

Abstract
Objective
The current pathological diagnostic criteria for sporadic inclusion body myositis (IBM) lack sensitivity. Using immunohistochemical techniques abnormal protein aggregates have been identified in IBM, including some associated with neurodegenerative disorders. Our objective was to investigate the diagnostic utility of a number of markers of protein aggregates together with mitochondrial and inflammatory changes in IBM.
Design
Retrospective cohort study. The sensitivity of pathological features was evaluated in cases of Griggs definite IBM. The diagnostic potential of the most reliable features was then assessed in clinically typical IBM with rimmed vacuoles (n=15), clinically typical IBM without rimmed vacuoles (n=9) and IBM mimics—protein accumulation myopathies containing rimmed vacuoles (n=7) and steroid-responsive inflammatory myopathies (n=11).
Setting
Specialist muscle services at the John Radcliffe Hospital, Oxford and the National Hospital for Neurology and Neurosurgery, London.
Results
Individual pathological features, in isolation, lacked sensitivity and specificity. However, the morphology and distribution of p62 aggregates in IBM were characteristic and in a myopathy with rimmed vacuoles, the combination of characteristic p62 aggregates and increased sarcolemmal and internal major histocompatibility complex class I expression or endomysial T cells were diagnostic for IBM with a sensitivity of 93% and specificity of 100%. In an inflammatory myopathy lacking rimmed vacuoles, the presence of mitochondrial changes was 100% sensitive and 73% specific for IBM; characteristic p62 aggregates were specific (91%), but lacked sensitivity (44%).
Conclusions
We propose an easily applied diagnostic algorithm for the pathological diagnosis of IBM. Additionally our findings support the hypothesis that many of the pathological features considered typical of IBM develop later in the disease, explaining their poor sensitivity at disease presentation and emphasising the need for revised pathological criteria to supplement the clinical criteria in the diagnosis of IBM.
Keywords: Neurology
Strengths and limitations of this study.
The present study is a multicentre retrospective evaluation of the diagnostic utility of pathological findings for differentiating inclusion body myositis (IBM) from myopathies important in the differential diagnosis—protein accumulation myopathies containing rimmed vacuoles and steroid-responsive inflammatory myopathies.
The main strength of our study was the systematic detailed analysis of well-defined cases. This enabled us to determine the sensitivity and specificity of individual pathological features and produce an easily applied pathological diagnostic algorithm for IBM for use in clinical practice.
Study limitations include the small number of cases and the retrospective design. Further prospective studies are now required in larger cohorts of patients.
Introduction
Sporadic inclusion body myositis (IBM) is the commonest acquired myopathy in those aged over 50 years.1 Although classified as an idiopathic inflammatory myopathy, muscle biopsy reveals both degenerative and inflammatory features. The widely used Griggs diagnostic criteria require the presence of several pathological findings,2 namely rimmed vacuoles, an inflammatory infiltrate with invasion of non-necrotic fibres by mononuclear inflammatory cells (partial invasion), and either amyloid deposits or 15–18 nm tubulofilaments identified by electron microscopy (EM), for a diagnosis of definite IBM. Although these features in combination are highly specific for IBM, individually they occur in other myopathies, including some important in the differential diagnosis for IBM.3–7 Moreover, cases of clinically typical IBM have been reported where the combination of these pathological features is absent causing diagnostic difficulty.8–11
Over the past two decades, pathological accumulation of many different proteins has been reported in muscle fibres in IBM.12 Proteins typically associated with neurodegenerative diseases such as β-amyloid (Aβ), hyperphosphorylated tau and ubiquitin and newer neurodegenerative markers such as p62 and transactivation response DNA binding protein-43 (TDP-43) have been identified, as well as proteins associated with myofibrillar myopathies (MFM), including desmin and αB-crystallin. However, not all observations have been consistently reproduced.13 14 Mitochondrial changes have also been proposed for inclusion in IBM diagnostic criteria.15 Clear guidelines for the incorporation of immunohistochemical findings and mitochondrial changes into diagnostic criteria for IBM have not been established.16
Previously, we have shown that the characteristic pattern of weakness associated with IBM is indicative of the diagnosis, even if the Griggs pathological features are absent.11 However, it is not invariably found at presentation. Here we sought to identify which pathological features, other than those included in the Griggs pathological criteria, add further support to the diagnosis of IBM. We systematically investigated which pathological features are present in Griggs pathologically definite IBM and then established the diagnostic utility of these features in cases of IBM lacking pathological features included in the Griggs criteria, using myopathies considered in the differential diagnosis of IBM as controls.
Materials and methods
Cases
All patients were followed by specialist muscle services at the John Radcliffe Hospital, Oxford and the National Hospital for Neurology and Neurosurgery, London. Biopsies were taken for diagnostic purposes from the deltoid or quadriceps muscles and prior to any treatment.
