

Abstract
Loss of cystic fibrosis transmembrane conductance regulator (CFTR) function reduces chloride secretion and increases sodium uptake, but it is not clear why CFTR mutation also results in progressive lung inflammation and infection. We previously demonstrated that CFTR-silenced airway cells migrate more slowly during wound repair than CFTR-expressing controls. In addition, CFTR-deficient cells and mouse models have been reported to have altered sphingolipid levels. Here, we investigated the hypothesis that reduced migration in CFTR-deficient airway epithelial cells results from altered sphingolipid composition. We used cell lines derived from a human airway epithelial cell line (Calu-3) stably transfected with CFTR short hairpin RNA (CFTR-silenced) or nontargeting short hairpin RNA (controls). Cell migration was measured by electric cell substrate impedance sensing (ECIS). Lipid analyses, addition of exogenous glycosphingolipids, and immunoblotting were performed. We found that levels of the glycosphingolipid, GM1 ganglioside, were ∼60% lower in CFTR-silenced cells than in controls. CFTR-silenced cells exhibited reduced levels of activated β1-integrin, phosphorylated tyrosine 576 of focal adhesion kinase (pFAK), and phosphorylation of Crk-associated substrate (pCAS). Addition of GM1 (but not GM3) ganglioside to CFTR-silenced cells restored activated β1-integrin, pFAK, and pCAS to near control levels and partially restored (∼40%) cell migration. Our results suggest that decreased GM1 in CFTR-silenced cells depresses β1-integrin signaling, which contributes to the delayed wound repair observed in these cells. These findings have implications for the pathology of cystic fibrosis, where altered sphingolipid levels in airway epithelial cells could result in a diminished capacity for wound repair after injury.
Keywords: pulmonary epithelial cells, sialo-lipids, chloride channels, cell attachment
cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cAMP-dependent chloride channel CF transmembrane conductance regulator (CFTR) (5). CFTR is expressed on the apical surface of epithelial cells of the lungs, liver, and pancreas (36, 43) as well in as some other cell types, such as macrophages and endothelial cells (37, 68). Many CFTR mutations have been identified that lead to CF. The most common mutation, a deletion of phenylalanine 508 (ΔF508), occurs in ∼70% of patients (26). Loss of CFTR function causes reduced chloride secretion and increased sodium uptake. Mutation of CFTR also results in progressive lung inflammation and bacterial infection; however, it is not clear how the loss of functional CFTR results in these pathologies (5, 26, 43). We recently demonstrated that lung epithelial cells depleted of CFTR by short hairpin RNA (shRNA) had a reduced capacity to migrate after wounding (46). Results from this report and other studies (20, 32, 54, 59) suggest that CFTR may play a role in epithelial cell repair after injury and that this function is impaired in CFTR-deficient cells.
Another feature noted to be defective in CF and CFTR-deficient cells is the regulation of sphingolipid levels. Sphingolipids include ceramide, sphingomyelin, glycosphingolipids, and gangliosides (sialic acid-containing glycosphingolipids) such as GM1 and GM3. These lipids play important roles in many cell functions and interact with cholesterol in the formation of membrane microdomains or lipid rafts (10, 49). Ganglioside composition has been reported to be altered in samples from CF patients and in cells expressing mutant CFTR. For example, asialo-GM1 is elevated in nasal polyp epithelial cell samples from CF patients relative to normal controls (44). An older study documented higher levels of glycosphingolipids, sphingomyelin, and cholesterol in tracheobronchial secretions from CF patients compared with controls (52). In addition, epithelial cells heterologously expressing ΔF508-CFTR have lower plasma membrane levels of GM1 and increased levels of asialo-GM1 relative to controls (12, 13). Finally, increased levels of the bioactive sphingolipid ceramide have been demonstrated in epithelial cells from CFTR-knockout mice, in cultured cells expressing ΔF508-CFTR, and in human CF patient alveolar cells compared with normal controls (21, 57, 60).
Given our observation that CFTR-silenced cells exhibit defective migration, and multiple studies that have shown that cell migration can be regulated by sphingolipids via their interactions with integrins (29, 35, 38, 55, 58), we investigated the hypothesis that reduced migration of CFTR-silenced cells is a result of altered sphingolipids in this cell type. We found that CFTR-silenced cells were significantly depleted of gangliosides. β1-Integrin activation and subsequent downstream phosphorylation of focal adhesion kinase (FAK) and p130/Crk-associated substrate (CAS) were suppressed in these cells. Addition of GM1, but not GM3 ganglioside, to CFTR-silenced cells restored β1-integrin activation and FAK and CAS phosphorylation and partially restored cell migration. These findings suggest that loss of gangliosides in CFTR-silenced cells results in downregulation of β1-integrin signaling which in turn decreases the potential for cell migration.1
MATERIALS AND METHODS
Lipids and antibodies.
N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoyl) (C5-BODIPY)-GM1 was synthesized as described previously (34). GM1, GM3, and ganglioside standards were from Matreya (State College, PA). Rabbit antibodies used were as follows: CFTR (Millipore, Billerica, MA), β-actin (Cell Signaling), p130/CAS (Santa Cruz Biotechnology), and tyrosine 576-phosphorylated (pY576)-FAK. Mouse antibodies were as follows: β1-integrin and FAK (BD Biosciences), GFP (Roche Applied Science, Indianapolis, IN), HUTS-4 (Millipore), tyrosine 397-phosphorylated (pY397)-FAK, and phospho-tyrosine (Cell Signaling). N-[7-(4-nitrobenzo-2-oxa-1,3-diazole)]-6-aminocaproyl sphingosine (C6-NBD-ceramide), C6-NBD-glucosylceramide, C6-NBD-sphingomyelin, AlexaFluor 488-conjugated secondary antibodies, and fluorescent and horseradish peroxidase (HRP)-labeled cholera toxin B subunit (CtxB) were purchased from Life Technologies. HRP-labeled secondary antibodies for immunoblots were from Millipore. All other reagents were from Sigma.
Cell culture.
CFTR-silenced and control cell lines (expressing CFTR shRNA and shRNA that lacks target specificity, respectively) were derived from the human lung adenocarcinoma cell line Calu-3, as described previously (46). Cells were maintained in minimal essential medium (MEM; Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan UT), nonessential amino acids (Life Technologies) and l-glutamine (Life Technologies), plus puromycin (4 μg/ml; Sigma) to maintain stable transfectants. In some cases cells were incubated with exogenous gangliosides, (5 μM GM1 or GM3) by addition in fresh media each day during incubation. All cell lines were maintained in a humidified 5% CO2 incubator at 37°C.
CFTR-adenovirus.
An adenoviral vector expressing human wild-type CFTR fused to green fluorescent protein (GFP-CFTR) was a kind gift from S. S. Hong (Université de Lyon, Lyon, France; 16). Adenoviral particles were incubated with cells at 109 particles/ml in normal culture media for 48 h. Infected cells were then cultured in serum-free medium for an additional 24 h and then used for experiments.
Cell migration.
