

Abstract
Evidence has accumulated that characterizes highly tumorigenic cancer cells residing in heterogeneous populations. The accepted term for such a subpopulation is cancer stem cells (CSC). While many questions still remain about their precise role in the origin, progression, and drug resistance of tumors, it is clear they exist. In this review, a current understanding of the nature of CSC, their potential usefulness in prognosis, and the need to target them will be discussed. In particular, separate studies now suggest that the CSC is plastic in its phenotype, toggling between tumorigenic and nontumorigenic states depending on both intrinsic and extrinsic conditions. Because of this, a static view of gene and protein levels defined by correlations may not be sufficient to either predict disease progression or aid in the discovery and development of drugs to molecular targets leading to cures. Quantitative dynamic modeling, a bottom up systems biology approach whereby signal transduction pathways are described by differential equations, may offer a novel means to overcome the challenges of oncology today. In conclusion, the complexity of cancer stem cells can be captured in mathematical models that may be useful for selecting molecular targets, defining drug action, and predicting sensitivity or resistance pathways for improved patient outcomes.
Keywords: cancer stem cells, prognosis, therapeutics, systems biology, plasticity, dynamic modeling, signal transduction pathways
Introduction
From 1994 to the present, there have been nearly 31800 scientific reports written about cancer stem cells (CSC) according to PubMed. Approximately 10% are reviews of some aspect of the field. For comparison, approximately 6000 scientific reports and reviews were written on the same topic in the previous 19 years. Therefore, it is an immense challenge to undertake a CSC review today, especially one evaluating prognosis and therapy. In order to contribute significantly to the swelling body of literature, this CSC review will peer into the topic with an emphasis on systems biology. This approach may bring the reader to the notion that the “devil is in the details”. Throughout this review, the reader will be directed to excellent ones already existing on the CSC topics addressed here.
The definition of systems biology can be as varied as the investigators who study it. To some, it is constructing networks from high-throughput data obtained from whole genome sequencing, gene expression assays, and proteomic and metabolomics studies and statistically analyzing the outcome for insight that is systems wide. Efforts are being made today to combine these vast databases in a meaningful way to understand disease in general and to discover new therapeutics. Thus, a global view of the parts list is an essential first step. But biology is dynamic and inferences made from these networks are limited. Therefore, a different perspective is offered here: quantitative dynamic modeling will be necessary to capture a changing landscape that is due to CSC. Evidence is building that CSCs are plastic in their phenotype, changing function dependent on intrinsic and extrinsic factors. If this state change can be modeled computationally and then tested, improved outcome for patients is possible.
Cancer Stem Cells - Brief History
In mid-1990’s, when John Dick and colleagues conducted a limiting dilution tumorigenicity assay with leukemic cells in immunocompromised mice and demonstrated that a single leukemic cell could recapitulate the original tumor, the cancer stem cell (CSC) concept of modern day began (1–3). History does show, though, that the concept of a highly tumorigenic cell that promotes progression was understood to exist for over 100 years. Following the initial study, CSCs were identified in solid human tumors of the breast, colon, brain, and prostate and in other histologies (4–8) (see (9) for an extensive list and details of those particular studies).
Many of the early controversies regarding CSCs centered on the definitions and misinterpretations of the very label; the use of “stem” seemed to suggest an origin for CSCs, the normal adult tissue stem cell, a concept also not well understood at the time. Later, many researchers concluded that the CSCs were solely responsible for all tumor heterogeneity via their ability to self-renew (as normal stem cells do) and to differentiate into nontumorigenic counterparts. At the time, little data supported this concept (10).
Today, the CSC concept is bolstered by many studies and the definition itself is being clarified (11) and limits it to malignant cells in neoplasia that maintain the tumor. The consensus workshop that offers this definition also relabels CSCs operationally as those cells that initiate tumors in xenografted immunocompromised mice utilizing a limiting dilution assay.
A number of exceptional recent reviews on CSCs provide details of their discovery in tumors (12), the methods utilized to isolate them (13,14) and how they might be targeted for therapy (9,15–17). In this review, I will emphasize the need to understand CSCs from a quantitative systems biology approach, from both the prognostic standpoint and the development of targeted therapy and its expected acquired drug resistance.
Laboratory Benchmarks of CSCs
Both molecular and functional assays characterize CSCs and include evaluation of selected surface antigens found on normal embryonic and adult tissue stem cells, clonogenic activity in soft agar, sphere formation in the presence of defined media and nonadherent conditions, and the limiting dilution tumorigenicity assay (Figure 1) (13,18). In some cases, the presence of an activated self-renewal pathway is thought to be important (19–21) and intrinsic drug and/or apoptosis resistance may also be a characteristic (22). It is critical to mention that molecular assays are not sufficient to define CSCs and functional studies are necessary with the limiting dilution tumorigenicity assays as the accepted standard (11).
Figure 1.
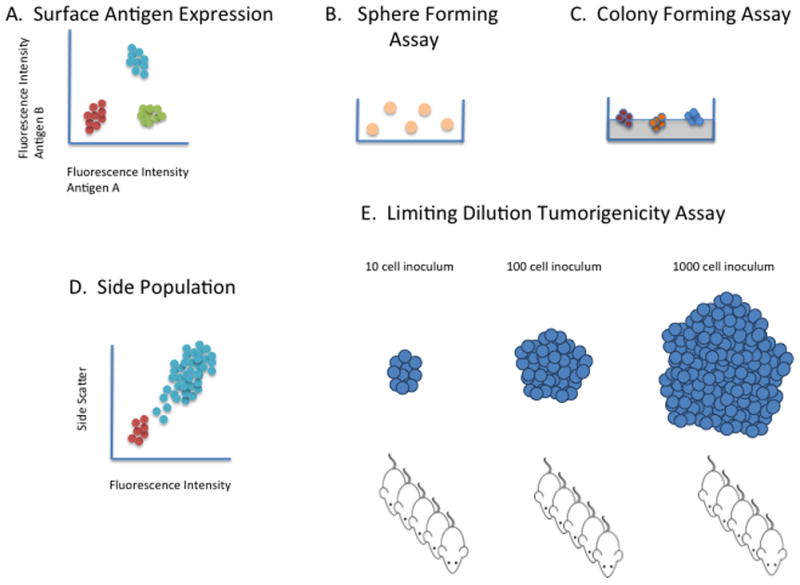
Laboratory Methods Evaluating CSCs. A. Surface Antigen Expression. A population of tumor cells is labeled with fluorescent antibody to antigens associated with stem-like activity. Frequently, two or more antigens may define CSC subpopulations (142). Common markers include CD44, CD24, CD133, and Epcam (CD326). B. Sphere Forming Assay. A population of tumor cells in placed in culture with defined media including certain growth factors (e.g. EGF, FGF) and no serum and sphere formation is evaluated. It is distinct from floating clusters of cells. If the spheres are aggregated and replated, propagation and reformation confirms the presence of CSCs (143). C. Colony Forming Assay. The ability to form colonies a clonal level in soft agar is a measure of tumorigenicity and therefore is considered useful in identifying CSC (144). It is thought that the cell division that occurs is frequently associated with differentiation and therefore, the larger the colony the less differentiated the initiating cell (CSC). D. Side Population Assay. Because CSC are thought to be intrinsically drug resistant, one measure in a population of cells is the ability to extrude drug through ABC transporters such as ABCB5 (145). Tumor cells are incubated with a Hoechst dye and the subpopulation without fluorescence, but above background levels are considered CSC. E. Limiting Dilution Tumorigenicity Assay. This is considered the gold standard assay for identifying CSC. As depicted, a population of tumor cells (presumably containing CSCs) is diluted many 10-fold logs prior to injection reaching levels as low as one to ten cells per mouse. At any given dilution, the fraction of mice possessing tumors can be used to calculate the ratio of CSCs (5). Immunocompromised mice are used and the site of injection may vary. Other factors also seem to vary the readout as the use of highly immunocompromised mice seems to improve tumor formation as does the addition of a commercially available extracellular matrix such as Matrigel (6).
