shown relative to control samples treated with vehicle alone. AICAR treatment induced a significant increase in pThr172-AMPKα signal compared with untreated cells. F: immunoblot of V-ATPase A subunit (top) and β-actin (bottom) in polarized S3 cells. G: quantification of the anti-V-ATPase A subunit signal normalized to β-actin revealed that AICAR treatment had no effect on the cellular expression of V-ATPase A subunit in S3 monolayers. Values are means (±SE) of three independent experiments normalized to β-actin (*P < 0.05; n = 3).
To further validate the immunofluorescence findings in this S3 cell line, we performed surface biotinylation assays to detect apical membrane expression of the V-ATPase. As a multisubunit complex, the V-ATPase is described as having an integral membrane domain (V0) and a cytoplasmic domain (V1). Therefore, we performed surface biotinylation of the polarized S3 cell monolayer after treatment in PBS buffer ± PKA agonists. We then immunoblotted the cell lysates for the transmembrane a4 V-ATPase subunit (ATP6V0A4), which has an important role in proximal tubule function. The a4 subunit was detected in S3 cell line lysates (Fig. 6D, bottom), as previously described (29). We also detected the a4 subunit in the biotinylated samples of these cells (Fig. 6D, top). Following PKA stimulation, we detected higher levels of a4 subunit in the biotinylated samples (Fig. 6, D and E). We did not detect significant changes in total cellular a4 expression in the absence or presence of the PKA activating compounds (Fig. 6D, bottom). Therefore, the data obtained by both quantifying the apical V-ATPase E subunit apical accumulation and by surface biotinylation of the a4 subunit are consistent with one another and describe the same trafficking phenomenon in polarized S3 cells: the first method by studying a subunit that belongs to the V1 cytoplasmic domain and the second method by studying a transmembrane subunit of the V-ATPase V0 sector.
AMPK activation prevents V-ATPase apical accumulation in an S3 segment-derived cell line.
To test whether AMPK activation inhibits V-ATPase apical accumulation in this polarized S3 segment-derived cell line, polarized cell monolayers were incubated in CCD cell culture media (see materials and methods) in the presence or absence of the AMPK activator AICAR (2 mM) for 4 h. The time period of 4 h incubation with AICAR for the S3 cells was chosen instead of the 75-min incubation period, which was used in kidney slices, because in the S3 cells 4 h of AICAR treatment was required to significantly activate AMPK (36). Immunofluorescence labeling was performed after treatment with AICAR or vehicle (water) in cell culture media followed by confocal fluorescence microscopy. Our results revealed that when cells were grown in medium containing HCO3− and hormones (potential PKA agonists), the V-ATPase has an apical distribution at baseline (Fig. 7A). However, V-ATPase E subunit immunolabeling in polarized S3 cells treated with AICAR for 4 h showed a more diffuse cytoplasmic localization (Fig. 7B). A significant cytoplasmic redistribution of the V-ATPase in the presence of AICAR was confirmed by quantification of the apical-to-cytoplasmic MPI ratio (Fig. 7C). Altogether, these results suggest that PKA and AMPK coregulate endogenous V-ATPase subcellular localization in polarized S3 cells. These findings are in agreement with our above results on proximal tubule V-ATPase regulation in kidney slices.
As a control we tested whether treatment with AICAR activated AMPK in this cell line by monitoring the levels of pThr172-AMPKα in S3 monolayer cell lysates (Fig. 7D, top). When normalized to the loading control immunoblot signal (Fig. 7D, bottom), we found that AICAR treatment increased the levels of the active AMPKα (Fig. 7E). Moreover, we noticed an apparent decrease in V-ATPase E-subunit expression by immunofluorescence staining in S3 cell monolayers treated with AICAR (Fig. 7B), as we had already observed in kidney slices treated with AICAR (Fig. 3B). We therefore tested whether protein expression of the V-ATPase A subunit, a direct target of AMPK phosphorylation (1, 23), was downregulated by immunoblotting in these cells as a result of AMPK activation (Fig. 7F, top). After normalization for a loading control (Fig. 7F, bottom), we did not detect any significant changes in the V-ATPase A subunit levels in monolayers treated with AICAR compared with untreated monolayers (Fig. 7G).
PKA and AMPK regulate V-ATPase activity in the S3 cells in culture.