Methods for demonstrating pathological features in IBM, additional to those defined by the Griggs criteria, were determined in six Griggs pathologically definite cases of IBM. Cases with no clinical or pathological evidence of neuromuscular disease were used as controls. The diagnostic utility of the pathological features identified was assessed in two groups of clinically typical IBM; one with rimmed vacuoles on muscle biopsy (IBM+RV; n=15), the other without rimmed vacuoles on muscle biopsy (IBM − RV; n=9). Disease controls were cases of steroid-responsive inflammatory myopathies (polymyositis and dermatomyositis; (PM&DM); n=11) and protein accumulation myopathies with rimmed vacuoles (PAM; n=7). Clinical characteristics and inclusion criteria are summarised in online supplementary tables S1 and S2. Tissue from brains donated to the Queen Square Brain Bank for Neurological Disorders was used as positive controls for protein aggregate staining.
Muscle biopsies
Muscles biopsies were snap frozen at the time of surgery in isopentane cooled liquid nitrogen. Until sectioning all samples were stored at −80°C. Serial tissue sections were cut to a thickness of 8 µm, allowed to air dry and stored at −80°C until staining. Prior to staining, tissue sections were allowed to dry at room temperature. Tissue sections were stained with haematoxylin and eosin (H&E), combined cytochrome oxidase (COX) succinate dehydrogenase (SDH) histochemistry and for amyloid using alkalinised Congo red, crystal violet and thioflavin S. Tissue sections for immunohistochemical staining were fixed for 10 min, if required, washed for 5 min in running water and incubated in 0.5% hydrogen peroxide to block endogenous peroxidase for 20 min. After further washing, tissue sections were incubated in 5% normal goat serum (Vector Laboratories, Burlingame, California, USA) for 30 min and then systematically stained for: (1) proteins classically associated with neurodegenerative disease: tau and hyperphosphorylated tau, ubiquitin, Aβ and α-synuclein; (2) proteins recently reported in neurodegenerative disease: p62, TDP-43, fused in sarcoma protein (FUS) and valosin containing protein (VCP); (3) nuclear membrane proteins: lamin A/C and emerin; (4) proteins associated with MFM: desmin, myotilin and αB-crystallin and (5) inflammatory cells and major histocompatibility complex class I (MHC class I): CD3 T cells, CD4 T cells, CD8 T cells, B cells and macrophages. Primary antibody binding was visualised using Dako REAL EnVision Detection System which contains horse-radish peroxidase (HRP) labelled goat anti-rabbit/mouse secondary and 3,3′-diaminobenzidine (DAB); following incubation with the relevant primary antibody, tissue sections were washed in phosphate-buffered saline (PBS), incubated with HRP labelled goat anti-rabbit/mouse secondary for 30 min, washed in PBS and incubated in a 1:50 solution of DAB for 3–5 min. Details of commercial antibodies and conditions used are provided in online supplementary table S3. Immunohistochemistry (IHC) for each antibody was performed on all cases simultaneously including positive and negative controls (see online supplementary figure S1).
Definitions and quantification
The total number of fibres and the number undergoing partial invasion, containing rimmed vacuoles, protein aggregates and COX-negative SDH-positive (COX−/SDH+) fibres were quantified using ImagePro V.6.2 (Media Cybernetics), to ensure that the whole biopsy was systematically analysed. Only transversely orientated fibres not undergoing necrosis or regeneration were quantified. Tissue sections stained with Congo red were visualised under fluorescent and polarised light. Areas of fluorescence were examined using both rhodamine red (excitation 512–546 nm and emission 600–640 nm) and fluorescein isothiocyanate (excitation 440–480 nm and emission 527–530 nm) filters to exclude autofluorescence. Online supplementary table S4 provides definitions of the pathological features assessed. The inflammatory infiltrate and MHC class I staining were analysed using a modified version of the semiquantitative juvenile dermatomyositis score tool (see online supplementary figure S2).17 Assessments were performed blind to clinical details and diagnosis by a single individual (SB). Ten per cent of slides were recounted to assess intra-observer reliability and 336 slides were assessed independently by two observers (SB and JLH) to determine inter-observer reliability.
Statistical analysis
Statistical analyses were performed using GraphPad PRISM V.5. Continuous and categorical variables were compared using Mann-Whitney U test and χ2 or Fisher's exact test, respectively. Spearman's rank order correlation was used to determine the strength and direction of associations between pathological findings. Linear regression was used to determine relationships between clinical features and pathological findings. Test characteristics were calculated using receiver operating characteristic (ROC) curves and 2×2 contingency tables. A test was considered diagnostic when sensitivity >75% and specificity >95% or sensitivity >95% and specificity >75%. Intra-observer and inter-observer agreement was calculated using Bland-Altman plots and Cohen's κ statistic (κ). Repeat counts were within 95% confidence intervals using Bland-Altman plots and κ was ≥0.7 indicating good intra-observer and good or excellent inter-observer reliability. Statistical significance was set at p<0.05.
Results
Pathological findings in Griggs pathologically definite IBM
p62, TDP-43, ubiquitin, myotilin and αB-crystallin immunoreactive aggregates were present in all six IBM cases but not in normal controls (figure 1A–E). p62 and αB-crystallin immunoreactive aggregates were present in a greater percentage of fibres than the pathological features required in the Griggs criteria (p<0.05; figure 2). Despite their abundance, αB-crystallin immunoreactive aggregates were difficult to quantify due to a significant variability in their morphology. No immunoreactive deposits were observed in IBM cases or normal controls with antibodies to tau and phosphorylated tau, Aβ, α-synuclein, desmin, emerin, lamin A/C, FUS or VCP. Alkalinised Congo red staining was more sensitive than crystal violet and thioflavin S staining for observing amyloid aggregates (figure 1F). Tissue sections containing congophilic deposits identified under fluorescence light showed no apple-green birefringence under polarised light. Mitochondrial changes and increased sarcolemmal and sarcoplasmic MHC class I staining were observed in all six IBM cases, but not in normal controls. The inflammatory infiltrate was predominantly composed of endomysial CD8 T cells and macrophages, with relatively few B cells.