An electric cell substrate impedance sensing (ECIS) system (Applied Biophysics, Troy, NY) was used to measure airway epithelial cell migration. Epithelial cells were grown on 8W1E ECIS arrays (Applied Biophysics) in growth media with serum until fully confluent, after which the media were replaced with serum-free media for 48 h. To induce wounding, cells in contact with the 250-μm electrode were subjected to lethal electroporation (900 μA, 6,400 Hz, 20 s) according to the manufacturer's instructions, leaving a uniform circular lesion at the electrode. Cells were then washed with MEM to dislodge any remaining dead cells from the electrode, and the treatment media (MEM plus 1 μg/ml epidermal growth factor with or without gangliosides) were added. Alternating current (∼1 μA, 15 kHz) was then continuously applied to the electrode to measure impedance (Z) and track wound repair. A frequency of 15 kHz was used to ensure near-continuous sampling of impedance changes throughout the experiments. At this frequency, changes in cell distribution on the electrode have the greatest measurable effect on impedance, with no noticeable effects on cell behavior or viability. Impedance before wounding was 12,000–14,000 Ω. Upon wounding, impedance fell to 2,000–3,000 Ω. The recovery of impedance over time after wounding to a plateau near the initial impedance value was indicative of wound closure. Wound repair was also monitored by video imaging, using a CO2 microscope stage incubator system (Applied Biophysics) attached to a Zeiss IM-35 microscope (Zeiss, Jena, Germany) that was equipped with a Hamamatsu C4742-95 digital camera (Hamamatsu, Bridgewater, NJ). The time at which complete wound closure could be observed visually corresponded closely to the plateau in the increase in impedance measured by the ECIS system. Experiments were performed in serum-free, HCO3−-buffered media in a humidified 5% CO2 incubator at 37°C. In some experiments, cells were cultured with 5 mM gangliosides for 4 days prior to wounding.
Preparation of cell lysates.
Cells were grown in 6-cm dishes with full growth media until 100% confluent and subsequently in basal media for 48 h. Cells were washed with PBS, lysed in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris·HCl, 150 mM NaCl, 5 mM NaF, 1 mM Na3VO4, 1% NP-40, 0.5% sodium deoxycholate, and 1% SDS, pH 7.5, supplemented with Complete protease inhibitor cocktail (Roche) and HALT phosphatase inhibitor cocktail (Thermo Scientific, Waltham, MA), and then centrifuged at 12,000 g and 4°C for 10 min. Supernatants (cell lysates) were collected and protein concentrations were measured using a bicinchoninic acid protein assay kit (Thermo).
Immunoblotting.
Proteins were separated by SDS-PAGE (7.5% TGX Mini-PROTEAN precast gels, Bio-Rad, Hercules, CA) under reducing conditions and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were probed with primary antibodies followed by secondary antibodies conjugated with HRP. Signals were visualized with enhanced chemiluminescence reagent (GE Healthcare, Little Chalfont, UK) and exposed to X-ray film. Protein bands were quantified using the ImageJ 1.42q image processing program (National Institutes of Health, Bethesda, MD; rsb.info.nih.gov). To detect the phosphorylation level of CAS, cell lysates were precleared by incubation with 40 μl of protein A-Sepharose (Sigma) for 1 h and then centrifugation at 325 g for 2 min. The cleared supernatant was incubated with antiphosphotyrosine antibody overnight and then with 20 μl of protein A-Sepharose for 3 h. Samples were then centrifuged as above, washed three times in RIPA buffer, eluted with Laemmli buffer containing 1.4 mM mercaptoethanol, and finally used for SDS-PAGE followed by immunoblotting with a CAS antibody.
Cell surface biotinylation.
The proportion of integrin on the plasma membrane was estimated by biotinylating the surface-exposed proteins using EZ-Link Sulfo-NHS-SS-biotin reagent (Thermo) for 30 min at 4°C according to the manufacturer's instructions. Samples were then lysed with RIPA buffer as above. Biotinylated proteins were isolated by incubation with streptavidin agarose beads (Thermo). SDS-PAGE sample buffer was then added to the beads, and SDS-PAGE and transfer to PVDF membranes were performed. Finally, β1-integrin was detected by immunoblotting as above.
Lipid analysis.
Lipid extraction and analysis were performed as described previously (40). Sphingolipids were separated by thin layer chromatography (TLC) using CHCl3/CH3OH/15 mM CaCl2 [11:9:2 (vol/vol/vol)] as the developing solvent and identified by comparison to known standards. Staining of GM1 on TLC plates was performed using a protocol for immunostaining of lipids (6) except that HRP-labeled CtxB was used instead of antibodies. Lipid samples were run on TLC plates as above. Plates were then dried and soaked in 0.02% polyisobutyl methacrylate for 1 min and air dried. Plates were then incubated in blocking buffer (1% bovine serum albumin/1% polyvinyl pyrolidine/0.02% sodium azide) at room temperature for 30 min. Plates were rinsed with washing buffer (PBS/1% Tween 20) and incubated with HRP-CtxB at 4°C overnight. After being rinsed with washing buffer, signals were visualized with enhanced chemiluminescence reagent. Gangliosides on TLC plates were also visualized by staining with resorcinol (28).
Assays for glucosylceramide and sphingomyelin synthases in cell lysates were performed using C6-NBD-ceramide as described previously (33). BODIPY-GM1 degradation was studied as previously described for BODIPY-lactosylceramide (9). Ceramide and its metabolites were analyzed using a modification of a previously described liquid chromatography-mass spectrometer method (7) by separating lipids on a Waters Acquity C8 UPLC BEH column 2.1 × 150 mm, 1.7 μm prior to introduction of compounds into a Thermo TSQ Quantum Ultra triple quadrupole mass spectrometer. Total unesterified cholesterol levels were measured using an Amplex red cholesterol assay kit (Life Technologies) according to the manufacturer's instructions.
Fluorescence microscopy.
Cells cultured on glass coverslips were fixed in 4% formaldehyde and then washed in PBS. For filipin staining, cells were incubated for 30 min at room temperature with 100 μg/ml filipin in PBS. For CtxB staining, fixed cells were permeabilized with 0.05% saponin in PBS and incubated for 30 min at room temperature with 2 μl/ml AlexaFluor 488-CtxB in PBS. To detect activated β1-integrin, living cells on coverslips were rinsed with PBS, incubated with 1:100 HUTS-4 antibody for 30 min at 10°C, rinsed and then fixed. Cells were then rinsed three times with PBS and incubated with 1:200 Alexa Fluor 594 anti-mouse antibodies for 2 h. In all cases, after staining, cells were washed in PBS, mounted in SlowFade Gold (Life Technologies), and observed by fluorescence microscopy using AX70 or IX70 microscopes (Olympus) equipped with Hamamatsu C4742-95 (Hamamatsu) or Photometrics (Tucson, AZ) QuantEM 512SC digital cameras, respectively. Digital images were acquired and analyzed using MetaMorph (version 7.3.2; Molecular Devices, Sunnyvale, CA). Quantitative analysis of HUTS-4 signal was performed by acquiring images of cells under identical exposure conditions. Cellular areas on images were digitally marked, and average intensity (total intensity/area) of these areas was measured using MetaMorph. Thus, average intensities included both diffuse and punctate signal. Average intensities of noncellular areas of the fields were acquired as background levels and were subtracted from the cell-associated average intensity values.
Confocal microscopy.
CFTR-silenced cells grown on coverglasses were infected with GFP-CFTR. After 3 days, the cells were fixed in 4% formaldehyde, permeabilized with 0.05% saponin in PBS, incubated for 30 min at room temperature with 2 μl/ml AlexaFluor 596-CtxB, and then incubated with DAPI for 5 min to stain nuclei. Specimens were mounted in Fluoro-Gel from Electron Microscopy Sciences (Hatfield, PA) and observed using a Zeiss LSM 510 confocal microscope.