To assess the role of PKA and AMPK in V-ATPase functional activity at the apical membrane, we measured pHo changes in S3 cells grown in a 24-well plate, using a protocol based on our previously described methods (2). The V-ATPase-dependent rate of pHo acidification was defined as the difference in the measured acidification rates in the absence or presence of the V-ATPase inhibitor bafilomycin during a 20-min incubation period. The Na+-free low buffering capacity solution was prepared to minimize the role of Na+/H+ exchangers to pHo. Also, the experiments were performed in the nominal absence of HCO3− to minimize buffering capacity or sAC activity, which could independently increase intracellular [cAMP] and PKA activity (35, 46, 68). When compared with untreated S3 cells, cells treated with the PKA-activating compounds demonstrated an increase in acidification rate, which could be attributed to V-ATPase-activity, while cells treated with the AMPK activator AICAR showed a near-significant decrease in the rate of V-ATPase-dependent acidification compared with buffer alone (Fig. 8A). Of note, the levels of various PKA-activating compounds in the 0 Na/0 bicarbonate solution compared with CCD medium are very low, and we surmise that baseline V-ATPase activity in these cells under these conditions was already low. However, the presence of AICAR inhibited but did not completely prevent V-ATPase activation by PKA activators. These results are consistent with our findings in kidney slices, where AMPK activation downregulated PKA-mediated V-ATPase apical accumulation. To confirm that PKA was activated in cells under the conditions of this experiment, we performed slot blots of cell lysates using an antibody directed against PKA phosphorylated substrates. (Fig. 8B). After quantification of these lysates we determined that the treatment of the S3 cells with the PKA activating drug cocktail for 30 min induced a significant increase of PKA activity (Fig. 8C).
Fig. 8.

AMPK inhibits the PKA effects on V-ATPase activity in cultured S3 cells. Cells were grown on 24-well plate for 5 days before incubation in 0 Na/0 Bicarbonate solution in the absence/presence of PKA and/or AMPK activators. The rate of extracellular acidification in each set of treatments was measured in a low buffering capacity solution before and after the addition of bafilomycin, a specific V-ATPase inhibitor. The mean (±SE) rate of extracellular acidification (−[final buffer pH − initial buffer pH]/Δt) was obtained in the presence and absence of bafilomycin for each treatment condition. There was negligible cell death after these experiments, as determined by a Trypan blue exclusion assay across conditions. Data were obtained from 4 separate experiments per condition (*P = 0.06 vs. 0 Na/0 Bicarbonate solution pH 7.4; **P < 0.05 vs. 0 Na/0 Bicarbonate solution at pH 7.4; ***P < 0.05 vs. 0 Na/0 Bicarbonate solution at pH 7.4 + AICAR and 0 Na/0 Bicarbonate solution at pH 7.4 + 6-MB-cAMP + IBMX; ANOVA followed by paired t-tests). B: slot blot using the PKA phosphorylation substrate-specific antibody (top) with β-actin antibody as loading control (bottom) of whole S3 cell lysates following treatment of polarized S3 cells with 0 Na/0 Bicarbonate solution at pH 7.4 in the absence/presence of the PKA-activating drug cocktail (6-MB-cAMP:IBMX; 1 mM:0.5 mM) for 20 min. C: PKA activator 6-MB-cAMP significantly increased the PKA phosphorylation substrate signal compared with the control 0 Na/0 Bicarbonate solution at pH 7.4. Values are means (±SE) of PKA phosphorylation substrate signal normalized to β-actin signal (*P < 0.05; n = 3). D: average rate of change in extracellular proton concentration as measured by extracellular pH (pHo) using the pH-sensitive dye SNARF-5F. These measurements were performed over a 25-min time period at room temperature on the apical solution of confluent S3 cell monolayers grown on filters. Prior to and during the pHo measurement period, the monolayers were treated with either vehicle or 2 mM AICAR apically and basolaterally. At the start of the measurement period, the media on the apical sides were replaced with 0 Na/0 Bicarbonate pH 7.4 containing the dye SNARF-5F in the presence or absence of 1 mM 6-MB-cAMP/0.5 mM IBMX and in the continued presence or absence of AICAR (gray bars). In addition for each of the 4 treatment conditions, the pHo measurements were repeated for filters treated with the V-ATPase inhibitor bafilomycin (50 nM; black bars; *P < 0.05 with respect to 0 Na/0 Bicarbonate pH 7.4 alone; **P < 0.05 respect to 0 Na/0 Bicarbonate pH 7.4 6-MB-cAMP/IBMX and with respect to 0 Na/0 Bicarbonate pH 7.4 plus bafilomycin; n = 6–12).