Figure 1 .

Protein aggregates and congophilic deposits in IBM. Stained cryostat sections, showing fibres, often in clusters, containing protein aggregates stained for p62 (A), TDP-43 (B), ubiquitin (C), αB-crystallin (D) and myotilin (E). Protein aggregates were present throughout fibres, and were observed in apparently normal fibres, vacuolated fibres and fibres surrounded by inflammatory infiltrates. In fibres containing TDP-43 aggregates, myonuclear TDP-43 staining was frequently reduced (B). Congophilic deposits were observed in vacuolated fibres using epifluorescence (F). Tissue sections were examined using both the rhodamine red and fluorescein isothiocyanate filters to exclude areas of autofluorescence (arrow). Combined fluorescent image is shown. Four patterns of immunoreactivity were observed in IBM and disease controls stained for p62 using IHC (G–J. Pattern I (G)—strongly stained, discreet and clearly delineated, round or angular aggregates, variable in number and size within a muscle fibre but rarely filling it and predominantly located subsarcolemmal, but also perinuclear and adjacent to vacuoles. Pattern II (H)—large aggregates of variable staining intensity. Pattern III (I)—fine granular aggregates dispersed throughout the fibre. Pattern IV (J)—fine granules and wisps of p62 immunoreactivity set within weakly basophilic inclusions. Scale bar represents 50 µm in A and D; 25 µm in B, C , E and F; 20 μm in G and H; 15 μm in I; and 10 μm in J. IBM, inclusion body myositis; IHC, immunohistochemistry; TDP-43, transactivation response DNA binding protein-43.
Figure 2 .
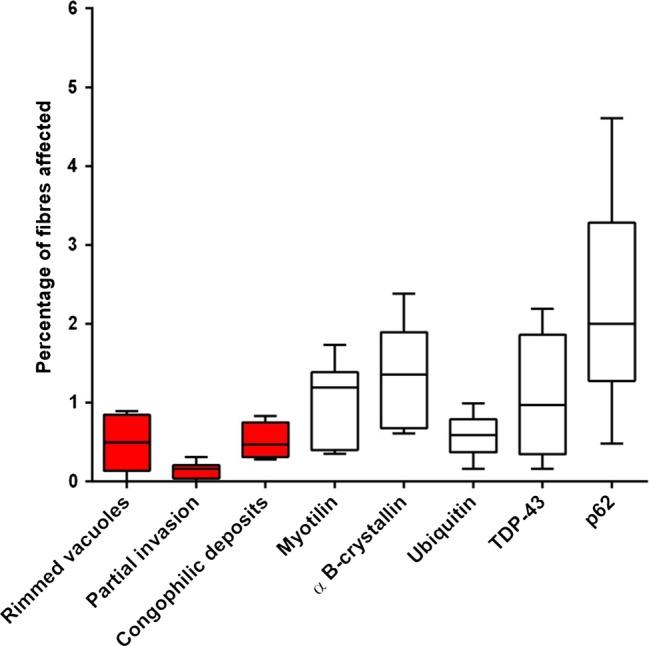
Box and whisker plot illustrating the percentage of muscle fibres containing pathological abnormalities included in the Griggs criteria and protein aggregates in Griggs pathologically definite inclusion body myositis (IBM). Fibres containing aggregates immunoreactive for p62 and αB-crystallin were more frequent than those containing the current diagnostic pathological features (red bars; p<0.05). Protein aggregates recognised by all antibodies were found in a significantly larger number of fibres than partial invasion (p<0.02). TDP-43, transactivation response DNA binding protein-43.
Quantitative analysis of pathological features in IBM and disease controls
Having shown that p62, TDP-43, ubiquitin and myotilin aggregates, congophilic deposits, MHC class I and inflammatory cells were prevalent in Griggs pathologically definite IBM, the presence of these abnormalities, together with mitochondrial changes were assessed in IBM+RV, IBM − RV and disease controls.
The percentage of fibres containing p62, TDP-43, myotilin and ubiquitin aggregates and congophilic deposits were greater in IBM+RV than in IBM − RV; there was no difference in the number of COX−/SDH+ fibres (figure 3A–F). Protein aggregates were observed in morphologically normal fibres and in fibres exhibiting pathological features included in the Griggs criteria. p62 and TDP-43 positive aggregates were present in a greater percentage of fibres in IBM+RV compared to PAM; however, there were no differences in the percentage of fibres containing myotilin and ubiquitin aggregates or congophilic deposits. The percentage of fibres containing p62, TDP-43 and ubiquitin aggregates or congophilic deposits were similar in IBM − RV and PM&DM; however, myotilin aggregates were present in a greater percentage of fibres in PM&DM and COX−/SDH+ fibres were more abundant in IBM − RV. Analysis of the total inflammatory infiltrate (the sum of the semiquantitative scores for T cells, B cells and macrophages) in the endomysium, perimysium and perivascular areas revealed that there were greater numbers of inflammatory cells in the endomysium and perimysium in IBM+RV than in PAM (p<0.03). The same analysis comparing the sum of the inflammatory cells in IBM − RV and PM&DM revealed that the distribution and intensity of the inflammatory infiltrate was similar.