Statistical analysis.
Data are expressed as means ± SE. Two-sample comparisons were made using unpaired two-tailed Student's t-tests; P < 0.05 was regarded as significant. Comparisons between multiple treatment conditions were made using a one-way analysis of variance (ANOVA) with Bonferroni post test. Gaussian distributions were determined using the method of Kolmogorov and Smirnov, and Bartlett's test was used to determine whether the standard deviations of the groups were equal. Analyses were performed using Prism 5 (version 5.04) software (GraphPad Software, La Jolla, CA).
RESULTS
We utilized the previously developed CFTR-silenced and control Calu-3 cell lines to study the potential role of sphingolipids in cell migration (46). We first verified the phenotype of CFTR-silenced and control cells. CFTR-silenced cells expressed no detectable CFTR, in contrast to controls that expressed significant levels of endogenous CFTR (Fig. 1A). As previously reported (46), after electrical wounding, control cells rapidly migrate into the wound region (circular electrode in Fig. 1B), with a T ½ (i.e., time at which impedance is one-half of impedance reached at upper plateau) of wound closure at ∼4 h (Fig. 1D) and full coverage of the electrode by 5–7 h (Fig. 1, B and C). CFTR-silenced cells migrated much more slowly after wounding (Fig. 1B), with a T ½ of ∼12 h (Fig. 1D), and did not reach a plateau until ∼15 h. (Fig. 1C).
Fig. 1.
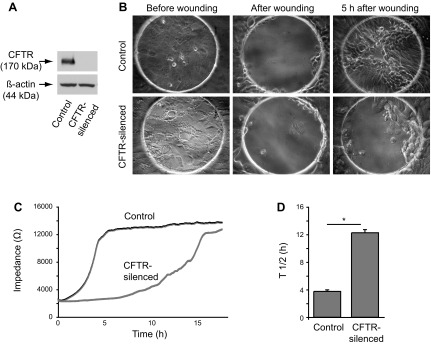
Decreased migration of CFTR-silenced cells after electrical wounding. Cells were cultured on electric cell substrate impedance sensing (ECIS) arrays in serum-free media for 2 days before wounding experiments. A: immunoblot of CFTR expression in Calu-3 control and CFTR-silenced stable cell lines. Equal protein quantities (20 μg) were loaded per lane. Immunoblot of β-actin levels on the same blot is shown to demonstrate equal protein loading. B: phase micrographs of control and CFTR-silenced cells on ECIS arrays. Cells are shown on 250-μm diameter circular electrodes before wounding, immediately after wounding by acute electric shock, and 5 h after wounding. C: impedance tracings of control and CFTR-silenced cells over time after electrical wounding. Lines are averages of 4 replicate samples. D: T ½ values for recovery of impedance after cell wounding for control and CFTR-silenced cells. T ½ values represent the time at which impedance was ½ of the impedance reached at upper plateau. Values are means ± SE of 4 replicates. *P < 0.01, significantly different in two tailed t-tests.
Gangliosides are decreased in CFTR-silenced cells.
Next, we investigated whether CFTR-silenced cells were altered in sphingolipid composition compared with control Calu-3 cells. We found that CFTR-silenced cells exhibited decreased binding by CtxB, a probe for GM1 ganglioside (Fig. 2A). Resorcinol staining of TLC of extracted lipids demonstrated that GM1 was the predominant ganglioside detected in cell extracts and that all detectable gangliosides were decreased in CFTR-silenced cells compared with controls (data not shown). GM1 detected by resorcinol was decreased by ∼60% in CFTR-silenced cells compared with controls (Fig. 2B). Analysis by TLC and CtxB-HRP staining confirmed the depletion of GM1 in CFTR-deficient cells (Fig. 2D).
Fig. 2.
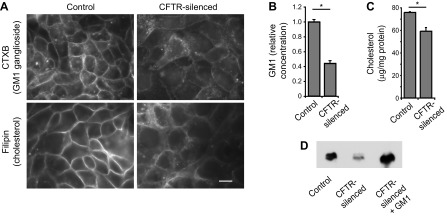
Reduced GM1 ganglioside and cholesterol in CFTR-silenced cells. A: confluent monolayers of control and CFTR-silenced cells were labeled with cholera toxin B subunit (CtxB) to detect GM1 ganglioside (top) or filipin to detect unesterified cholesterol (bottom). Images were processed identically for the two micrographs shown for each fluorophore so that intensities can be compared. Note decreased overall filipin and CtxB labeling and increased punctate fluorescence in CFTR-silenced cells. Bar, 10 μm. B: GM1 levels in cells were measured biochemically by lipid extraction followed by thin layer chromatography (TLC), detection of gangliosides with resorcinol, and quantitation by image analysis. GM1 was identified by comparison to known standards. Results are expressed relative to control levels. C: total unesterified cholesterol levels in cells were measured biochemically using a cholesterol oxidase-based assay. In B and C, values are means ± SE for 3 samples. *P < 0.05, significantly different in two-tailed t-tests. D: control, CFTR-silenced cells, and CFTR-silenced cells treated for 2 days with 5 μM GM1 ganglioside were extracted, and lipids were separated by TLC. GM1 was detected by CtxB-horseradish peroxidase staining of TLC plates.
We investigated the distribution of unesterified cholesterol in CFTR-silenced and control cells using filipin. In control cells, filipin predominantly stained the plasma membrane; however, in CFTR-silenced cells, plasma membrane staining was decreased and more filipin staining was observed on intracellular structures (Fig. 2A). Biochemical analysis showed that unesterified cholesterol was decreased by ∼20% in CFTR-silenced cells compared with controls (Fig. 2C).
CFTR regulates GM1 levels.
We performed a series of experiments to determine whether the low level of GM1 detected in CFTR-silenced cells compared with control cells was due to the loss of CFTR expression rather than an artifact of cell line selection. We infected CFTR-silenced cells with an adenoviral vector that expresses wild-type GFP-CFTR and has previously been shown to restore Cl− transport in CFTR-deficient cells (16). When examined by confocal microscopy, GFP-CFTR-infected cells exhibited higher levels of GM1 than silenced cells without GFP-CFTR, as detected by fluorescent-CtxB staining (Fig. 3A). GM1 (CtxB) and GFP-CFTR intensities were measured by image analysis in randomly chosen individual cells from a series of micrographs. We found that GM1 fluorescence intensity of individual cells was significantly positively correlated with GFP-CFTR levels (Fig. 3B). When data were grouped by GFP-CFTR intensity into low (25% of cells), intermediate (50% of cells), and high (25% of cells) GFP-CFTR intensity, those cells with the high GFP-CFTR intensity had significantly higher GM1 levels than either the intermediate or low categories (Fig. 3C).
Fig. 3.