We also tested the effects of PKA and AMPK activators on V-ATPase activity in polarized S3 cells grown on filters by measuring extracellular pH (pHo) changes, and then converting these values to reflect changes in extracellular H+ concentration ([H+]). The rate of change in extracellular [H+] in response to various agonists was measured as changes in fluorescence emission by SNARF-5F dissolved in the low-buffering capacity (0 Na/0 Bicarbonate) solution bathing the apical surface of the polarized S3 cells. The results we obtained with this pH sensor (Fig. 8D; gray bars) revealed that the rate of change in extracellular [H+] increased in the presence of the PKA activating cocktail containing 6-MB-cAMP/IBMX compared with 0 Na/0 Bicarbonate alone. In the presence of the AMPK activator AICAR, the rate of change in extracellular [H+] was lower than 0 Na/0 Bicarbonate alone, whether PKA activators were present or not. When compared with the measurements performed in the presence of the V-ATPase inhibitor bafilomycin (Fig. 8D; black bars) the rate of change in extracellular [H+] was significantly decreased by bafilomycin only in cells that had been treated with the PKA activator cocktail. In all other conditions, the presence of bafilomycin did not change the rate of change in extracellular [H+]. These findings indicate that under the conditions of the assay, baseline V-ATPase activity in 0 Na/0 Bicarbonate was negligible, and that the PKA activator cocktail increased V-ATPase activity. Of note, the PKA activators also inhibited a V-ATPase-independent pathway of proton secretion in the S3 cell monolayers, as the bafilomycin-resistant component of the rate of change in extracellular [H+] was reduced with PKA activation compared with untreated cells. In the presence of the AMPK activator AICAR, the V-ATPase-independent component contributing to changes in extracellular [H+] was also inhibited relative to untreated controls. This bafilomycin-resistant, V-ATPase-independent pathway may represent H+ efflux via a proton-monocarboxylate cotransporter (50). In summary, the findings obtained using the SNARF-5F technique (Fig. 8D) are qualitatively similar to the findings using buffer exchange and pHo measurements with a pH meter (Fig. 8A). However, the conditions used for the SNARF-5F measurements, which include performing the experiment at room temperature, were such that the baseline bafilomycin-sensitive H+ secretion from the S3 cells was negligible.
DISCUSSION
The goal of these studies was to investigate the role of kinases in the regulation of proximal tubule H+ transport by the V-ATPase. Our group has previously studied the regulation of this protein complex in epithelia derived from the Wolffian duct, which include the epididymis, vas deferens, and the kidney collecting duct.
The composition of the V-ATPase in the proximal tubule differs from that in kidney collecting duct intercalated cells. For example, the B subunit isoform expressed in rodent collecting duct intercalated cells and in epididymal clear cells is the B1 subunit (ATP6V1B1) (16, 43, 63). Mutations in this subunit can cause distal RTA in human patients (32). However, mice with knockout of the B1 subunit failed to develop RTA unless challenged with a dietary acid load (18). It was later found that the B2 subunit in these mice, which is the B subunit isoform expressed in proximal tubule, was expressed in the collecting duct of the B1-knockout animals. It was concluded that this adaptive ectopic B2 subunit expression in intercalated cells was in part responsible for the initial lack of a phenotype in the V-ATPase B1-knockout animals (48). Therefore, because of the different subunit isoform compositions of the V-ATPase in the proximal and distal tubule, we were curious to determine whether the PKA and AMPK-mediated regulation of this pump would be observed in the proximal tubule. In other published work from our laboratory, we have identified that the A subunit, a conserved isoform between the proximal and distal tubule, is the target of both of these kinases. As the A subunit is common to the V-ATPase expressed in both proximal tubule and collecting duct intercalated cells, we thought it was feasible that PKA could stimulate the apical insertion of the V-ATPase while acute AMPK activation would decrease the activity of the pump at the membrane in the proximal tubule cells.