Figure 3 .

Box and whisker plots illustrating the percentage of fibres in each diagnostic category containing p62 (A), TDP-43 (B), myotilin (C) and ubiquitin (D) immunoreactive aggregates, congophilic deposits (E) and COX−/SDH+ fibres (F). All protein aggregates were present in a greater percentage of fibres in IBM+RV than in IBM - RV. There was no difference in the percentage of COX−/SDH+ muscle fibres between these groups. IBM+RV biopsies had a greater percentage of fibres containing p62 (A) and TDP-43 (B) immunoreactive aggregates and COX−/SDH+ fibres (F) than PAM. Pathological findings were similar in IBM - RV and PM&DM, with no differences in the percentage of fibres containing p62 (A), TDP-43 (B) and ubiquitin (D) immunoreactive aggregates or congophilic deposits (E). However, there was a greater percentage of COX−/SDH+ fibres (F) in IBM - RV than PM&DM and a greater percentage of fibres containing myotilin immunoreactive aggregates (C) in PM&DM than IBM - RV. *Statistically significant results. COX, cytochrome oxidase; IBM, inclusion body myositis; PAM, protein accumulation myopathies with rimmed vacuoles; PM&DM, steroid-responsive inflammatory myopathies; SDH, succinate dehydrogenase; TDP-43, transactivation response DNA binding protein-43.
Diagnostic utility of pathological features in IBM and disease controls
To mimic the diagnostic difficulty encountered in clinical practice, the ability of each test to differentiate between myopathies containing rimmed vacuoles (IBM+RV and PAM) and between inflammatory myopathies (IBM − RV and PM&DM) was assessed.
Diagnostic utility determined using ROC curves
Individually, the presence of p62 immunoreactive inclusions and COX−/SDH+ fibres had the highest sensitivity and specificity for differentiating IBM+RV from PAM, (see online supplementary figure S3 and table 1). Differentiating between IBM − RV and PM&DM, myotilin positive inclusions and COX−/SDH+ fibres had the highest sensitivity and specificity (see online supplementary figure S4 and table 1). Only the presence of myotilin positive inclusions satisfied criteria to be considered suitable as a diagnostic test (<0.01% of fibres containing myotilin aggregates had a sensitivity of 100% and specificity of 82% for IBM − RV).
Table 1.
Test characteristics
| IBM+RV vs PAM |
IBM - RV vs PM&DM |
|||||||
|---|---|---|---|---|---|---|---|---|
| Test feature | AUC | Cut-off (% of affected fibres) | Sensitivity | Specificity | AUC | Cut-off (% of affected fibres) | Sensitivity | Specificity |
| Rimmed vacuoles | 0.60 | >0.28 | 0.53 | 0.71 | – | – | – | – |
| p62 aggregates | 0.87 | >0.48 | 0.87 | 0.86 | 0.60 | >0.21 | 0.22 | 0.91 |
| TDP-43 aggregates | 0.80 | >0.34 | 0.80 | 0.86 | 0.53 | <0.01 | 0.89 | 0.18 |
| Ubiquitin aggregates | 0.68 | >0.18 | 0.53 | 0.86 | 0.64 | <0.01 | 1.00 | 0.27 |
| Myotilin aggregates | 0.55 | <2.50 | 1.00 | 0.29 | 0.91 | <0.01 | 1.00 | 0.82 |
| Congophilic deposits | 0.56 | >0.24 | 0.73 | 0.71 | 0.56 | >0.03 | 0.11 | 1.00 |
| COX−/SDH+ fibres | 0.87 | >0.04 | 0.86 | 0.86 | 0.93 | >0.1 | 0.78 | 0.91 |
AUC, area under the curve; COX, cytochrome oxidase; IBM, inclusion body myositis; PAM, protein accumulation myopathies with rimmed vacuoles; PM&DM, steroid-responsive inflammatory myopathies; RV, rimmed vacuoles; SDH, succinate dehydrogenase; TDP-43, transactivation response DNA binding protein-43.
Diagnostic utility determined by comparing proportion of affected cases in each diagnostic group
In the aforementioned experiments, the number of fibres within each muscle biopsy was quantified. However, this is impractical for routine clinical use. Thus, the proportions of affected cases in each group were compared (table 2). This revealed that neither staining for protein aggregates nor congophilic deposits could differentiate between IBM+RV and PAM. The pathological findings in IBM − RV and PM&DM were also similar, except that the absence of myotilin immunoreactive aggregates was sensitive and specific for IBM − RV. COX−/SDH+ fibres were also suggestive of IBM − RV; one or more COX−/SDH+ fibres had a sensitivity of 100% and a specificity of 73% for IBM − RV.
Table 2.