Regulation of GM1 levels by CFTR. CFTR-silenced cells were infected with green fluorescent protein (GFP)-CFTR adenovirus for 3 days. A–C: cells cultured on coverslips were fixed, stained with fluorescent CtxB to determine GM1 distribution, and observed by confocal microscopy. A: micrographs show the distribution of GFP-CFTR and GM1 and DAPI (to identify nuclei) in a field of cells. Bar, 10 μm. Note that the cell with highest GFP-CFTR level also has the strongest signal for GM1. B: fluorescence intensities for GFP-CFTR and CtxB (GM1) of individual cells from several micrographs were measured by image analysis and plotted. n = 64 cells. GM1 and GFP-CFTR levels were significantly correlated (Pearson product-moment R = 0.6942, P < 0.0001). C: using data in B, cells were categorized on the basis of GFP-CFTR intensity into low (lowest 25% of cells), intermediate (middle 50% of cells), and high (25% of cells with the highest GFP-CFTR intensity). Levels of GFP-CFTR and CtxB (GM1) in these 3 groups are compared in the bar graph. GM1 levels were significantly different between each group in two-tailed t-tests, as indicated by P values. D: lysates of control, CFTR-silenced cells, and CFTR-silenced cells infected with GFP-CFTR adenovirus were immunoblotted with antibodies against CFTR and GFP to show the expression of GFP-CFTR in adenovirus-infected CFTR-silenced cells. Equal protein quantities (40 μg) were loaded per lane. E: CFTR-silenced cells and CFTR-silenced cells infected with GFP-CFTR were extracted and analyzed for GM1 by TLC and CtxB staining as in Fig. 2D. Cell lysates equivalent to 1 mg protein were extracted for each lane. Two replicates for each condition are shown. A GM1 standard is also shown. F: control cells were treated with vehicle or with 30 μM CFTRinh-172 for 2 days and analyzed for GM1 levels as in E. Two replicates for each condition are shown.
We also examined the effects of CFTR expression and activity on GM1 levels using biochemical methods. GFP-CFTR expression in CFTR-silenced cells was detectable on western blots using both GFP and CFTR antibodies (Fig. 3D). GFP-CFTR expression in CFTR-silenced cells increased GM1 levels compared with CFTR-silenced cells not expressing GFP-CFTR, as determined by TLC with CtxB staining (Fig. 3E). Finally, inhibition of CFTR function in control cells (with endogenous CFTR) using the pharmacologic inhibitor CFTRinh-172 decreased GM1 levels (Fig. 3F). Together, the results in Fig. 3 support the concept that CFTR function is required to maintain normal levels of GM1 in Calu-3 cells.
Alterations in sphingolipid metabolism in CFTR-silenced cells.
Studies were performed to try to understand the mechanisms underlying the depletion of gangliosides in CFTR-silenced cells. We examined the possibility that ceramide levels are altered in CFTR-silenced cells, since several reports have demonstrated changes in ceramide in CFTR-deficient cells and mouse models (21, 57, 60). Using mass spectrophotometry to detect individual ceramide species, we found that CFTR-silenced cells had significantly higher levels of several ceramide species, the most prominent being C16 and C24:1 ceramide (Fig. 4A). CFTR-silenced cells exhibited a 70% increase in total ceramide (all species combined) compared with normal cells (Fig. 4B). The findings that ceramide levels are elevated in CFTR-silenced cells (Fig. 4, A and B) suggest that it is unlikely that the decrease in gangliosides is due to decreased amounts of the precursor, ceramide.
Fig. 4.

Ceramide levels and sphingolipid metabolism in control and CFTR-silenced cells. A: ceramide species and metabolites. Samples of cell lysates (100 μg protein) were analyzed by mass spectrophotometry. Values are means ± SE of 3 replicates for each sample. Sphingosine-1-phosphate levels were also assessed but were undetectable. B: total ceramide levels. Individual ceramide species values from replicates in A were summed and are expressed as means ± SE. C: activities of glucosylceramide synthase and sphingomyelin synthase were tested in cell lysates using C6-NBD-ceramide as a substrate. Values are means ± SE of 3 replicates per sample and are expressed relative to control values. D: BODIPY-GM1 degradation. Cells were incubated with BODIPY-GM1 for 2, 24, or 48 h. Lipids were then extracted from cell samples and extracts were analyzed by TLC using BODIPY-sphingolipid standards. Metabolites produced from BODIPY-GM1 included BODIPY-GM3, BODIPY-ceramide, and BODIPY-sphingomyelin. BODIPY-GM1 remaining is expressed as a percentage of total BODIPY-lipids detected for each sample. Values are means ± SE of 3 replicates. *P < 0.05 and **P < 0.01, significant differences in t-tests comparing values for control and CFTR-silenced cells.
We next investigated the activity of glucosylceramide synthase, which catalyzes the first glycosylation reaction leading to the formation of higher glycosphingolipids such as GM1 (31). For this assay, we incubated cell lysates with C6-NBD-ceramide, a fluorescent ceramide analog, for 1 h. However, we found no difference in glucosylceramide synthase activity between control and CFTR-silenced cells (Fig. 4C). This same assay allowed the detection of sphingomyelin synthase activity, which was also not significantly different in CFTR-silenced cells (Fig. 4C).
Finally, we incubated cells with BODIPY-GM1 for up to 2 days to assess the possibility that ganglioside degradation might be altered in CFTR-silenced cells. We found that CFTR-silenced cells exhibited a small (∼20%) but significant increase in BODIPY-GM1 degradation compared with controls (Fig. 4D). These results suggest that the loss of endogenous gangliosides in CFTR-silenced cells is not due to decreased biosynthetic activity at an early step in ganglioside synthesis but may in part result from an increase in ganglioside catabolism.
β1-Integrin signaling is reduced in CFTR-silenced cells and restored by GM1 addition.
We and others have shown that gangliosides are able to regulate the activation of β1-integrins (35, 50, 51, 55, 61). Since altered integrin signaling could play a role in the reduced migration observed in CFTR-silenced cells, we investigated β1-integrin activation and signaling in these cells. Total and plasma membrane levels of β1-integrin were similar in control and CFTR-silenced cells as determined by immunoblotting (Fig. 5A). β1-Integrin activation was measured by the binding to cells of HUTS-4, an antibody that is specific for the activated conformation of β1-integrin. HUTS-4-immunolabeled cells exhibited punctate fluorescent structures, which may indicate focal complexes containing activated β1-integrin, as well as diffusely fluorescent membranes (Fig. 5B). We quantitated the HUTS-4 signal associated with individual cells by image analysis. By this measure, β1-integrin activation was depressed >50% in CFTR-silenced cells compared with controls (Fig. 5, B and C). To determine whether this loss of activation was due to decreased gangliosides, CFTR-silenced cells were preincubated with 5 μM GM1 for 4 days. This treatment caused an increase of GM1 in CFTR-silenced cells to levels similar to those observed in controls (data not shown). Incubation of CFTR-silenced cells with GM1 restored β1-integrin activation to a level that was not significantly different from control cells (Fig. 5, B and C).
Fig. 5.

CFTR silencing reduces β1-integrin activation. Control and CFTR-silenced cells were cultured for 2 days in serum-free media. CFTR-silenced cells were also cultured in the presence of 5 μM GM1 as indicated. A: plasma membrane and total levels of β1-integrin as assessed by immunoblotting. Live control and CFTR-silenced cells were incubated with activated biotin reagent for 30 min at 4°C to label plasma membrane proteins. Cell lysates were prepared, and biotinylated proteins were captured using streptavidin-coated beads. Samples of biotinylated proteins recovered from beads and total lysates were then analyzed for β1-integrin by immunoblotting. B: β1-Integrin activation in control and CFTR-silenced cells was assessed by immunofluorescence using the HUTS-4 antibody, which only recognizes activated β1-integrin. Two examples are shown of control, CFTR-silenced, and CFTR-silenced + GM1 cells. Within each horizontal set, each image was processed identically so that intensities can be compared. Note that HUTS-4 stained diffuse membrane as well as punctate structures. Bar, 10 μm. C: quantitation of activated β1-integrin levels by image analysis of HUTS-4 staining as shown in B. Values were normalized to control levels and are means ± SE of 10–15 micrographs containing ≥20 cells per micrograph for each condition. *P < 0.01, significant differences between indicated groups as calculated using one-way ANOVA with a Bonferroni post test.