Signal Transduction Pathways of CSC
Figure 2 provides an overview of relevant signal transduction pathways under study in the CSC field. The variety of receptors and ligands is formidable and the literature is replete with biochemical connections between any given pathway and therefore not complete. The emphasis of Figure 2 is on the output of any given signal transduction pathway, listed at the arrows. The output, too, is not complete, but this selected view is intended to depict crosstalk.
Figure 2.
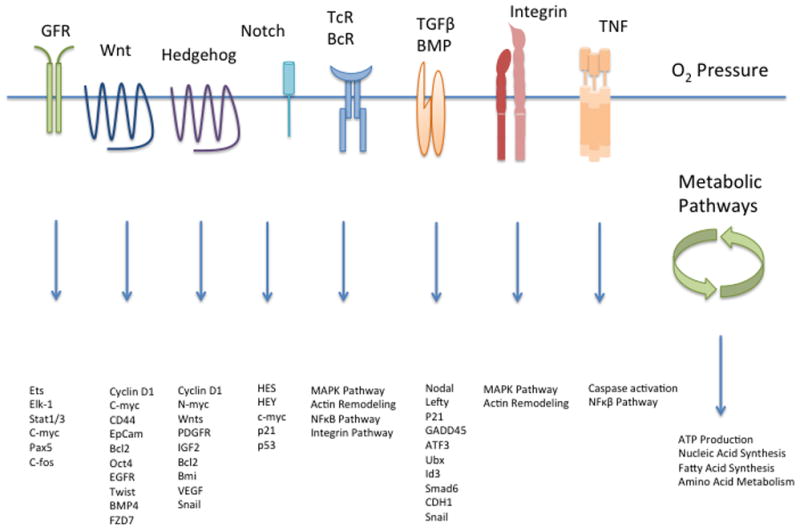
Signaling Pathways. A simplified overview of signal transduction pathways altered in CSCs. In the CSC literature, an emphasis has been placed on those with a role to play in normal development (Wnt, Hedgehog, Notch). Gene expression that follows stimulation or activation of a given receptor is shown (e.g., Wnt). In some instances, subsequent crosstalk with particular pathways is noted (TNF). Additionally, a tumor cell must produce energy and essential metabolites such as nucleotides, fatty acids, and amino acids as building blocks for maintenance. Oxygen gradients are also crucial and likely vary within any given region of a tumor. Growth factors, extracellular matrix and hormones are not depicted, but provide a context in which the signal transduction pathways act.
Because it seemed likely that CSCs derive from normal adult tissue stem cells and/or their progenitors, many efforts have been made to link signal transduction pathways to CSC self-renewal through symmetric and asymmetric cell division. Those pathways would be the same ones that coordinate embryonic development and tissue repair, in particular, the Wnt, Hedgehog, and Notch pathways. Salient examples of studies implicating the pathways will be described here as space limits an extensive survey and other recent reviews exist and are cited below.
Wnt Pathway
Growth factors comprising the Wnt family bind to GPCRs of the Frizzled family and other co-receptors such as LRP5/6 and signal stabilization of the transcriptional co-activator, β-catenin. This occurs through inhibition of constitutive degradation of β-catenin and the phosphorylation steps that signal ubiquitination and transport to the proteasome. Once β-catenin replaces transcriptional repressors in the nucleus, it regulates gene expression essential for cell proliferation, survival, and differentiation. Additionally, the Wnt pathway functions in establishing the mitotic spindle (23) and can also regulate asymmetric cell division that may lead to preservation of an adult stem cell and production of differentiated progeny (24).
Many studies have implicated a role for Wnt signaling in CSCs. Normal hematopoietic differentiation is regulated by Wnt signaling and likewise, a role for its signaling in leukemeogenesis has been demonstrated (25,26). In addition, enhanced β-catenin transcriptional activity was shown to promote glioma tumor growth due to CSCs (27,28). Recently, strong evidence for role of Wnt signaling in colon adenoma formation from murine intestinal stem cells was provided with lineage tracing studies (29). Finally, Malanchi and colleagues ablated β-catenin during in vivo tumor formation in dermal CSC and prevented growth (30). In summary, the role for Wnt signaling in CSC mediated tumor growth is far-reaching and most certainly complex since in some instances the pathway can promote differentiation and elsewhere, proliferation in normal stem cells. Tissue context may explain which predominates.
Hedgehog Pathway
The Hedgehog (HH) pathway regulates body patterning, cell fate, and segment polarity during embryogenesis through secretion of morphogens called hedgehog-type ligands. After appropriate processing and release from neighboring cells, HH ligands bind to Patched which is then released from its constitutive function, inhibiting Smoothened, a seven transmembrane domain receptor. Following this, the Gli family of transcriptional activators and inhibitors are released to promote or repress transcription. Gli transcription factors are also exquisitely regulated in the cytoplasm by SUFU (suppressor of fused) through post-translational modifications that further protein stability, activation, and localization (31).
Hedgehog signaling has been associated with diverse tumor histologies and their CSCs contained therein (in particular, breast cancer (32), glioma (33), basal cell carcinoma (34), gastric cancer (35), and colon carcinoma (36)). CSCs express functional HH components and modification of such alters sphere formation and/or tumorigenicity. One report (36) is notable because it demonstrates that patient derived CD133 expressing colon carcinoma cells are driven by GLI1 transcription and not the subpopulation lacking CD133. These authors also emphasize the critical understanding of crosstalk with Wnt signaling, a classical understanding of colon carcinoma pathogenesis.
Notch Pathway
The Notch pathway signals developmental processes such as cell fate decisions and morphogenesis through cell-cell contact of two membrane bound receptors, Delta and Notch. With engagement, the intracellular domain of Notch (NICD) is released by the action of proteases, TACE and γ-secretase. Once in the nucleus, depression of transcription factors and other epigenetic regulators promotes target gene expression. Interestingly, Notch signaling has intimate contacts with both Wnt and HH signaling and appears to promote differentiation by interceding at certain points. For example, GSK3β, a kinase that when active, has dual roles in suppressing Wnt and HH is inactivated upon Notch signaling and therefore may promote tumorigenesis (37).
Notch signaling has been implicated and correlated with CSCs in diverse tumor histologies as well. The solid and hematologic tumors that demonstrate active Notch signaling in CSC include acute myeloid leukemia (38), breast cancer (39), pancreatic cancer (40), embryonal brain tumors (41), and colon carcinoma (42). It is important to note that a role for Notch signaling in these tumor types is largely associative, e.g. increased gene expression of one or several components of the pathway is found in the CSC subpopulation. Furthermore, in some cases, an inhibitor of Notch signaling is employed as a means to implicate the pathway in CSC function. Confirmatory studies that include lineage tracing of cancer initiating cells are needed to solidify the role of Notch signaling in CSCs. This is vitally important since Notch signaling may be oncogenic or suppressive.