Comparison of the proportion of positive cases in each group
| Pathological features | IBM+RV | PAM | IBM+RV vs PAM |
IBM - RV | PM&DM | IBM - RV vs PM&DM |
IBM+RV vs IBM - RV | ||
|---|---|---|---|---|---|---|---|---|---|
| n (%) | n (%) | Sensitivity | Specificity | n (%) | n (%) | Sensitivity | Specificity | p value | |
| Number of cases | 15 (100) | 7 (100) | 9 (100) | 11 (100) | |||||
| Aggregated proteins, n (%) | |||||||||
| p62 | 15 (100) | 6 (86) | 1.00 | 0.14 | 4 (44) | 3 (27)* | 0.44 | 0.73 | 0.003† |
| TDP-43 | 13 (87) | 5 (71) | 0.87 | 0.29 | 1 (11) | 2 (18)* | 0.11 | 0.82 | 0.001† |
| Ubiquitin | 11 (73) | 4 (57) | 0.73 | 0.43 | 0 (0) | 3 (27)* | 0.00 | 0.73 | 0.001† |
| Myotilin | 10 (67) | 5 (71) | 0.67 | 0.29 | 0 (0) | 9 (82) | 0.00 | 0.18 | 0.002† |
| Congophilic deposits | 13 (87) | 7 (100) | 0.87 | 0.00 | 1 (11) | 0 (0) | 0.11 | 1.00 | 0.001† |
| COX−/SDH+ fibres‡, n (%) | |||||||||
| Any | 12 (86) | 2 (29) | 0.86 | 0.71 | 9 (100) | 3 (27) | 1.00 | 0.73 | 0.50 |
| Inflammatory features, n (%) | |||||||||
| MHC class I upregulation | 15 (100) | 3 (43) | 1.00 | 0.57 | 7 (78) | 11 (100) | 0.78 | 0.00 | 0.13 |
| Strong MHC class I upregulation | 14 (93) | 0 (0) | 0.93 | 1.00 | 9 (100) | 10 (91) | 1.00 | 0.09 | 1.00 |
| Partial invasion | 10 (67) | 0 (0) | 0.67 | 1.00 | 3 (33) | 2 (18) | 0.33 | 0.82 | 0.21 |
| Endomysial CD3 T-cell score >1 | 13 (87) | 0 (0) | 0.87 | 1.00 | 4 (44) | 7 (64) | 0.44 | 0.36 | 0.06 |
| Endomysial CD4 T-cell score >1 | 12 (80) | 0 (0) | 0.80 | 1.00 | 2 (22) | 5 (45) | 0.22 | 0.54 | 0.01† |
| Endomysial CD8 T-cell score >0 | 14 (93) | 0 (0) | 0.93 | 1.00 | 7 (78) | 9 (82) | 0.78 | 0.18 | 0.53 |
| Endomysial CD68 macrophage score >1 | 12 (80) | 0 (0) | 0.80 | 1.00 | 4 (44) | 8 (73) | 0.44 | 0.27 | 0.10 |
*Pathological features present in DM, but not PM cases.
†Statistically significant results.
‡In IBM with rimmed vacuoles n=14.
COX, cytochrome oxidase; IBM, inclusion body myositis; MHC, major histocompatibility complex; PAM, protein accumulation myopathies with rimmed vacuoles; PM&DM, steroid-responsive inflammatory myopathies; RV, rimmed vacuoles; SDH, succinate dehydrogenase; TDP-43, transactivation response DNA binding protein-43.
Increased MHC class I expression lacked specificity. However, strong (diffuse sarcolemmal and sarcoplasmic) MHC class I upregulation was diagnostic for IBM+RV, differentiating it from PAM, as were the presence of either endomysial CD3 T-cell, CD4 T-cell, or CD68 macrophage scores >1 or an endomysial CD8 T-cell score >0. Partial invasion was specific for IBM+RV, but lacked sensitivity. Although the sum of the inflammatory infiltrate was similar in IBM − RV and PM&DM, analysis of the inflammatory cell subtypes revealed greater numbers of perimysial CD3 T cells, CD8 T cells and endomysial B cells (were observed) in PM&DM than in IBM − RV (p≤0.02), however, this was not diagnostically useful. There was no difference in the proportion of cases with fibres undergoing partial invasion between IBM − RV and PM&DM.
As IBM − RV is more pathologically akin to PM than DM, analyses were repeated comparing IBM − RV and PM cases (n=6). No p62, TDP-43 or ubiquitin immunoreactive aggregates were observed in PM cases and the diagnostic utility of tests for differentiating between IBM − RV and PM yielded similar results to prior analyses between IBM − RV and PM&DM.