We next investigated the possibility that signaling downstream from β1-integrin is also disrupted in CFTR-silenced cells. Upon integrin activation, FAK becomes autophosphorylated at tyrosine 397 (Y397). Y397 phosphorylation of FAK induces the binding of src family kinases to FAK, which then causes the phosphorylation of FAK at tyrosine 576 and 577 by src, at which point FAK is fully activated (67). We found that FAK phosphorylation at Y397 and Y576 was reduced by 26 and 43%, respectively, in CFTR-silenced cells compared with controls (Fig. 6, A–C). Total FAK levels were similar in control and CFTR-silenced cells (Fig. 6A). Treatment of CFTR-silenced cells with GM1 significantly increased FAK Y397 and Y576 phosphorylation compared with untreated CFTR-silenced cells and restored FAK Y397 and Y576 phosphorylation to control levels (Fig. 6, A–C). In contrast, treatment of CFTR-silenced cells with ganglioside GM3 had no significant effect on FAK phosphorylation (Fig. 6, A–C). Incubation of control cells with GM1 did not alter pFAK576 levels (data not shown). Another downstream event of integrin signaling is the phosphorylation of CAS by src kinases (23, 67). We found that tyrosine-phosphorylated CAS (pY-CAS) levels were inhibited (∼40%) in CFTR-silenced cells compared with controls (Fig. 6, A and D). Similar to our findings with FAK, pY-CAS levels were restored by GM1 but not by GM3 (Fig. 6, A and D). Since we also found that CFTR-silenced cells had elevated ceramide levels, we investigated the possibility that this excess ceramide was interfering with integrin signaling. However, decreasing ceramide in CFTR-silenced cells by inhibition of acid sphingomyelinase with amitriptylene (20 or 60 μM for 1 h), which decreased total ceramide levels (from 170% of controls in CFTR-silenced cells to 130% of controls in CFTR-silenced cells treated with amitriptylene), did not affect pFAK levels (data not shown).
Fig. 6.

Focal adhesion kinase (FAK) and Crk-associated substrate (CAS) phosphorylation is reduced in CFTR silencing and restored by GM1. Control and CFTR-silenced cells were cultured in serum-free medium for 2 days. In separate samples, CFTR-silenced cells were cultured over this time with 5 μM GM1 or GM3. Cell lysates were collected and immunoblotted for total FAK, Y397, and Y576 phospho-FAK, β-actin, and total CAS. Equal protein quantities (20 μg) were loaded per lane. Phosphorylated CAS (pY-CAS) was detected by immunoprecipitating with a phospho-tyrosine antibody and probing with an antibody against CAS. A: representative immunoblots. B–D: quantitation of pY397-FAK (B), pY576-FAK (C), and pY-CAS (D). Signals on immunoblots were quantified by image analysis. Ratios of pY-FAK/total FAK and pY-CAS/total CAS were calculated for each replicate sample. Values were normalized by dividing the phospho-/total ratios for each replicate by (the sum of phospho-/total protein ratios for controls, CFTR-silenced, and CFTR-silenced + GM1 from the same blot pair)/3. Results are means ± SE of ≥3 replicates per condition. *P < 0.01 and **P < 0.001, significant differences between indicated groups as calculated using one-way ANOVA with a Bonferroni post test.
GM1 partially restores migration in CFTR-silenced cells.
Since incubation of CFTR-silenced cells with GM1 was able to restore β1-integrin signaling, we tested the possibility that GM1 would stimulate migration after wounding in CFTR-silenced cells. We found that GM1 addition led to a significantly increased rate of migration from a T ½ of 12 h in CFTR-silenced cells to a T ½ of 9 h in CFTR-silenced cells plus GM1, but not to the rate (T ½ = 4 h) observed in controls (Fig. 7). No effect on migration was observed when CFTR-silenced cells were incubated with GM3 ganglioside instead of GM1 (Fig. 7D).
Fig. 7.
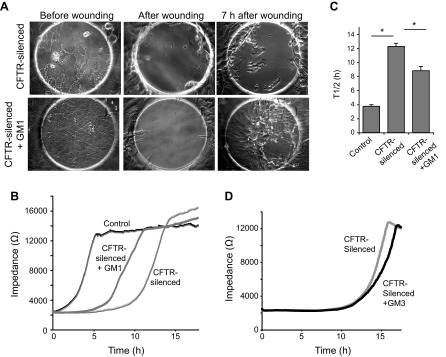
Stimulation of cell migration in CFTR-silenced cells by GM1. Migration of CFTR-silenced and control cells was measured on ECIS arrays as in Fig. 1. A: micrographs of CFTR-silenced cells cultured for 4 days ± 5 μM GM1. Cells are shown before wounding, immediately after electrical wounding, and 7 h after wounding. B: impedance over time traces for cells on ECIS arrays. Note partial recovery of migration in CFTR-silenced cells cultured with GM1. C: T ½ values for recovery of impedance after cell wounding. Values are means ± SE of 4 replicates. *P < 0.01, significantly different in two-tailed t-tests. D: impedance tracing of CFTR-silenced cells pretreated for 4 days ± 5 μM GM3 before wounding. Unlike GM1, GM3 did not stimulate recovery after wounding.
DISCUSSION
We investigated the hypothesis that delayed migration observed in CFTR-silenced cells was a result of altered sphingolipid composition. We found that CFTR-silenced cells had a 60% decrease in the glycosphingolipid GM1, reduced β1-integrin activation, and decreased phosphorylation of FAK and CAS. Addition of GM1 to CFTR-silenced cells was able to restore β1-integrin activation and phosphorylation of FAK and CAS to control levels. GM1 supplementation also enhanced migration of CFTR-silenced cells, although it did not fully restore the migration rate to the levels seen in controls. These findings suggest that a reduction in GM1 in CFTR-silenced cells contributes to delayed wound repair in these cells via inhibition of β1-integrin activation and signaling.
Our study agrees with several previous reports documenting alterations in sphingolipid composition in CFTR-deficient cells. Our finding of decreased GM1 in CFTR-silenced cells is consistent with studies demonstrating lower GM1 in CFTR-deficient cells compared with normal cells (1, 12, 13). Although a few studies have reported lower levels of ceramide in serum of CF patients and in serum and whole tissue preparations from CFTR-knockout mice compared with controls (17, 18), others have found higher ceramide levels in epithelial cells from patient samples (8, 60), cultured cells with silenced CFTR or expressing mutant CFTR (21), and in cells and tissue from CFTR-knockout mice (4, 57, 69) compared with appropriate controls. Our results are consistent with the latter reports, as we found a significant increase in ceramide levels in CFTR-silenced cells compared with controls. Elevations of ceramide in CF models have been linked to increased inflammation and cell death in CFTR-deficient cells (2, 57, 60). In our studies, however, decreasing ceramide levels using an inhibitor of acid sphingomyelinase did not restore pFAK to control levels, suggesting that elevated ceramide may not play a role in the suppressed β1-integrin signaling that we observed in CFTR-silenced cells. Instead, we found that supplementation of CFTR-silenced cells with GM1 was able to restore integrin and FAK signaling to control levels and partially restored cell migration. Taken together, these results suggest that different pathological phenotypes (e.g., decreased migration, inflammation) observed in CFTR-deficient cells might be linked to different alterations in sphingolipids.