Tumor Microenvironment
Understanding the role of the tumor microenvironment (TME) has come to the forefront of CSC research and adds another layer of complexity to the CSC concept. Fundamentally, the TME is composed of both acellular (growth factors, chemokines, matrix proteins, and coagulation factors) and cellular components (including but not limited to fibroblasts, endothelial cells, and immune cells). If the primary tumor is considered, it is postulated that the TME will derive from and/or resemble the normal stem cell niche (43). It would be further expected that both tumorigenic and nontumorigenic cells modify the TME. Hence, one could consider the TME as a dynamic microenvironment that supports and promotes the tumor, changing with tumor progression (44). Vast efforts have also defined how the TME modifies tumor cells. Many of the TME factors have been defined and have led to an understanding of the biology of CSCs per se. For example, a role for CXCR4, a chemokine receptor was found on CSCs and it may regulate CSC metastatic processes (45).
Inflammatory mediators found in the TME, such as IL-6 and the vast array of growth factors (e.g. EGF, PDGF, IGF, Wnt, VEGF), have been well studied and documented to promote CSC survival, proliferation, and self-renewal (44). These mediators may act on the tumor cells themselves or promote angiogenesis. Therefore, efforts have been directed towards understanding the role of hypoxia in maintaining CSCs. In normal embryonic development, low oxygen tension is common and if applied to CSCs, is thought to promote self-renewal and stem-like characteristics. For example, in glioblastoma, it has been shown that in exceptionally hypoxic regions in the central portions of the tumor mass, there are a significant number of tumor cells possessing the CSC markers, CD133 and nestin when compared to the periphery (46). This contrasts, though, with studies detailing the presence of CSC in perivascular niches, locations expected to contain sufficient oxygen (47). Therefore, the role of oxygen tension in regulating CSC function is complex and may well depend on particular signaling pathways, the quality and quantity of crosstalk, and genetic and epigenetic processes underlying the individual CSC.
Tumor Heterogeneity due to Clonal Evolution vs. CSC Hierarchical Self-renewal and Differentiation
The CSC concept offered a counterpoint to the understanding of tumor origins and progression (Figure 3). In particular, the clonal evolution hypothesis stated that critical genetic abnormalities occurring stochastically that modify certain genes such as those that regulate the cell cycle and survival would be selected for within the tumor microenvironment, i.e. certain disrupted genomes would provide a selection advantage. Accumulation of these abnormalities would leave behind clones that are all equally tumorigenic. Tumor heterogeneity could be explained by a multiplicity of clones. Furthermore, additional mutations would underlie the acquisition of a metastatic phenotype. The landmark review by Hanahan and Weinberg in 2000 capsulizes this understanding (48). Today, a significant number of studies that sequence whole genomes either on the single cell level or in bulk tumors continue to support the idea that clonal evolution underpins tumor heterogeneity (see (49,50) for representative examples). For a detailed discussion on the present day understanding of clonal evolution, see (51).
Figure 3.
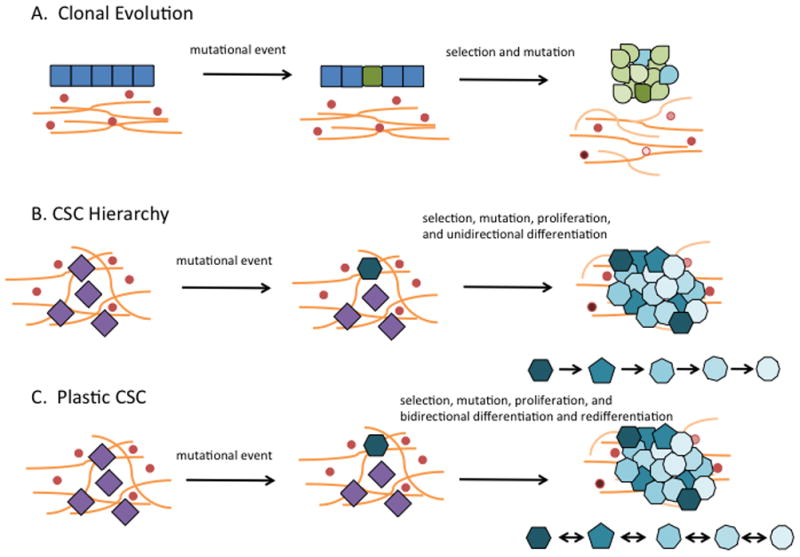
Tumor Heterogeneity. At present, the development of tumor heterogeneity may be three mechanisms. While depicted singly, some aspects of each may overlap. A. Clonal evolution. Accumulation of sufficient mutations in a differentiated epithelial cell (green) brings uncontrolled growth. Continuation of this mutational process may be aided by genomic instability while selection pressure for survival occurs. Different clones result (variable shades of green and blue). B. CSC hierarchy. CSCs are expected to reside in a niche reflecting their origin, the normal adult tissue stem cell. If this is true, then abnormal self-renewal (hexagonal blue cell) leads to differentiation of the CSC which then comprises the bulk of the tumor and is nontumorigenic. C. Plastic CSC. CSCs may derive from normal adult tissue cells (hexagonal blue cell), but as they divide and differentiate, it is also possible that subsequent progenitor cells may reacquire a more “stem-like” phenotype. This plasticity could explain tumor heterogeneity. The reversion from a differentiated CSC back to a more “stem-like” phenotype may result from environmental cues or therapy.
Since hematologic cancers were the first reported evidence of a CSC and an understanding of the hierarchical processes of normal hematopoiesis existed, it seemed possible that leukemic CSCs had similar properties that resided at the apex of multipotent stem cells. Because it resembled a normal hematopoietic stem cell with aberrant signaling (due to its underlying genetic abnormalities) proliferation could result in self-renewal and/or differentiation (into nontumorigenic cells) within the TME; therefore, populating the tumor with heterogeneity. Some evidence accumulated to support this and the notion of a tumor hierarchy was applied to solid tumors as well (12,52,53). For example, putative colon CSCs from patient biopsies could recapitulate the tumor histology in murine xenografts and these newly generated tumors demonstrated lineage differentiation (54). Similar findings were found for CNS tumors (55). Furthermore, for some tumors (neuroblastoma, for example) greater differentiation status of the tumor bulk was a favorable prognostic factor (56). Thus, it is likely that if genetics is destiny, differentiation potential of CSCs will supply some of the observed intratumor heterogeneity (57).
It is important to note that the origin of CSCs, when encountered in clinical samples, is largely unknown. Therefore, it is possible that CSCs may have resulted from a dedifferentiation process of a progenitor. The subsequent aberrant proliferation and differentiation process in this instance could produce tumor heterogeneity and has been shown to occur, at least for mammary epithelial cells (58).
Tumor Heterogeneity due to CSC Plasticity
Several lines of evidence are now indicating that a flexible and readily modifiable phenotype exists in normal cells, tumor cells, and CSCs in particular. For example, it is not uncommon for tumor formation to occur after transplantation of induced pluripotent stem (iPS) cells (59). iPS cells are those that can dedifferentiate when supplied with four transcription factors that maintain pluripotency. These findings suggest that CSCs can arise from highly differentiated cells, seed a tumor de novo, and possess a pluripotent phenotype.