Diagnostic utility of categorising the pattern of p62 staining
The pattern of p62 staining could be categorised into four distinct groups (figure 1G–J). Aggregates observed in IBM were present in vacuolated and non-vacuolated fibres and were strongly stained, discreet and clearly delineated, round or angular and typically located subsarcolemmal, perinuclear and perivacuolar (pattern I). This pattern was observed in every IBM case with p62 aggregates, one (9%) case of DM and three (43%) cases of PAM (hereditary inclusion body myopathy, dystrophinopathy and genetically undefined MFM). Defining the pattern of immunoreactivity increased the discriminative value of p62 IHC for differentiating IBM+RV from PAM; pattern I p62 aggregates compared to any p62 aggregates increased the specificity from 14% to 57%, with no loss of sensitivity. Differentiating IBM − RV and PM&DM, pattern I p62 aggregates were highly specific (91%), but lacked sensitivity (44%). Patterns II, III and IV were not observed in any IBM cases. Patterns II and III appeared to be specific for PAM (n=2; 26%), both were cases of myotilinopathy (n=2; 67%), and DM (n=2; 40%), respectively. Pattern IV occurred in a genetically undefined case of MFM. No differences were observed in the morphology of TDP-43, myotilin or ubiquitin aggregates between biopsies.
Clinicopathological correlation
In IBM+RV, IBM − RV and pathologically definite IBM, there were no correlations in individual biopsies between pathological features. No relationships were identified between the pathological findings and age at symptom onset, age at biopsy, disease duration or serum creatine kinase. The same results were obtained when the IBM groups were analysed separately and as one.
Proposed diagnostic algorithm
On the basis of our pathological findings, we propose a diagnostic algorithm for differentiating IBM from its disease mimics (figure 4).
Figure 4 .

Proposed diagnostic algorithm for IBM based on pathological findings. Increased MHC class I staining was observed in all cases of IBM and pattern I p62 aggregates in all cases of IBM+RV making them good initial screening tests. Their absence rules out a diagnosis of IBM and IBM+RV, respectively. The presence of endomysial CD3 T-cell score >1, endomysial CD8 T-cell score >0 or strong MHC class I staining in a biopsy with rimmed vacuoles and pattern I p62 aggregates secures a diagnosis of IBM+RV. Differentiating IBM - RV and PM&DM pathologically is more challenging. The presence of COX−/SDH+ fibres is not specific to IBM - RV; although COX−/SDH+ fibres were not present in every case of IBM - RV their absence casts doubt on the diagnosis of IBM - RV. Pattern I p62 aggregates may enable IBM - RV to be differentiated from PM when present. However, they may lack sensitivity for IBM - RV and therefore, their absence does not rule out the diagnosis. COX, cytochrome oxidase; GT, Gomori trichrome; IBM, inclusion body myositis; MHC, major histocompatibility complex; PM&DM, steroid-responsive inflammatory myopathies; SDH, succinate dehydrogenase.
The algorithm was tested in a further 23 cases that fulfilled the criteria for IBM+RV (n=12) and IBM − RV (n=11). The algorithm correctly diagnosed 20 (87%) cases: 12 (100%) cases of IBM+RV and 8 (73%) cases of IBM − RV. In IBM − RV, COX−/SDH+ fibres were present in 8 (73%) cases, pattern I p62 aggregates in 8 (73%) cases and both in 6 (55%) cases.
Discussion
While the Griggs pathological criteria have been accepted as diagnostic of IBM, many patients who, observed over time undoubtedly have IBM, lack one or more of the Griggs pathological features at presentation, even on repeat biopsy.8 11 Despite IBM being associated with a characteristic pattern of finger flexor and knee extensor weakness, not all patients have this pattern at disease onset, and muscle biopsy remains an important tool in differentiating IBM from its mimics. We sought to determine which additional pathological features support a diagnosis of IBM, demonstrating that characteristic p62 immunoreactive aggregates, strong MHC class I upregulation, endomysial CD3 T-cell score >1, CD8 T-cell score >0 and COX−/SDH+ fibres are features with sufficient sensitivity and specificity to differentiate IBM from pathologically similar myopathies and we propose an easily applied pathological algorithm for the diagnosis of IBM (figure 4).
In agreement with previous studies, we observed p62,18 TDP-43,19 ubiquitin13 and αB-crystallin20 immunoreactive aggregates and a predominantly endomysial inflammatory infiltrate3 in Griggs pathologically definite IBM. Diagnostic pathological studies of IBM have concentrated on differentiating IBM from other inflammatory myopathies and two recent quantitative studies have found that TDP-43 and markers of autophagy such as p62 and LC3 may be of diagnostic use.21 22 However, in these studies only a fraction of each biopsy was analysed. We have found this limited quantification does not correlate with the percentage of affected fibres in a biopsy nor does it reflect the way in which a muscle biopsy is assessed. Additionally, studies have lacked protein accumulation myopathies with rimmed vacuoles control cases as it is believed that the inflammatory changes present in IBM enable it to be easily differentiated from other vacuolar myopathies.22 However, inflammatory changes are frequently observed in muscular dystrophies and the degree of inflammatory change necessary to confidently diagnose IBM is currently unknown.