We demonstrated that reduced GM1 in CFTR-silenced cells was a result of loss of CFTR function, as reexpression of GFP-CFTR in CFTR-silenced cells significantly increased GM1 levels, and pharmacologic inhibition of CFTR in control cells decreased GM1 (Fig. 3). Similar reductions in GM1 have been previously reported for CF cells and cells overexpressing ΔF508-CFTR (1, 12, 13); however, the mechanisms underlying this alteration in gangliosides have never been identified. Possible reasons for a decrease in GM1 in CFTR-silenced cells are decreased synthesis or increased degradation of GM1. We found that the levels of ceramide species are elevated by ∼70% in CFTR-silenced cells. Thus, it is not likely that the reduction in gangliosides is due to a shortage of ceramide required for glycosphingolipid synthesis. No loss in glucosylceramide synthase activity was detected in CFTR-silenced cells, suggesting that there is no block in the initial glycosylation step of glycosphingolipid synthesis. Previous studies have reported that in CFTR-deficient cells, reduced GM1 is accompanied by an increased expression of asialo-GM1 (13, 44), suggesting defects in the terminal (sialic acid) glycosylation of gangliosides. A similar defect in the terminal glycosylation of proteins is reported in cystic fibrosis mutant cells vs. wild-type counterparts (15, 42). However, investigations have not detected any changes in the overall expression of the glycosyltransferases involved in terminal glycosylation in cystic fibrosis mutant cells (15, 42, 53). Thus, it has been proposed that the loss of function of the CFTR protein may lead to an alteration in the organization of the Golgi apparatus, which interferes with the normal function of glycosyltransferases resident in different regions of the Golgi (42). Support for this hypothesis comes from a study showing that Golgi stacks are disrupted in cystic fibrosis mutant cells and that this defect is corrected by expression of wild-type CFTR (25).
It is also possible that lysosomal function is altered with the loss of CFTR function. Our finding of increased BODIPY-GM1 degradation in CFTR-silenced cells (Fig. 4D) suggests possible changes in lysosomal organization or activity in these cells, since GM1 and other gangliosides are degraded mainly in the lysosomes (45). Similarly, the activity of lysosomal enzymes involved in sphingolipid degradation is reported to be altered in CFTR-deficient cells, leading to increased ceramide levels (2, 57). Thus, CFTR-deficient cells have been observed to exhibit defects in biosynthetic steps (e.g., terminal glycosylation) in the Golgi apparatus and alterations in catabolic activity (e.g., sphingolipid and ganglioside degradation) in the lysosomes. Both of these metabolic alterations could contribute to the observed reductions in GM1 that we observed in CFTR-silenced cells. Together, all of these observations point to a possible generalized defect in membrane trafficking in CFTR-deficient cells. One hypothesis put forth to explain these defects is that the CFTR protein contributes to the regulation of pH in acidic organelles such as the Golgi and lysosomes (1, 69). Since both the Golgi apparatus and lysosomes require establishment of acidic luminal pH for normal function (30, 65), abnormal regulation of pH might disrupt the functions of these organelles. Much evidence exists supporting the idea that CFTR is involved in Golgi and lysosomal pH regulation (1, 3, 11, 57, 69); however, other studies have called into question this concept (14, 19). A second possible mechanism for altered organelle organization in CFTR mutant or knockout cells is that CFTR might affect the trafficking machinery directly. CFTR has been shown to interact with some proteins that regulate endocytosis and membrane traffic (56, 66). Thus, it is possible that in cells that normally express CFTR, loss of CFTR could disrupt membrane trafficking machinery, leading to defects in Golgi and lysosomal function. Further work is needed to determine the potential role of CFTR in the regulation of membrane trafficking.
Many studies have demonstrated the ability of gangliosides to regulate integrin activation and downstream signaling, as well as cell migration (22, 24, 47, 58, 62). The modification of integrin function by gangliosides has been demonstrated to occur by several mechanisms, including alteration of the organization of integrins in plasma membrane microdomains and modulation of the interactions between integrins and other proteins, e.g., growth factor receptors (41, 50, 51, 58, 62, 64). Here, we show for the first time that CFTR silencing leads to decreased integrin signaling that is a result of reduced GM1. Cell surface levels of total β1-integrin were unaltered in CFTR-silenced cells compared with controls; however, activated β1-integrin was significantly decreased (Fig. 5). Addition of GM1 to CFTR-silenced cells could restore β1-integrin activation. Although not further investigated, immunofluorescence images of CFTR-silenced cells treated with GM1 appear to have increased punctate structures containing activated β1-integrin (Fig. 5B). Thus, it is possible that the depletion of GM1 from CFTR-silenced cells leads to loss of β1-integrin localization to microdomains, and hence decreased integrin activation. Addition of GM1 to CFTR-silenced cells may restore the organization of integrin-containing microdomains, thus increasing integrin activation.
Although β1-integrin activation in CFTR-silenced cells could be fully returned to control levels by GM1 addition, cell migration after wounding was only partially restored by GM1. This result suggests that other factors besides GM1 concentration may be important for the restoration of wound repair function in these cells. For example, other glycosphingolipids besides GM1 may be needed to optimally restore migration. Unlike GM1, GM3 addition was unable to restore FAK signaling or wound repair in CFTR-silenced cells. These findings may be related to previously observed differences in the effects of specific gangliosides on integrins and cell migration (39, 63, 64). Other gangliosides (e.g., GM2, GD3) or combinations of gangliosides might promote integrin signaling or cell migration after wounding in CFTR-silenced cells, but we have not tested these possibilities.
Other determinants (e.g., other signaling cascades) that are not controlled by ganglioside levels also may contribute to defective integrin signaling and migration in CFTR-silenced cells. Recently, Sun et al. (54) found that airway epithelial cell migration in response to wounding was dependent on intrinsically generated electrical currents that required CFTR. Thus, it is likely that CFTR channel activity plays a major role in migration and that the stimulation of integrins by GM1 cannot completely overcome this requirement.
Finally, our study may have relevance for the behavior of CFTR-deficient cells in human patients. Some early work suggests that glycosphingolipid composition is altered in CF patients (44, 52); however, this area has not been well studied. Owing to persistent inflammation and bacterial infections, epithelial cells of CF patient lungs are challenged with the need for continuous repair (27, 48). Thus, the possibility that altered glycosphingolipid composition and integrin signaling inhibit epithelial cell repair in these patients warrants further investigation. In conclusion, our studies strongly support a role for altered glycosphingolipids contributing to reduced cellular migration in cells deficient in CFTR function.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants GM60934 (to R. E. Pagano and A. H. Limper) and HL-095811 (to S. M. O'Grady), and the Mayo Foundation. Ceramide analysis was performed by the Mayo Metabolomics Core Facility and was supported by NIH/National Center for Research Resources/Center for Translational Science Award Grant UL1 RR024150.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.I., R.E.P., A.S.S., S.M.O., A.H.L., and D.L.M. conception and design of research; Y.I. and A.S.S. performed experiments; Y.I., S.M.O., and D.L.M. analyzed data; Y.I., A.H.L., and D.L.M. interpreted results of experiments; Y.I. and D.L.M. prepared figures; Y.I., S.M.O., A.H.L., and D.L.M. edited and revised manuscript; Y.I., A.S.S., S.M.O., A.H.L., and D.L.M. approved final version of manuscript; D.L.M. drafted manuscript.