While recent studies suggest that AMPK activation is known to protect the proximal tubule in the setting of ischemia, it is less clear whether PKA activation is physiologically relevant to V-ATPase activity in the proximal tubule. Proton secretion in the proximal tubule by NHE3 is downregulated by pathways that involve activation of PKA, such as PTH and dopamine (31, 45, 49, 66). Most of the bicarbonate recovered from the filtrate in the proximal tubule occurs via H+ secretion into the proximal tubule by NHEs. However, in NHE3 KO mice the V-ATPase that takes over can contribute up to 40% of total H+ secretion (65). In distal tubule intercalated cells we have previously shown how a sAC/cAMP/PKA cascade activates the V-ATPase at the apical membrane. However, in our previous work, we had failed to detect sAC in the proximal tubule (25). New studies using anti-sAC antibodies against different epitopes suggest that this bicarbonate sensor may be present in the proximal tubule (59). Our data indicate that in Ringer buffer or bicarbonate-containing cell culture media the proximal tubule V-ATPase is at the apical membrane of S3 segment cells in kidney slices or in the S3 cell line. These findings support the potential role of sAC in the regulation of the V-ATPase in the proximal tubule. One important question that will need to be elucidated in further studies is whether the high levels of bicarbonate reabsorbed in this nephron segment result in a basal levels of sAC activation and cAMP production, or whether sAC activity can be acutely modulated in this setting.
Our findings reveal that the regulation of V-ATPase subcellular localization and function by PKA and AMPK in the proximal tubule is similar to the regulation in the A-type intercalated cells of the collecting duct and in epididymal clear cells. This regulation in the proximal tubule may also be mediated by PKA phosphorylation at residue Ser-175 and AMPK phosphorylation at residue Ser-384 of the ubiquitous V-ATPase A subunit (ATP6V1A) (1, 2).
Although the activating stimuli for PKA-mediated regulation of the V-ATPase in the proximal tubule remain a subject for further study, it is clear that the effects of this kinase with respect to the V-ATPase can be counteracted in the setting of AMPK activation. AMPK has been shown to activate Na+/H+ exchange in a cell line of kidney origin (53). Our findings suggest that under conditions of metabolic depletion AMPK could downregulate the V-ATPase while upregulating Na+-dependent proton extrusion pathways. Of note, agonists such as ANG II stimulate the rat proximal tubule V-ATPase by a PKA-independent mechanism (12). ANG II activation of the V-ATPase via PKC supersedes the effects of PKA activation of the V-ATPase, as ANG II-induced signaling decreases cAMP. In our system, the effects of PKA were examined independently of ANG II, and in the absence of this hormone PKA does have a relevant role in proximal tubule V-ATPase activation. In view of our current findings, we propose that the V-ATPase is activated at the plasma membrane of the proximal tubule by PKA, which could play an important role in proton secretion and bicarbonate reabsorption in the proximal tubule under conditions of volume expansion (i.e., when the Na+-dependent proton extrusion pathways are downregulated), and when ANG II plasma concentrations are low. Investigating the interrelationship of ANG II and AMPK in the proximal tubule is an important goal for future study.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant R01-DK-084184 to N. M. Pastor-Soler; Sanofi Fellowship Award and NIH Grant F32-DK-097889 to M. M. Al-bataineh; NIH Grant R01-HL-112863 to M. M. Myerburg; and the Epithelial Imaging Subcore of the NIH Grant P30-DK-079307 “Pittsburgh Kidney Research Center” (P30 Pittsburgh Center for Kidney Research).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.M.A.-b. and N.M.P.-S. conception and design of research; M.M.A.-b., F.G., A.M., M.M.M., and N.M.P.-S. performed experiments; M.M.A.-b., M.M.M., and N.M.P.-S. analyzed data; M.M.A.-b., M.M.M., and N.M.P.-S. interpreted results of experiments; M.M.A.-b. and N.M.P.-S. prepared figures; M.M.A.-b. and N.M.P.-S. drafted manuscript; M.M.A.-b., A.M., M.M.M., and N.M.P.-S. edited and revised manuscript; M.M.A.-b., F.G., A.M., M.M.M., and N.M.P.-S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank C. Smolak and J. Bruns for technical assistance. We thank Drs. K. Hallows and R. Hughey for critical reading of this manuscript.