Recently, though more compelling evidence has been provided to support a plastic CSC phenotype. Gupta et al., in characterizing CSCs in selected breast tumor lines, found that once purified, non-CSCs can revert back to a mixed population in culture that contained newly generated CSCs. This process was thought to occur stochastically (60). Fundamentally, these findings suggest CSCs can arise via appropriate signals as limited and simplistic as those provided in tissue culture. Certainly genetic lesions underlie some of the state changes described in the report, but the rapidity at which the change occurs belies the explanation that mutations are responsible.
Another recent report underscores the spontaneous nature of CSC plasticity. Weinberg and colleagues transformed a nontumorigenic subpopulation of mammary epithelial cells in vitro and found that they became CSC-like once injected in vivo (58). These findings suggest that the tumor microenvironment may promote CSC plasticity.
Functional plasticity following treatment of xenografted colon tumors was also demonstrated recently (61). In their report, Kreso and colleagues demonstrated that genetically identical, tumorigenic clonal populations (in murine xenografts) varied in tumorigenic potential upon serial transplantation and that oxaliplatin treatment did not alter the genetic component as measured by DNA sequencing and copy number analysis. While no CSC markers were measured, this study points to a phenotypically dynamic population is responsive to TME factors and that following chemotherapy, plasticity can persist and it is likely due to factors other than genetic alterations.
The concept of CSC plasticity can now be extended and may explain a well described phenomenon observed previously, i.e., that of vasculomimicry in tumors (62). When first described, these irregular vascular channels were attributed to abnormal leaky endothelial cells induced by growth factors supplied by the surrounding tumor cells; however, further extensive study revealed their origin to be the tumor cells induced by hypoxia. Furthermore, their signaling apparatus was consistent with an endothelial cell. In summary, this body of literature underscores the adaptive nature of tumor cells in general, their ability to transdifferentiate in specific, and their capacity to recapitulate embryonic development. Since the channels were first associated with aggressive melanoma, it remains a formal possibility that they derive from CSCs.
Finally, a team at the Developmental Therapeutics Program at the National Cancer Institute reported that a small molecule probe could induce the hallmarks of CSCs in the colon tumor lines of the NCI60 panel (63). At submicromolar concentrations and 5 day exposures of SC-1, colon tumor lines acquired characteristics consistent with the CSC phenotype including increased tumorigenicity, clonogenicity, and sphere formation. SC-1, a dual kinase and GTPase inhibitor, is able to maintain normal murine embryonic stem cells in culture with out the addition of standard growth factors such as LIF (lymphocyte inhibitory factor) (64). It is likely that modulation of SC-1’s molecular targets, ERK1/2 and RasGAP brought about newly acquired CSC functions. At this time, SC-1’s mechanism of action is unclear though and even contradictory because when inhibited, RasGAP should promote MAPK signaling but when ERK 1/2 is inhibited, MAPK signaling is diminished. It is notable that in normal embryonic stem cells, MAPK signaling is thought to regulate differentiation. These puzzling observations may be reconciled with a systems biology approach to be described in detail below.
CSC and Prognosis
As the CSC concept has been supported by ample amounts of data in preclinical models and sample biopsies, research has progressed to understand their clinical role in prognosis. Since many biomarkers have been associated with CSC function, several investigators pursued them as indicators of disease progression and outcome. But, heterogeneity in expression and methods utilized prevents identification of any clear biomarker. Efforts have also been directed to capture a CSC-gene signature but, this too presents difficulties because CSC may be rare in some tumors (65,66). However, several reviews have been recently written on prognostic indicators and address how certain surface antigens in both breast and liver cancer may be utilized in this context (67,68). A sampling of the current literature is presented below.
Markers as prognostic factors
LGR5, a GPCR that marks normal colon stem cells and appears to function in CSC has been identified as a prognostic marker for glioblastoma and is associated with poor outcome as measured by immunohistochemistry (69). A more commonly studied CSC marker, ALDH1 (aldehyde dehydrogenase) has been evaluated in patients with breast cancer and other malignancies and found to be associated with poor outcomes (70–75). ALDH1 is thought to regulate stemness by suppressing differentiation. CD133 has also been evaluated extensively (76). The presence of complex networks in CSCs suggests that a multiplex strategy might be more successful. For example, Neumeister and colleagues evaluated ALDH1, CD44, and cytokeratin expression in 642 breast cancer patient and found a highly significant worse outcome for those with the putative CSC marker display (77).
Prognostic role for meta-analyses of CSC markers
While there is a surfeit of studies supporting the notion that the presence and frequency of CSC in clinical samples is associated with worse outcome, there are studies that find no such connection (see (78–80) for examples). Thus, the usefulness of meta-analyses may help resolve the conflicting data that most certainly depend on methods of tissue acquisition, choice of technique, or reagents such as antibodies. Two recent studies that aggregate many therefore solidify the notion that CD133 expression has prognostic value for both hepatocellular carcinoma and gastric cancer (81,82). CD133’s role in CSC biology is not clear at this time and further study will be needed to further its prognostic value in the clinic.
CSC gene signature prognostic value
As it is likely that gene expression signatures and whole genome sequencing will form the basis of precision therapy for individual patients, several groups have utilized CSC gene profiling to characterize a vast array of markers for prognostic use. In the first of these reports, Liu and colleagues identified an “invasiveness” gene signature in a CSC subpopulation defined by high CD44 and low CD24 expression in breast cancer (65). The CSC subpopulation was compared to normal breast epithelium and a significant association with worsened overall survival was identified. Another study supports the utilization of microarray technology for prognostic purposes in the context of CSC biology (66). Recently, a more compelling and sophisticated prognostic model has been developed and it provides a role for gene expression signatures of CSC (83). In particular, one of three profiles defines the epithelial-mesenchymal transition (EMT) that may characterize the stem-like features in CSC (see (58,84) for recent reviews on CSC and EMT). In a rigorous test, this profile held significant prognostic value for breast cancer patients.
CSC function and prognosis
The clinical utility of prognostic CSC gene signatures and biomarkers is undisputed, but at this time, a definitive approach is unavailable and will require further study. In addition, these static endpoint type assays may not be sufficient to fully understand CSC in vivo (11). Therefore, investigators have utilized CSC function as a prognostic marker. Indeed, for malignant glioma, sphere forming ability and tumorigenicity offered prognostic value for 32 patients (85). Importantly, neurosphere formation was an independent factor for poor outcome. Future studies will be needed to define what pathways might contribute to sphere formation and what role EMT might play.
Much progress has been made implicating a role for CSC in overall outcome for cancer patients. But direct evidence for the presence of functional but plastic CSCs in patients is lacking and it is remains possible that future studies in imaging may offer a means to quantify and study the CSC subpopulation (86–88).
CSC and Therapy
It has been apparent since the earliest reports proposing a role for CSCs in hematologic and solid tumors that, if CSCs are responsible for tumor maintenance and progression and can explain the common relapse of most tumors following standard chemotherapy, then, CSCs need to be targeted (9). Any unique properties residing in such cells would become the likely targets. However, it is frequently mentioned that, if CSCs originate from normal adult tissue stem cells, a great challenge lies ahead to avoid detrimental side effects of CSC targeted therapy (89,90). The strategies to develop therapeutics are diverse and are listed here in no particular order: 1. Target components of the developmental signaling pathways (Wnt, Hedgehog, and Notch) 2. Target surface antigens marking CSCs; 3. Promoting cell death by overcoming resistance to apoptosis; 4. Promoting tumor cell differentiation; 5. Targeting components of EMT; 6. Targeting TME including stromal cells themselves and their interactions with CSC; 7. Targeting survival pathways of CSCs (PI3K, mTOR, MAPK); 8. Targeting angiogenesis. The means to accomplish these goals include a diversity of molecular entities such as small molecules, antibodies (with and without conjugated toxins), immunotherapy (via cytokines and chemokines), aptamers (small nucleotides with high affinity binding possibilities), gene therapy, oncolytic viruses, and microRNAs (91,92).