To mimic the typical diagnostic conundrums encountered in clinical practice, we evaluated the ability of the pathological findings to differentiate IBM+RV from other vacuolar myopathies and IBM − RV from steroid-responsive inflammatory myopathies. We found that quantitative analysis of protein aggregates, congophilic deposits and COX−/SDH+ fibres was of limited diagnostic use. Analysing the biopsies dichotomously and using a semiquantitative score-tool to assess the inflammatory changes revealed that increased MHC class I labelling was sensitive for IBM making it a good initial screening test, its absence excluding the diagnosis. In agreement with an earlier study, we found that p62 aggregates identified the largest number of affected fibres in IBM.23 Additionally, as a novel finding, the morphology and distribution of p62 aggregates was characteristic in IBM. This characteristic pattern of p62 immunoreactive aggregates was highly sensitive for IBM+RV (100%); their absence from a biopsy containing rimmed vacuoles effectively ruling-out a diagnosis of IBM. We confirmed that the most diagnostically useful pathological findings in IBM+RV were evidence of an immune-mediated process; strong MHC class I staining, endomysial CD3 T-cell score >1 or an endomysial CD8 T-cell score >0 were diagnostic. Having identified any of these features in a biopsy containing rimmed vacuoles no extra diagnostic certainty was gained from observing partial invasion, COX−/SDH+ fibres or congophilic deposits.
The most discriminative pathological tests for differentiating between IBM − RV and PM&DM were COX/SDH staining and myotilin IHC. Consistent with a recent study,9 we found that the absence of mitochondrial changes casts doubt on a diagnosis of IBM. There was no difference in the median age between IBM − RV and PM&DM cases to account for the difference observed in COX−/SDH+ fibres. The presence of myotilin and ubiquitin immunoreactive aggregates appeared to rule out a diagnosis of IBM − RV. However, we believe that the presence of these features in IBM+RV indicates that they are unlikely to be diagnostically reliable features for differentiating between IBM − RV and steroid-responsive inflammatory myopathies. Although no pathological feature was able to differentiate IBM − RV from steroid responsive inflammatory myopathies with certainty the presence of characteristic p62 aggregates and the absence of COX−/SDH+ fibres may help in supporting and opposing a diagnosis of IBM − RV, respectively. Pattern I p62 immunoreactive aggregates were only present in 44% of the initial IBM − RV cases tested, but they were not observed in PM cases and were very rare in DM. Although pattern I p62 aggregates appear to lack sensitivity their specificity was 91% making their presence highly suggestive of a diagnosis of IBM − RV. However, we identified pattern I p62 aggregates in 8 of 11 (73%) further cases of IBM − RV that were assessed indicating that they may have a greater sensitivity and that p62 IHC warrants further investigation and validation in a larger, independent series. The diagnostic utility of the other patterns of p62 staining is uncertain. Although pattern II appeared to have some specificity for myotilinopathy the small number of cases makes drawing any conclusion problematic. In addition to p62 other autophagic proteins have been found in IBM and suggested as diagnostic markers.22 Autophagy is a cellular mechanism for degrading and recycling cellular proteins and organelles. Therefore, altered autophagy could lead to the accumulation of abnormal mitochondria and misfolded aggregation-prone proteins and may also result in altered antigen presentation leading to the widespread increase of MHC class I. The presence of mitochondrial changes, aggregated proteins and MHC class I upregulation in IBM suggests that altered autophagy may play an important role in the pathogenesis.
A number of pathological features—protein aggregates, congophilic deposits and some inflammatory changes—were more abundant in IBM+RV than IBM − RV. Despite using slightly different inclusion criteria, similar differences have been reported between pathologically typical and pathologically atypical IBM.21 However, we found no differences in the number of COX−/SDH+ fibres, the degree of MHC class I upregulation, the morphology and distribution of p62 immunoreactive aggregates or the pattern of the inflammation between IBM+RV and IBM − RV, supporting our clinical observations that these are the same disease. We believe that the pathological differences between IBM+RV and IBM − RV are, in part, due to differences in disease duration. Two studies have shown that rimmed vacuoles are more common in patients who are older at the time of muscle biopsy,11 24 suggesting that they are associated with chronologically more advanced disease. Therefore, the pathological findings which are more abundant in IBM+RV and thought to be typical of IBM may instead be indicative of chronologically more advanced disease explaining their limited sensitivity at disease presentation. However, possibly due to the number of cases analysed, we were unable to confirm a relationship between pathological features and clinical findings. It could be argued that biopsies from different muscles may have affected the pathological findings observed and differences between IBM groups. However, in a recent review of 59 muscle biopsies from IBM cases in our clinical archive with quadriceps (n=31) and deltoid (n=28) biopsies we found no significant difference in the frequency of pathological findings.
A robust clinicopathological definition of IBM is of paramount importance for diagnosis and for selection and entry of patients into clinical trials. We have shown that certain pathological findings are more abundant than those included in the current pathologically focused diagnostic criteria. Moreover, p62 immunoreactive deposits, increased MHC class I expression, endomysial CD3 T cells and CD8 T cells and COX−/SDH+ fibres have sufficient sensitivity and specificity to aid in the histological differentiation of IBM from disease mimics, supporting their inclusion in future diagnostic criteria for IBM alongside clinical criteria. Both CD3 T cells and CD8 T cells are included in the diagnostic algorithm as there was little difference in their sensitivity and specificity for differentiating IBM+RV from PAM. However, IHC staining for CD3 T cells is likely to be more widely available and avoids the costs of extra staining to subtype the inflammatory infiltrate enabling the diagnostic algorithm to be used by a greater number diagnostic laboratories. Using our diagnostic algorithm, we found that there would be little additional diagnostic security in identifying partial invasion, performing EM or staining for amyloid deposits. Finally, mitochondrial changes and MHC class I upregulation were the most consistent findings in our IBM cases suggesting that they are central to the pathogenesis and that further investigation and therapeutic intervention should be directed towards these features.