REFERENCES
- 1.Alzamora R, Al-Bataineh MM, Liu W, Gong F, Li H, Thali RF, Joho-Auchli Y, Brunisholz RA, Satlin LM, Neumann D, Hallows KR, Pastor-Soler NM. AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am J Physiol Renal Physiol 305: F943–F956, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzamora R, Thali RF, Gong F, Smolak C, Li H, Baty CJ, Bertrand CA, Auchli Y, Brunisholz RA, Neumann D, Hallows KR, Pastor-Soler NM. PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the a subunit in kidney cells. J Biol Chem 285: 24676–24685, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol 10: 923–934, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Bouley R, Breton S, Sun T, McLaughlin M, Nsumu NN, Lin HY, Ausiello DA, Brown D. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest 106: 1115–1126, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouley R, Pastor-Soler N, Cohen O, McLaughlin M, Breton S, Brown D. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra). Am J Physiol Renal Physiol 288: F1103–F1112, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Breton S, Brown D. Cold-induced microtubule disruption and relocalization of membrane proteins in kidney epithelial cells. J Am Soc Nephrol 9: 155–166, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Breton S, Brown D. New insights into the regulation of V-ATPase-dependent proton secretion. Am J Physiol Renal Physiol 292: F1–F10, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Brown D, Breton S. Mitochondria-rich, proton-secreting epithelial cells. J Exp Biol 199: 2345–2358, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Brown D, Hirsch S, Gluck S. Localization of a proton-pumping ATPase in rat kidney. J Clin Invest 82: 2114–2126, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cano A, Miller RT, Alpern RJ, Preisig PA. Angiotensin II stimulation of Na-H antiporter activity is cAMP independent in OKP cells. Am J Physiol Cell Physiol 266: C1603–C1608, 1994 [DOI] [PubMed] [Google Scholar]
- 11.Carraro-Lacroix LR, Girardi AC, Malnic G. Long-term regulation of vacuolar H+-ATPase by angiotensin II in proximal tubule cells. Pflügers Arch 458: 969–979, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Carraro-Lacroix LR, Malnic G. Signaling pathways involved with the stimulatory effect of angiotensin II on vacuolar H+-ATPase in proximal tubule cells. Pflügers Arch 452: 728–736, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Chan YL, Giebisch G. Relationship between sodium and bicarbonate transport in the rat proximal convoluted tubule. Am J Physiol Renal Fluid Electrolyte Physiol 240: F222–F230, 1981 [DOI] [PubMed] [Google Scholar]
- 14.Chen SH, Bubb MR, Yarmola EG, Zuo J, Jiang J, Lee BS, Lu M, Gluck SL, Hurst IR, Holliday LS. Vacuolar H+-ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J Biol Chem 279: 7988–7998, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Constantinescu A, Silver RB, Satlin LM. H-K-ATPase activity in PNA-binding intercalated cells of newborn rabbit cortical collecting duct. Am J Physiol Renal Physiol 272: F167–F177, 1997 [DOI] [PubMed] [Google Scholar]
- 16.Da Silva N, Shum WW, El-Annan J, Paunescu TG, McKee M, Smith PJ, Brown D, Breton S. Relocalization of the V-ATPase B2 subunit to the apical membrane of epididymal clear cells of mice deficient in the B1 subunit. Am J Physiol Cell Physiol 293: C199–C210, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Du Z, Yan Q, Wan L, Weinbaum S, Weinstein AM, Wang T. Regulation of glomerulotubular balance. I. Impact of dopamine on flow-dependent transport. Am J Physiol Renal Physiol 303: F386–F395, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, Giebisch G, Geibel JP, Lifton RP. The B1-subunit of the H+ ATPase is required for maximal urinary acidification. Proc Natl Acad Sci USA 102: 13616–13621, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–929, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Fraser S, Mount P, Hill R, Levidiotis V, Katsis F, Stapleton D, Kemp BE, Power DA. Regulation of the energy sensor AMP-activated protein kinase in the kidney by dietary salt intake and osmolality. Am J Physiol Renal Physiol 288: F578–F586, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Gluck S, Nelson R. The role of the V-ATPase in renal epithelial H+ transport. J Exp Biol 172: 205–218, 1992 [DOI] [PubMed] [Google Scholar]