A number of investigators have written CSC reviews with emphasis on a particular tumor histology and aspects of therapeutics and are listed here: breast (93,94), colon (95), glioblastoma (96,97), head and neck squamous cell carcinoma (91), hepatocellular carcinoma (98), non-small cell lung cancer (99), ovarian cancer (92) and pancreatic cancer (100). For a general overview of CSC and therapeutics, three excellent reviews can be cited (17,101,102). It is a common feature of these reviews to suggest the both tumorigenic and non-tumorigenic cells must be targeted and a combination of CSC-directed agents and standard cytotoxic therapy will be needed. At present, no such clinical trials have been reported although one group plans to target CSC cytokine receptors (CXCR1/2) with a small molecule and also treat with standard chemotherapy in women with advanced breast cancer (103). Preclinical studies, on the other hand, are supporting this therapeutic strategy. Olive and coworkers presented evidence that in a genetically engineered mouse model for pancreatic cancer, inhibition of HH signaling and the standard chemotherapy, gemcitabine, improved survival, possibly through stromal effects brought about the HH inhibitor, IPI-926 (104).
Support for a CSC role in drug resistance and relapse
While the presence of CSCs in human tumors is generally accepted, their role in intrinsic and acquired drug resistance is less clear and supported by little evidence. However, 2 reports utilizing clinical samples do provide corroboration for this hypothesis. For example, Creighton and colleagues found that a CSC signature is enriched in tumors derived from breast cancer patients treated with docetaxel (105). In addition, Yu et al. show that there is a significant increase in in vitro mammosphere formation in cells from breast tumor biopsies post neoadjuvant chemotherapy (106).
A spontaneous mouse mammary tumor model (BRCA1/p53-) further supports the contention that CSCs are resistant to chemotherapy and that they underpin relapse. For example, Shafee et al. demonstrated that such tumors can be successfully treated with cisplatin, but 2–3 months later relapse with increased percentages of cells expressing CSC markers. These cells also have increased colony forming capacity, suggesting critical functional preservation of CSCs (107). And recently, Chen et al. identified murine glioma CSCs that resupply the tumor after temozolomide treatment in careful tracking studies (108). Similar results were found for AML (109) and colorectal cancer CSCs (110).
Selected drug candidates
Small molecule inhibitors of CSCs are being developed at a rapid rate and generally focus on the developmental pathways regulating CSCs (Wnt, HH, and Notch) and survival pathways (PI3K, Akt, and MAPK) (see (17,89,99) for tables with extensive lists of potential therapeutic small molecules). It should be noted though, that most of these lead compounds have not been specifically discovered in assays that readout CSC characteristics. A recent report that does consider sphere formation in its screening campaign is notable (111). A brief description of recent drug discoveries that offer the promise as CSC targeted agents follows.
Salinomycin, an antibiotic produced by a Streptomycete, was identified in a screen that targeted mesenchymal and immortal breast cell lines. It was shown to reduced mammosphere formation, the percentage of CD44+/CD24− breast cancer cells after treatment in vivo in a preclinical model, and altered a CSC gene signature usually associated with poor prognosis (112). How this ionophore acts to directly inhibit CSCs is still an open question and several groups are investigating its mechanism of action (113–115). Anecdotal reports of clinical use in oncology suggest some efficacy, but highlight potential toxicities that may preclude clinical use (116). It remains possible that congeners may be developed that overcome unwanted side effects.
Sulforaphane, a natural product found in foodstuffs (broccoli, for example), also appears to target CSCs because it reduces mammosphere formation in vitro and CSCs were eliminated in preclinical models of breast cancer (117). Inhibition of Wnt signaling may be responsible for the observed preclinical effects. No reports of clinical studies are present in the literature, but the molecule is being considered as a chemopreventative agent (118).
A final example of potential CSC directed therapeutic is metformin, an anti-diabetic drug in use. In two separate tumor histologies (pancreatic cancer and glioblastomas), metformin selectively inhibited proliferation of the CD133 expressing subpopulation (119,120). Furthermore, reduced spherogenesis was observed and the number of CSCs in murine mammary xenografts was reduced significantly when metformin was utilized as a single agent (121). Of note, metformin does reduce cancer risk in diabetic patients (122). Because there is sufficient clinical experience, the drug was able to move rapidly to clinical trials and a Phase 1 trial has been reported (123). A description of future trials can be found in (124). The mechanism of action of metformin is likely to be multifactorial and include action on the mTOR and NFκ-B pathways and the cell cycle.
Preclinical studies of new therapeutic strategies
Xenografted or transgenic murine models are the basis of studies that target CSC using two of the strategies mentioned above. An antibody to CD44, a common marker of CSCs, was shown to reduce tumor growth in human breast cancer xenografts (125) and inhibitors of Notch signaling eliminated CSC in a EGFR transgenic mouse model (126). Thus, the proposed strategies offer avenues for successful chemotherapy. But is does seem likely that single agent modalities encourage rapid escape from inhibition and downstream effects may be surprising. A report by Conley et al. shows that antiangiogenic agents promote the formation of CSCs, largely through HIF 1α signaling and indirectly, through the activation of survival signaling by Akt and β-catenin (127).
Systems Biology
Systems biology is the study of complex biological processes with properties, which emerge from the fundamental laws of physics, chemistry and biology. Those processes constitute the life of a cell and each level of organization up to and including the whole organism. It is an evolving field and commonly studied in cancer research through genomics, proteomics, and metabolomics. Resulting data from these studies form network interaction maps that are represented by nodes (biological components) and edges (a connection/a reaction/or some biological relatedness). Largely two approaches of network analysis have been undertaken, a top down approach that employs statistics and inferential analyses and a bottom up approach that is more quantitative in nature, and often requires advanced mathematics because it attempts to capture dynamic relationships (Figure 4). There are advantages and disadvantages of each modeling type and in some laboratories, a combined approach is being taken (128). In this review, quantitative dynamic modeling will be the focus (129).
Figure 4.
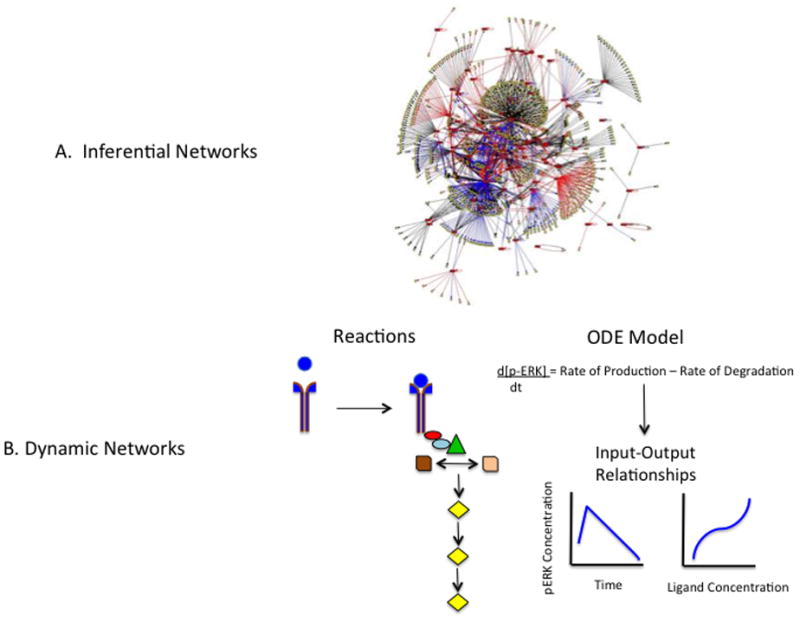
Systems Biology Approaches. A. Inferential Networks. It is commonplace to evaluate genomic, proteomic and metabolic findings construct networks of connections. The connections are based on statistical correlations and known networks residing in databases. Networks like the one depicted here is typically static in nature. (From: http://www.yaneslab.com/research/ with permission). B. Dynamic Networks. MAPK signaling is illustrated in symbols and from this, mathematical representations of the biology are determined through assignment of reaction rates and other parameters. Input/output relationships over time can be graphed and new connections may emerge.