Footnotes
This article is the topic of an Editorial Focus by Jada C. Domingue and Mrinalini C. Rao (11a).
REFERENCES
- 1.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature 352: 70–73, 1991 [DOI] [PubMed] [Google Scholar]
- 2.Becker KA, Riethmuller J, Luth A, Doring G, Kleuser B, Gulbins E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol 42: 716–724, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Becker KA, Riethmuller J, Zhang Y, Gulbins E. The role of sphingolipids and ceramide in pulmonary inflammation in cystic fibrosis. Open Respir Med J 4: 39–47, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker KA, Tummler B, Gulbins E, Grassme H. Accumulation of ceramide in the trachea and intestine of cystic fibrosis mice causes inflammation and cell death. Biochem Biophys Res Commun 403: 368–374, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Belcher CN, Vij N. Protein processing and inflammatory signaling in cystic fibrosis: challenges and therapeutic strategies. Curr Mol Med 10: 82–94, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhaumik SK, Singh MK, Karmakar S, De T. Immuno stimulating glycophosphosphingolipid antigen from Leishmania donovani is recognized by visceral leishmaniasis patient sera. Mol Biochem Parasitol 159: 121–129, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39: 82–91, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Brodlie M, McKean MC, Johnson GE, Gray J, Fisher AJ, Corris PA, Lordan JL, Ward C. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am J Respir Crit Care Med 182: 369–375, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Chen CS, Bach G, Pagano RE. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci USA 95: 6373–6378, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng ZJ, Singh RD, Marks DL, Pagano RE. Membrane microdomains, caveolae, and caveolar endocytosis of sphingolipids. Mol Membr Biol 23: 101–110, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8: 933–944, 2006 [DOI] [PubMed] [Google Scholar]
- 11a.Domingue JC, Rao MC. CFTR and GM1 “gangl-ing” up to heal thy wound. Focus on “Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased β1-integrin signaling and delayed wound repair.” Am J Physiol Cell Physiol (March 13, 2014). 10.1152/ajpcell.00075.2014 [DOI] [PubMed] [Google Scholar]
- 12.Dosanjh A, Lencer W, Brown D, Ausiello DA, Stow JL. Heterologous expression of ΔF508 CFTR results in decreased sialylation of membrane glycoconjugates. Am J Physiol Cell Physiol 266: C360–C366, 1994 [DOI] [PubMed] [Google Scholar]
- 13.Dosanjh A, Muchmore EA. Expression of DeltaF508 cystic fibrosis transmembrane regulator (CFTR) decreases membrane sialylation. Open Respir Med J 3: 79–84, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn KW, Park J, Semrad CE, Gelman DL, Shevell T, McGraw TE. Regulation of endocytic trafficking and acidification are independent of the cystic fibrosis transmembrane regulator. J Biol Chem 269: 5336–5345, 1994 [PubMed] [Google Scholar]
- 15.Glick MC, Kothari VA, Liu A, Stoykova LI, Scanlin TF. Activity of fucosyltransferases and altered glycosylation in cystic fibrosis airway epithelial cells. Biochimie 83: 743–747, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Granio O, Norez C, Ashbourne Excoffon KJ, Karp PH, Lusky M, Becq F, Boulanger P, Zabner J, Hong SS. Cellular localization and activity of Ad-delivered GFP-CFTR in airway epithelial and tracheal cells. Am J Respir Cell Mol Biol 37: 631–639, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Guilbault C, De Sanctis JB, Wojewodka G, Saeed Z, Lachance C, Skinner TA, Vilela RM, Kubow S, Lands LC, Hajduch M, Matouk E, Radzioch D. Fenretinide corrects newly found ceramide deficiency in cystic fibrosis. Am J Respir Cell Mol Biol 38: 47–56, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Guilbault C, Wojewodka G, Saeed Z, Hajduch M, Matouk E, De Sanctis JB, Radzioch D. Cystic fibrosis fatty acid imbalance is linked to ceramide deficiency and corrected by fenretinide. Am J Respir Cell Mol Biol 41: 100–106, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Haggie PM, Verkman AS. Defective organellar acidification as a cause of cystic fibrosis lung disease: reexamination of a recurring hypothesis. Am J Physiol Lung Cell Mol Physiol 296: L859–L867, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hajj R, Lesimple P, Nawrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J Pathol 211: 340–350, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Hamai H, Keyserman F, Quittell LM, Worgall TS. Defective CFTR increases synthesis and mass of sphingolipids that modulate membrane composition and lipid signaling. J Lipid Res 50: 1101–1108, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamamura K, Tsuji M, Ohkawa Y, Nakashima H, Miyazaki S, Urano T, Yamamoto N, Ueda M, Furukawa K. Focal adhesion kinase as well as p130Cas and paxillin is crucially involved in the enhanced malignant properties under expression of ganglioside GD3 in melanoma cells. Biochim Biophys Acta 1780: 513–519, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem 271: 13649–13655, 1996 [DOI] [PubMed] [Google Scholar]
- 24.Hashiramoto A, Mizukami H, Yamashita T. Ganglioside GM3 promotes cell migration by regulating MAPK and c-Fos/AP-1. Oncogene 25: 3948–3955, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Hollande E, Salvador-Cartier C, Alvarez L, Fanjul M. Expression of a wild-type CFTR maintains the integrity of the biosynthetic/secretory pathway in human cystic fibrosis pancreatic duct cells. J Histochem Cytochem 53: 1539–1552, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacquot J, Tabary O, Clement A. Hyperinflammation in airways of cystic fibrosis patients: what's new? Expert Rev Mol Diagn 8: 359–363, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Kirk KL. CFTR channels and wound healing. Focus on “Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair.” Am J Physiol Cell Physiol 299: C888–C890, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Ledeen RW, Yu RK. Gangliosides: structure, isolation, and analysis. Methods Enzymol 83: 139–191, 1982 [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Yan S, Wondimu A, Bob D, Weiss M, Sliwinski K, Villar J, Notario V, Sutherland M, Colberg-Poley AM, Ladisch S. Ganglioside synthase knockout in oncogene-transformed fibroblasts depletes gangliosides and impairs tumor growth. Oncogene 29: 3297–3306, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luzio JP, Rous BA, Bright NA, Pryor PR, Mullock BM, Piper RC. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci 113: 1515–1524, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Maccioni HJ, Quiroga R, Ferrari ML. Cellular and molecular biology of glycosphingolipid glycosylation. J Neurochem 117: 589–602, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Maille E, Trinh NT, Prive A, Bilodeau C, Bissonnette E, Grandvaux N, Brochiero E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-α after injury. Am J Physiol Lung Cell Mol Physiol 301: L945–L955, 2011 [DOI] [PubMed] [Google Scholar]