- 22.Gong F, Alzamora R, Smolak C, Li H, Naveed S, Neumann D, Hallows KR, Pastor-Soler NM. Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 298: F1162–F1169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hallows KR, Alzamora R, Li H, Gong F, Smolak C, Neumann D, Pastor-Soler NM. AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells. Am J Physiol Cell Physiol 296: C672–C681, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol 298: F1067–F1077, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallows KR, Wang H, Edinger RS, Butterworth MB, Oyster NM, Li H, Buck J, Levin LR, Johnson JP, Pastor-Soler NM. Regulation of epithelial Na+ transport by soluble adenylyl cyclase in kidney collecting duct cells. J Biol Chem 284: 5774–5783, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamm LL, Nakhoul NL. Renal acidification. In: Brenner and Rector's The Kidney (8th ed.), edited by Brenner BM. Philadelphia, PA: Saunders Elsevier, 2008, p. 248–279 [Google Scholar]
- 27.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774–785, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 67: 821–855, 1998 [DOI] [PubMed] [Google Scholar]
- 29.Hennings JC, Picard N, Huebner AK, Stauber T, Maier H, Brown D, Jentsch TJ, Vargas-Poussou R, Eladari D, Hubner CA. A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubule. EMBO Mol Med 4: 1057–1071, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Hinton BT, Turner TT. Is the epididymis a kidney analogue? Physiology 3: 28–31, 1988 [Google Scholar]
- 30.Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 8: 124–136, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Kahn AM, Dolson GM, Hise MK, Bennett SC, Weinman EJ. Parathyroid hormone and dibutyryl cAMP inhibit Na+/H+ exchange in renal brush border vesicles. Am J Physiol Renal Fluid Electrolyte Physiol 248: F212–F218, 1985 [DOI] [PubMed] [Google Scholar]
- 32.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Kaunitz JD, Cummins VP, Mishler D, Nagami GT. Inhibition of gentamicin uptake into cultured mouse proximal tubule epithelial cells by l-lysine. J Clin Pharmacol 33: 63–69, 1993 [DOI] [PubMed] [Google Scholar]
- 34.Krapf R, Seldin DW, Alpern RJ. Clinical syndromes of metabolic acidosis. In: Selding and Giebisch's the Kidney: Physiology and Pathophysiology (4th ed.), edited by Alpern RJ, Herbert SC. Boston: Elsevier Academic, 2008, p. 1667–1720 [Google Scholar]
- 35.Lang K, Wagner C, Haddad G, Burnekova O, Geibel J. Intracellular pH activates membrane-bound Na+/H+ exchanger and vacuolar H+-ATPase in human embryonic kidney (HEK) cells. Cell Physiol Biochem 13: 257–262, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Li H, Thali RF, Smolak C, Gong F, Alzamora R, Wallimann T, Scholz R, Pastor-Soler NM, Neumann D, Hallows KR. Regulation of the creatine transporter by AMP-activated protein kinase in kidney epithelial cells. Am J Physiol Renal Physiol 299: F167–F177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lieberthal W, Zhang L, Patel VA, Levine JS. AMPK protects proximal tubular cells from stress-induced apoptosis by an ATP-independent mechanism: potential role of Akt activation. Am J Physiol Renal Physiol 301: F1177–F1192, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu M, Sautin YY, Holliday LS, Gluck SL. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar H+-ATPase. J Biol Chem 279: 8732–8739, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Madsen KM, Verlander JW, Kim J, Tisher CC. Morphological adaptation of the collecting duct to acid-base disturbances. Kidney Int Suppl 33: S57–S63, 1991 [PubMed] [Google Scholar]
- 40.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol 298: R851–R861, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murata F, Tsuyama S, Suzuki S, Hamada H, Ozawa M, Muramatsu T. Distribution of glycoconjugates in the kidney studied by use of labeled lectins. J Histochem Cytochem 31: 139–144, 1983 [DOI] [PubMed] [Google Scholar]
- 42.Nelson N, Harvey WR. Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol Rev 79: 361–385, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Nelson RD, Guo XL, Masood K, Brown D, Kalkbrenner M, Gluck S. Selectively amplified expression of an isoform of the vacuolar H(+)-ATPase 56-kilodalton subunit in renal intercalated cells. Proc Natl Acad Sci USA 89: 3541–3545, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastor-Soler NM, Gong F, Alzamora R, Smolak C, Thali R, Li H, Neumann D, Hallows KR. Role of AMPK and PKA in the trafficking of V-ATPase in kidney intercalated cells. FASEB J 24: 1002–1011, 2010 [Google Scholar]