Cancer could be considered a disease of abnormal signal transduction as this assumption underlies most drug development efforts. It is reductionist science that has allowed us a vast understanding of the pathways that appear to cascade in a linear fashion. But the literature is replete with reports demonstrating crosstalk among canonical understanding of any given pathway. For example, we understand Wnt signaling leading to β-catenin translocation and regulation of gene transcription and similarly, activation of the MAPK pathway from RAS to ERK, but recently, it has been shown that RAS protein levels (and expected function) is downregulated if the Wnt pathway is active. This was found to be so because β-catenin and RAS share the same protein degradation pathway through the scaffold protein, AXIN (130–132). Furthermore, this varies for tumor histology: in melanoma, Wnt signaling has positive prognostic value and in colon cancer, Wnt signaling predicts for negative outcome. Most certainly, this is only one aspect of the crosstalk between Wnt and MAPK. How does one capture the changing landscape of signal transduction components? Does the level of Wnt activation effect the degree of RAS degradation? What is the baseline degradation rate of RAS? Is there a threshold? Does it matter if RAS is mutated? What net function will result? And while some wet lab experiments might approach answers to the questions, a quantitative in silico and dynamic approach will bring a fuller and richer understanding of cellular function and perhaps, more solid predictions of outcome when therapies are applied.
Quantitative and dynamic models are representations of biological function at the nanoscale level and include both time and spatial aspects (133). In constructing such models, one typically begins with a network diagram of a signal transduction pathway or some portion thereof. In the instances described here, expected crosstalk will be needed. There are no limitations in size, but initially, confining the model is a good start. Following this, parameters need to be assigned and include but not limited to, protein concentrations, binding affinities, reaction rates, and diffusion constants. These details are found experimentally and are frequently reported already. When that is not the case, estimations can be made and the range of estimations can be scanned to help constrain the model. It is important to note that these models follow the Laws of Mass Action and therefore consider there are random molecular collisions occurring in the cell as if it were a well-stirred test tube.
Once parameters are defined, ordinary differential equations can be programmed into commercially available software and are the basis of simulations. Partial differential equations can take into account spatial aspects of biological function, such as transport across and diffusion to membranes (134). Results from simulations can be compared to existing literature and the model can be adjusted. Laboratory validation will be key to a successful (useful) model.
The earliest models constructed in this fashion revealed unexpected and emergent properties (135). For example, MAPK signaling is bistable, which implies that there are two separate cell states with different configurations of pathway components (e.g. phosphorylation states), but both architectures equally reach a steady state. This kind of information may be essential for understanding drug action. For instance, which configuration will be affected by a drug? Does it matter for therapy? Other emergent properties followed from dynamic modeling including ultrasensitivity (defined as the large changes in output following small changes in input) and robustness (defined as the ability to maintain unaltered function even in the instance of changing stimuli). It has been shown that ultrasensitivity is intricately related to protein levels (136) and robustness depends on coupling (137). Only dynamic modeling, which captures time and space, will reveal fundamental systems properties. Surely, new emergent properties are to be found as this is an infant field.
Applying Dynamic Modeling to CSC Plasticity
CSC plasticity may be modeled. If plasticity means differences in cell state such as tumorigenic vs. nontumorigenic cells, then quantitative dynamic modeling may capture the switch. If the EMT phenomenon is a representative shift, then it is expected that gene expression programs are altered and new proteins are expressed and other nonessential ones repressed. However, it is not simply the existence of proteins that define the transition, but rather the function of each protein, its post-translational modifications, and the spatial and temporal context in which it acts. Sufficient literature exists to build these models, set them in motion, and define outcomes. Importantly, the in silico model could be validated in the laboratory in an iterative process.
This is just one example of what may underlie the concept of plasticity. Another view could take into account what now seems to be fundamental to all cells: the ability to dedifferentiate and redifferentiate to new cell types, i.e. iPS cells. It is likely that what drives CSCs are signal transduction pathways that promote long-term survival in normal adult stem cells and the replicative potential that aids tissue repair. It follows then, that the differentiated cell has lost, at least for the short term, some of these functions. Bearing in mind that a CSC may derive from anywhere along the continuum of stem cell to differentiated cell, it is likely to be an admixture of a normal stem cell phenotype and a differentiated cell phenotype. In silico modeling of cellular differentiation and its connection to cell survival and proliferation is another approach to understand CSCS. As with EMT, the epigenetic and genetic changes associated with this type of plasticity may not be sufficient to predict tumorigenic vs. nontumorigenic function.
Applying Dynamic Modeling to CSC Prognosis
Because CSCs are heterogeneous even within tumors, may vary in proportion, and shift phenotype given a multiplicity of stimuli, it is not likely a single biomarker will provide a high degree of prognostic value. Here, dynamic modeling, may offer improved indicators. For example, a quiescent CSC may require certain survival pathways functioning and by modeling, could be found to be more stable than ones subject to various degrees of regulation. Furthermore, because of the complexity, several biomarkers may be needed to offer prognostic value. This is to say, a systems wide approach that takes connectivity within a changing internal milieu into account may be useful. This would be an advance over static measures from gene expression or immunohistochemistry.
Applying Dynamic Modeling to CSC Therapy
Identifying molecular targets
Once an in silico model is developed and validated, it can be the basis of many aspects of drug discovery and development. For example, using the methods represented in Figure 4B, mathematical representations of drug inhibition (EC50 values or dissociation constants (Kd)) can be inserted prior to a simulation run. The desired outcome could be evaluated. Thus, of the pictured MAPK pathway, one can ask whether it is better to inhibit ERK1/2 or MEK. To provide a deeper understanding of the system, parameters can be altered including but not limited to mutants with continuous activity (e.g. mutant RAS), variable levels of growth factors, and even interactions with nearby cellular components. Furthermore, if the model is sufficiently well developed, the desired outcome (cell death) may be determined. Or in other words, apoptosis pathways could similarly be modeled, connected to the pathway of interest and interrogated to determine if modulating the presumptive molecular targets brings the desired outcome. While this type of systems analysis would not be unique to CSCs, it is presumed that certain pathways may be in a different architecture that leads to tumorigenicity. In sum, quantitative dynamic modeling can aid in the selection of molecular targets prior to costly experimentation.