Supplementary Material
Acknowledgments
We acknowledge the Oxford Brain Bank, supported by the Medical Research Council (MRC), Brains for Dementia Research (BDR) and the NIHR Oxford Biomedical Research Centre.
Footnotes
Contributors: SB participated in acquisition of the data, analysis and interpretation of the data and drafting of the manuscript. WS participated in critical revision of the manuscript for important intellectual content. CS participated in study concept and design and critical revision of the manuscript for important intellectual content. MH and DH-J participated in critical revision of the manuscript for important intellectual content. JLH participated in study concept and design, critical revision of the manuscript for important intellectual content and study supervision.
Funding: SB is funded by the Myositis Support Group. JLH is supported by the Reta Lila Weston Institute for Neurological Studies and the Myositis Support Group. This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Competing interests: None.
Ethics approval: The study received ethical approval from the Departments of Research and Development at Oxford University Hospitals NHS Trust, Oxford and University College London Hospitals NHS Foundation Trust, London.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All additional data can be found in supplementary tables and figures.
References
- 1.Needham M, Corbett A, Day T, et al. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci 2008;15:1350–3 [DOI] [PubMed] [Google Scholar]
- 2.Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995;38:705–13 [DOI] [PubMed] [Google Scholar]
- 3.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol 1984;16:193–208 [DOI] [PubMed] [Google Scholar]
- 4.Mhiri C, Gherardi R. Inclusion body myositis in French patients. A clinicopathological evaluation. Neuropathol Appl Neurobiol 1990;16:333–44 [DOI] [PubMed] [Google Scholar]
- 5.Villanova M, Kawai M, Lübke U, et al. Rimmed vacuoles of inclusion body myositis and oculopharyngeal muscular dystrophy contain amyloid precursor protein and lysosomal markers. Brain Res 1993;603:343–7 [DOI] [PubMed] [Google Scholar]
- 6.Van der Meulen MF, Hoogendijk JE, Moons KG, et al. Rimmed vacuoles and the added value of SMI-31 staining in diagnosing sporadic inclusion body myositis. Neuromuscul Disord 2001;11:447–51 [DOI] [PubMed] [Google Scholar]
- 7.Ferrer I, Olivé M. Molecular pathology of myofibrillar myopathies. Expert Rev Mol Med 2008;10:e25. [DOI] [PubMed] [Google Scholar]
- 8.Amato AA, Gronseth GS, Jackson CE, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol 1996;40:581–6 [DOI] [PubMed] [Google Scholar]
- 9.Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008;70:418–24 [DOI] [PubMed] [Google Scholar]
- 10.Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain 2011;134:3176–84 [DOI] [PubMed] [Google Scholar]
- 11.Brady S, Squier W, Hilton-Jones D. Clinical assessment determines the diagnosis of inclusion body myositis independently of pathological features. J Neurol Neurosurg Psychiatry 2013;84:1240–6 [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SA. Theories of the pathogenesis of inclusion body myositis. Curr Rheumatol Rep 2010;12:221–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherriff FE, Joachim CL, Squier MV, et al. Ubiquitinated inclusions in inclusion-body myositis patients are immunoreactive for cathepsin D but not β-amyloid. Neurosci Lett 1995;194:37–40 [DOI] [PubMed] [Google Scholar]
- 14.Greenberg SA. How citation distortions create unfounded authority: analysis of a citation network. BMJ 2009;339:b2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007;6:620–31 [DOI] [PubMed] [Google Scholar]
- 16.Benveniste O, Hilton-Jones D. International workshop on inclusion body myositis held at the Institute of Myology, Paris, on 29 May 2009. Neuromuscul Disord 2010;20:414–21 [DOI] [PubMed] [Google Scholar]
- 17.Wedderburn LR, Varsani H, Li CKC, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthritis Rheum 2007;57:1192–201 [DOI] [PubMed] [Google Scholar]
- 18.Nogalska A, Terracciano C, D'Agostino C, et al. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 2009;118:407–13 [DOI] [PubMed] [Google Scholar]
- 19.Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 2008;79:1186–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banwell BL, Engel AG. Alpha B-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 2000;54:1033–41 [DOI] [PubMed] [Google Scholar]
- 21.Dubourg O, Wanschitz J, Maisonobe T, et al. Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol 2011;30:103–8 [PMC free article] [PubMed] [Google Scholar]
- 22.Hiniker A, Daniels BH, Lee HS, et al. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013; 1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Agostino C, Nogalska A, Engel WK, et al. In sporadic inclusion-body myositis muscle fibres TDP-43-positive inclusions are less frequent and robust than p62-inclusions, and are not associated with paired helical filaments. Neuropathol Appl Neurobiol 2010; http://www.ncbi.nlm.nih.gov/pubmed/20626631 [DOI] [PubMed] [Google Scholar]
- 24.Momma K, Noguchi S, Malicdan MCV, et al. Rimmed vacuoles in Becker muscular dystrophy have similar features with inclusion myopathies. PLoS ONE 2012;7:e52002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.