- 45.Olsen NV. Effects of dopamine on renal haemodynamics tubular function and sodium excretion in normal humans. Dan Med Bull 45: 282–297, 1998 [PubMed] [Google Scholar]
- 46.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem 278: 49523–49529, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21: 523–533, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Paunescu TG, Russo LM, Da Silva N, Kovacikova J, Mohebbi N, Van Hoek AN, McKee M, Wagner CA, Breton S, Brown D. Compensatory membrane expression of the V-ATPase B2 subunit isoform in renal medullary intercalated cells of B1-deficient mice. Am J Physiol Renal Physiol 293: F1915–F1926, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Pollock AS, Warnock DG, Strewler GJ. Parathyroid hormone inhibition of Na+-H+ antiporter activity in a cultured renal cell line. Am J Physiol Renal Fluid Electrolyte Physiol 250: F217–F225, 1986 [DOI] [PubMed] [Google Scholar]
- 50.Poole RC, Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Physiol Cell Physiol 264: C761–C782, 1993 [DOI] [PubMed] [Google Scholar]
- 51.Preisig PA, Ives HE, Cragoe EJ, Jr, Alpern RJ, Rector FC., Jr Role of the Na+/H+ antiporter in rat proximal tubule bicarbonate absorption. J Clin Invest 80: 970–978, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 298: F177–F186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rotte A, Pasham V, Eichenmuller M, Bhandaru M, Foller M, Lang F. Upregulation of Na+/H+ exchanger by the AMP-activated protein kinase. Biochem Biophys Res Commun 398: 677–682, 2010 [DOI] [PubMed] [Google Scholar]
- 54.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 19: 282–285, 1998 [DOI] [PubMed] [Google Scholar]
- 55.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7: 189–200, 2011 [DOI] [PubMed] [Google Scholar]
- 56.Steinmetz PR. Cellular organization of urinary acidification. Am J Physiol Renal Fluid Electrolyte Physiol 251: F173–F187, 1986 [DOI] [PubMed] [Google Scholar]
- 57.Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol 2001. May; Appendix 3: Appendix 3B. 10.1002/0471142735.ima03bs21 [DOI] [PubMed] [Google Scholar]
- 58.Takiar V, Nishio S, Seo-Mayer P, King JD, Jr, Li H, Zhang L, Karihaloo A, Hallows KR, Somlo S, Caplan MJ. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci USA 108: 2462–2467, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int 79: 1277–1288, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Truong LD, Phung VT, Yoshikawa Y, Mattioli CA. Glycoconjugates in normal human kidney. A histochemical study using 13 biotinylated lectins. Histochemistry 90: 51–60, 1988 [DOI] [PubMed] [Google Scholar]
- 62.van Balkom BW, van Raak M, Breton S, Pastor-Soler N, Bouley R, van der Sluijs P, Brown D, Deen PM. Hypertonicity is involved in redirecting the aquaporin-2 water channel into the basolateral, instead of the apical, plasma membrane of renal epithelial cells. J Biol Chem 278: 1101–1107, 2003 [DOI] [PubMed] [Google Scholar]
- 63.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar-ATPase. Physiol Rev 84: 1263–1314, 2004 [DOI] [PubMed] [Google Scholar]
- 64.Wagner CA, Giebisch G, Lang F, Geibel JP. Angiotensin II stimulates vesicular H+-ATPase in rat proximal tubular cells. Proc Natl Acad Sci USA 95: 9665–9668, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang T, Yang CL, Abbiati T, Schultheis PJ, Shull GE, Giebisch G, Aronson PS. Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. Am J Physiol Renal Physiol 277: F298–F302, 1999 [DOI] [PubMed] [Google Scholar]
- 66.Weinman EJ, Minkoff C, Shenolikar S. Signal complex regulation of renal transport proteins: NHERF and regulation of NHE3 by PKA. Am J Physiol Renal Physiol 279: F393–F399, 2000 [DOI] [PubMed] [Google Scholar]
- 67.Zimolo Z, Montrose MH, Murer H. H+ extrusion by an apical vacuolar-type H+-ATPase in rat renal proximal tubules. J Membr Biol 126: 19–26, 1992 [DOI] [PubMed] [Google Scholar]
- 68.Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17: 82–84, 2003 [DOI] [PubMed] [Google Scholar]