- 33.Marks DL, Paul P, Kamisaka Y, Pagano RE. Methods for studying glucosylceramide synthase. Methods Enzymol 311: 50–59, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Martin OC, Pagano RE. Internalization and sorting of a fluorescent analog of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanisms. J Cell Biol 125: 769–781, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miura Y, Kainuma M, Jiang H, Velasco H, Vogt PK, Hakomori S. Reversion of the Jun-induced oncogenic phenotype by enhanced synthesis of sialosyllactosylceramide (GM3 ganglioside). Proc Natl Acad Sci USA 101: 16204–16209, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mogayzel PJ, Jr, Flume PA. Update in cystic fibrosis 2009. Am J Respir Crit Care Med 181: 539–544, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Noe J, Petrusca D, Rush N, Deng P, VanDemark M, Berdyshev E, Gu Y, Smith P, Schweitzer K, Pilewsky J, Natarajan V, Xu Z, Obukhov AG, Petrache I. CFTR regulation of intracellular pH and ceramides is required for lung endothelial cell apoptosis. Am J Respir Cell Mol Biol 41: 314–323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohkawa Y, Miyazaki S, Hamamura K, Kambe M, Miyata M, Tajima O, Ohmi Y, Yamauchi Y, Furukawa K. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J Biol Chem 285: 27213–27223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono M, Handa K, Sonnino S, Withers DA, Nagai H, Hakomori S. GM3 ganglioside inhibits CD9-facilitated haptotactic cell motility: coexpression of GM3 and CD9 is essential in the downregulation of tumor cell motility and malignancy. Biochemistry 40: 6414–6421, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Puri V, Jefferson JR, Singh RD, Wheatley CL, Marks DL, Pagano RE. Sphingolipid storage induces accumulation of intracellular cholesterol by stimulating SREBP-1 cleavage. J Biol Chem 278: 20961–20970, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Regina Todeschini A, Hakomori SI. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta 1780: 421–433, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhim AD, Stoykova LI, Trindade AJ, Glick MC, Scanlin TF. Altered terminal glycosylation and the pathophysiology of CF lung disease. J Cyst Fibros 3, Suppl 2: 95–96, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Rottner M, Freyssinet JM, Martinez MC. Mechanisms of the noxious inflammatory cycle in cystic fibrosis. Respir Res 10: 23, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest 92: 1875–1880, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci 33: 10195–10208, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schiller KR, Maniak PJ, O'Grady SM. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am J Physiol Cell Physiol 299: C912–C921, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shibuya H, Hamamura K, Hotta H, Matsumoto Y, Nishida Y, Hattori H, Furukawa K, Ueda M. Enhancement of malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci 103: 1656–1664, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shute J, Marshall L, Bodey K, Bush A. Growth factors in cystic fibrosis - when more is not enough. Paediatr Respir Rev 4: 120–127, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 11: 688–699, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Singh RD, Holicky EL, Cheng Z, Kim SY, Wheatley CL, Marks DL, Bittman R, Pagano RE. Inhibition of caveolar uptake, SV40 infection, and β1-integrin signaling by a nonnatural glycosphingolipid stereoisomer. J Cell Biol 176: 895–901, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh RD, Marks DL, Holicky EL, Wheatley CL, Kaptzan T, Sato SB, Kobayashi T, Ling K, Pagano RE. Gangliosides and β1-integrin are required for caveolae and membrane domains. Traffic 11: 348–360, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slomiany A, Murty VL, Aono M, Snyder CE, Herp A, Slomiany BL. Lipid composition of tracheobronchial secretions from normal individuals and patients with cystic fibrosis. Biochim Biophys Acta 710: 106–111, 1982 [DOI] [PubMed] [Google Scholar]
- 53.Stoykova LI, Liu A, Scanlin TF, Glick MC. α1,3Fucosyltransferases in cystic fibrosis airway epithelial cells. Biochimie 85: 363–367, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Sun YH, Reid B, Fontaine JH, Miller LA, Hyde DM, Mogilner A, Zhao M. Airway epithelial wounds in rhesus monkey generate ionic currents that guide cell migration to promote healing. J Appl Physiol 111: 1031–1041, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sung CC, O'Toole EA, Lannutti BJ, Hunt J, O'Gorman M, Woodley DT, Paller AS. Integrin α5β1 expression is required for inhibition of keratinocyte migration by ganglioside GT1b. Exp Cell Res 239: 311–319, 1998 [DOI] [PubMed] [Google Scholar]
- 56.Tang BL, Gee HY, Lee MG. The cystic fibrosis transmembrane conductance regulator's expanding SNARE interactome. Traffic 12: 364–371, 2011 [DOI] [PubMed] [Google Scholar]
- 57.Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, Gulbins E. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14: 382–391, 2008 [DOI] [PubMed] [Google Scholar]
- 58.Toledo MS, Suzuki E, Handa K, Hakomori S. Effect of ganglioside and tetraspanins in microdomains on interaction of integrins with fibroblast growth factor receptor. J Biol Chem 280: 16227–16234, 2005 [DOI] [PubMed] [Google Scholar]
- 59.Trinh NT, Bardou O, Prive A, Maille E, Adam D, Lingee S, Ferraro P, Desrosiers MY, Coraux C, Brochiero E. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur Respir J 40: 1390–1400, 2012 [DOI] [PubMed] [Google Scholar]
- 60.Ulrich M, Worlitzsch D, Viglio S, Siegmann N, Iadarola P, Shute JK, Geiser M, Pier GB, Friedel G, Barr ML, Schuster A, Meyer KC, Ratjen F, Bjarnsholt T, Gulbins E, Doring G. Alveolar inflammation in cystic fibrosis. J Cyst Fibros 9: 217–227, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valentino LA, Ladisch S. Tumor gangliosides enhance α2β1 integrin-dependent platelet activation. Biochim Biophys Acta 1316: 19–28, 1996 [DOI] [PubMed] [Google Scholar]
- 62.Van Slambrouck S, Hilkens J, Bisoffi M, Steelant WF. AsialoGM1 and integrin α2β1 mediate prostate cancer progression. Int J Oncol 35: 693–699, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang XQ, Sun P, Paller AS. Ganglioside GM3 inhibits matrix metalloproteinase-9 activation and disrupts its association with integrin. J Biol Chem 278: 25591–25599, 2003 [DOI] [PubMed] [Google Scholar]
- 64.Wang XQ, Sun P, Paller AS. Gangliosides inhibit urokinase-type plasminogen activator (uPA)-dependent squamous carcinoma cell migration by preventing uPA receptor/αβ integrin/epidermal growth factor receptor interactions. J Invest Dermatol 124: 839–848, 2005 [DOI] [PubMed] [Google Scholar]
- 65.Weisz OA. Acidification and protein traffic. Int Rev Cytol 226: 259–319, 2003 [DOI] [PubMed] [Google Scholar]
- 66.Weixel KM, Bradbury NA. Endocytic adaptor complexes bind the C-terminal domain of CFTR. Pflügers Arch 443, Suppl 1: S70–S74, 2001 [DOI] [PubMed] [Google Scholar]
- 67.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692: 103–119, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Xu Y, Krause A, Hamai H, Harvey BG, Worgall TS, Worgall S. Proinflammatory phenotype and increased caveolin-1 in alveolar macrophages with silenced CFTR mRNA. PLos One 5: e11004, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Li X, Grassme H, Doring G, Gulbins E. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J Immunol 184: 5104–5111, 2010 [DOI] [PubMed] [Google Scholar]