Defining drug mechanism of action
With a lead drug candidate in hand, computational modeling may be another tool that helps unravel its mechanism of action. This would also hold true for molecular probes. Our recent work with a small molecule probe, SC-1, offered an opportunity to interrogate an in silico MAPK pathway as it raised questions on paradoxical findings (63). SC-1 promoted CSC characteristics in colon tumor lines and its presumptive molecular targets are RasGAP and ERK1/2. If RasGAP alone is inhibited, MAPK signaling would be expected to be on and if ERK1/2 alone is inhibited, MAPK signaling would be expected to be off. This was further complicated by the fact that MAPK signaling promotes tumorigenesis but is also understood to promote differentiation. Therefore, a computational model (138) was interrogated to determine the net signaling effect due to the differential action on two molecular targets by a molecular probe with known binding affinities. In general, laboratory results verified a time course of decreasing protein levels of phospho-ERK1/2 after exposure to SC-1 (Figure 5, see (63) and unpublished results). It is important to note that some aspects of the model were not verified by laboratory results and an iterative process will be necessary to fine-tune the model. This example is illustrative and computational modeling predicting drug MOA has been attempted before where a theoretical inhibitor was modeled to inhibit NFκ-B signaling (139).
Figure 5.
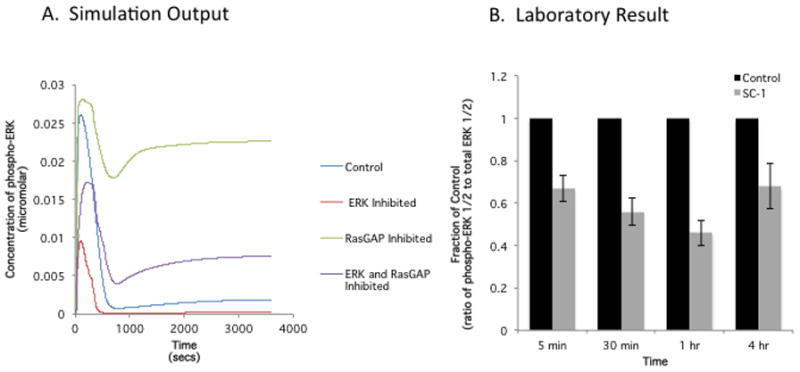
Dynamic Modeling and Laboratory Result: Effect of a small molecule probe, which promotes CSC characteristics. on its molecular target, ERK. A. A dynamic model of MAPK signaling was constructed and included the inhibition of the presumptive targets (RasGAP and ERK1/2) of SC-1 using in vitro dissociation constants previously determined. B. HT29 colon tumor line was incubated with SC-1 and phospho-ERK1/2 was determined by western blot over time. The steady decrease of phospho-ERK1/2 up to 1 hr and the rebound at 4 hrs is reflected in the simulation results. However, the initial increase phospho-ERK1/2 in simulation is not reflected in the laboratory results. Adjustments to the model will be needed.
The iterative process necessary to align the in silico model with laboratory results depends on the parameters and limits residing in the differential equations utilized. In the example shown here (Figure 5), the quantitative aspects were derived from a previously published study evaluating the MAPK pathway and its role in regulating both proliferation and differentiation in the pheochromocytoma cell line, PC12 (138). Notably, the protein expression levels, rates of reactions, and other parameters of the pathway components may not reflect the colon tumor line HT29 used in this study. Therefore, initial follow-up laboratory studies can include evaluation of these measurements which can then be reinserted into the in silico model. Alternatively, a wide range of parameters can be easily substituted in the model to determine if any one predicts the laboratory results. Of course, if the adjusted parameter predicts the laboratory result, it too, would need to be confirmed experimentally. While in some respects, the process described here is not very different from hypothesis driven studies, the adjustments made in the in silico model may eliminate some hypotheses and point the investigator into more promising areas of study. Importantly, the in silico model may predict unexpected results that are only apparent when systems are functioning together and are beyond human intuition.
Prescribing precision therapy
It is expected that whole genome sequencing, gene expression studies, either those utilizing microarray analysis or RNA sequencing of tumors, and proteomic and metabolic evaluations will define network models for patients prior to decisions about therapies that will uniquely target that tumor. But, these network building efforts only provided a static view of active cellular processes, are inferred, and will not reflect any perturbations by drugs or stimuli (e.g. autocrine or paracrine growth factors and metabolites). Furthermore, inferred networks cannot account for dose levels, drug affinity, or unexpected downstream consequences that result when pathways interact. In silico modeling of a patient’s tumor holds promise as this author can envision a desktop application for oncologists which allows input of selected therapies at selected doses for certain time courses for a particular patient.
Predicting drug resistance mechanisms
The onset of drug resistance (whether slowly or rapidly) by some of today’s molecularly targeted therapies (e.g. imantinib and vemurafenib) demonstrates the need to predict crosstalk resulting from treatment with single agents or in combination with others. For example, in two months time, patients relapse while being treated with vemurafenib for late stage melanoma (mediated by mutant BRAF) despite significant tumor regression (140,141). As mentioned above, perturbation of a dynamic and quantitative model might allow the appearance of drug resistance mechanisms not otherwise predicted from a linear cascade of signal transduction. It would be interesting to determine if different isoforms of RAF affected drug resistance, does the amount of protein effect outcome, and would inhibiting MEK and ERK result in feedback loops rather than induction of apoptosis. At present, simple in silico models addressing these types of questions can be readily developed and validated.
Conclusion
Tumor heterogeneity, as defined by CSCs, is one barrier that impedes successful prognosis and therapy for precision medicine in oncology. Furthermore, the underlying mechanisms of heterogeneity, including both clonal evolution and cellular plasticity, indicate a need to consider the complexity of tumor progression. To adequately describe abnormal signal transduction pathways of both intratumoral and intertumoral heterogeneity will require significant computing power. A systems biology approach may uncover unexpected bundles of prognostic markers based on functioning pathways, not static moments. And equally important, perturbation with drug imposes another shifting aspect and computational modeling may point to more durable outcomes.
Acknowledgments
This work was supported by the Developmental Therapeutics Program, Division of Cancer Diagnosis and Treatment, National Cancer Institute
The author would like to thank Drs. Beverly Teicher and Kathy Jung for reviewing the manuscript.
ABBREVIATIONS
- ALDH1
Aldehyde dehydrogenase 1
- CSC(s)
Cancer stem cell(s)
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial mesenchyme transition
- FGF
Fibroblast growth factor
- GPCR
G protein coupled receptor
- GSK3β
glycogen synthase kinase 3β
- HH
Hedgehog
- IGF
Insulin-like growth factor
- iPS
Induced pluripotent stem (cells)
- LGR5
Leucine rich repeat G protein coupled receptor
- LIF
Lymphocyte inhibitory factor
- LRP5/6
Low density lipoprotein receptor-relate protein 5
- MAPK
MAP kinase
- PDGF
Platelet derived growth factor
- TACE
TNFα converting enzyme
- TME
Tumor microenvironment
- VEGF
Vascular endothelial growth factor
Footnotes
The author has no conflicts of interest to declare.
The views in this paper are those of the author and do not necessarily reflect the position of the US Government.
References
- 1.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Wang JC, Lapidot T, Cashman JD, Doedens M, Addy L, Sutherland DR, et al. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood. 1998;91:2406–14. [PubMed] [Google Scholar]
- 4.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 6.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: The CD44+a2b1+ cell population is enriched in tumor-initiating cells. Cancer Research. 2007;67:6796–805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Kotilarova S, Kotliarova Y, Li A, Sun Q, Donin NM, et al. Tumor stem cells derived from gliobastomas cultured in bFGF and EGF closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 9.Kvinlaug BT, Huntly BJP. Targeting cancer stem cells. Expert opinion on therapeutic targets. 2007;11:915–27. doi: 10.1517/14728222.11.7.915. [DOI] [PubMed] [Google Scholar]
- 10.Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer research. 2006;66:1891–5. doi: 10.1158/0008-5472.CAN-05-3450. discussion 1890. [DOI] [PubMed] [Google Scholar]
- 11.Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nature reviews. Cancer. 2012;12:767–75. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 12.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 13.Sugihara E, Saya H. Complexity of cancer stem cells. International journal of cancer Journal international du cancer. 2013;132:1249–59. doi: 10.1002/ijc.27961. [DOI] [PubMed] [Google Scholar]
- 14.Medema JP. Cancer stem cells: the challenges ahead. Nature cell biology. 2013;15:338–44. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- 15.Prud’home GJ. Cancer stem cells and novel targets for antitumor strategies. Current Pharmaceutical Design. 2012;18:2838–49. doi: 10.2174/138161212800626120. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert CA, Ross AH. Cancer stem cells: cell culture, markers, and targets for new therapies. Journal of cellular biochemistry. 2009;108:1031–8. doi: 10.1002/jcb.22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou B-BS, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nature reviews. Drug discovery. 2009;8:806–23. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 18.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL, et al. Cancer stem cells - Perspectives on current status and future directions: AACR workshop on Cancer Stem Cells. Cancer Research. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 19.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS biology. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira S-M, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proceedings of the National Academy of Sciences USA. 2009;106:268–73. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strizzi L, Abbott DE, Salomon DS, Hendrix MJ. Potential for cripto-1 in defining stem cell-like characteristics in human malignant melanoma. Cell Cycle. 2008;7:1931–5. doi: 10.4161/cc.7.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alkatout I, Kabelitz D, Kalthoff H, Tiwari S. Prowling wolves in sheep’s clothing: the search for tumor stem cells. Biological Chemistry. 2008;389:799–811. doi: 10.1515/BC.2008.094. [DOI] [PubMed] [Google Scholar]
- 23.Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signalling. The EMBO journal. 2012;31:2705–13. doi: 10.1038/emboj.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Current opinion in cell biology. 2013;25:254–64. doi: 10.1016/j.ceb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell stem cell. 2012;10:412–24. doi: 10.1016/j.stem.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, et al. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer cell. 2010;18:606–18. doi: 10.1016/j.ccr.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 27.Zhang N, Wei P, Gong A, Chiu W-T, Lee H-T, Colman H, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer cell. 2011;20:427–42. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Augustin I, Goidts V, Bongers A, Kerr G, Vollert G, Radlwimmer B, et al. The Wnt secretion protein Evi/Gpr177 promotes glioma tumourigenesis. EMBO molecular medicine. 2012;4:38–51. doi: 10.1002/emmm.201100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science (New York, NY) 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 30.Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452:650–3. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 31.Coni S, Infante P, Gulino A. Control of stem cells and cancer stem cells by Hedgehog signaling: pharmacologic clues from pathway dissection. Biochemical pharmacology. 2013;85:623–8. doi: 10.1016/j.bcp.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Dontu G, Mantle ID, Patel S, Ahn N, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer research. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt S-N, Borges I, Ruiz i Altaba A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. The EMBO journal. 2010;29:2659–74. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grachtchouk M, Pero J, Yang SH, Ermilov AN, Michael LE, Wang A, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. The Journal of clinical investigation. 2011;121:1768–81. doi: 10.1172/JCI46307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Z, Yue W, Wei B, Wang N, Li T, Guan L, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PloS one. 2011;6:e17687. doi: 10.1371/journal.pone.0017687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO molecular medicine. 2009;1:338–51. doi: 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nature reviews. Cancer. 2003;3:756–67. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 38.Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia. 2006;20:2147–54. doi: 10.1038/sj.leu.2404401. [DOI] [PubMed] [Google Scholar]
- 39.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010;70:709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PloS one. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li Y-M, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer research. 2006;66:7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 42.Hoey T, Yen W-C, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell stem cell. 2009;5:168–77. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer research. 2006;66:4553–7. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 44.Castaño Z, Fillmore CM, Kim CF, McAllister SS. The bed and the bugs: interactions between the tumor microenvironment and cancer stem cells. Seminars in cancer biology. 2012;22:462–70. doi: 10.1016/j.semcancer.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche--there goes the neighborhood? International journal of cancer. Journal international du cancer. 2011;129:2315–27. doi: 10.1002/ijc.26312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pistollato F, Abbadi S, Rampazzo E, Persano L, Della Puppa A, Frasson C, et al. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem cells (Dayton, Ohio) 2010;28:851–62. doi: 10.1002/stem.415. [DOI] [PubMed] [Google Scholar]
- 47.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 48.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 49.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng CKY, Pemberton HN, Reis-Filho JS. Breast cancer intratumor genetic heterogeneity: causes and implications. Expert review of anticancer therapy. 2012;12:1021–32. doi: 10.1586/era.12.85. [DOI] [PubMed] [Google Scholar]
- 52.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 53.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–9. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 54.Vermeulen L, Todaro M, Mello F, de S, Sprick MR, Kemper K, Alea MP, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proceedings of the National Academy of Sciences; USA. 2008. pp. 13427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 56.Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system) Cancer. 1999;86:364–72. [PubMed] [Google Scholar]
- 57.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer cell. 2012;21:283–96. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 60.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 61.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AMK, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science (New York, NY) 2013;339:543–8. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seftor REB, Hess AR, Seftor EA, Kirschmann DA, Hardy KM, Margaryan NV, et al. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. The American journal of pathology. 2012;181:1115–25. doi: 10.1016/j.ajpath.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mertins SD, Scudiero DA, Hollingshead MG, Divelbiss RD, Alley MC, Monks A, et al. A small molecule (pluripotin) as a tool for studying cancer stem cell biology: proof of concept. PloS one. 2013;8:e57099. doi: 10.1371/journal.pone.0057099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen S, Do JT, Zhang ZQ, Yao S, Yan F, Peters EC, et al. Self-renewal of embryonic stem cells by a small molecule. Proceedings of the National Academy of Sciences USA. 2006;103:17266–71. doi: 10.1073/pnas.0608156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England journal of medicine. 2007;356:217–26. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 66.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer cell. 2007;11:259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 67.Gökmen-Polar Y, Nakshatri H, Badve S. Biomarkers for breast cancer stem cells: the challenges ahead. Biomarkers in medicine. 2011;5:661–71. doi: 10.2217/bmm.11.57. [DOI] [PubMed] [Google Scholar]
- 68.Liu L-L, Fu D, Ma Y, Shen X-Z. The power and the promise of liver cancer stem cell markers. Stem cells and development. 2011;20:2023–30. doi: 10.1089/scd.2011.0012. [DOI] [PubMed] [Google Scholar]
- 69.Nakata S, Campos B, Bageritz J, Bermejo JL, Becker N, Engel F, et al. LGR5 is a marker of poor prognosis in glioblastoma and is required for survival of brain cancer stem-like cells. Brain pathology (Zurich, Switzerland) 2013;23:60–72. doi: 10.1111/j.1750-3639.2012.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasheed ZA, Yang J, Wang Q, Kowalski J, Freed I, Murter C, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. Journal of the National Cancer Institute. 2010;102:340–51. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Laboratory investigation; a journal of technical methods and pathology. 2010;90:234–44. doi: 10.1038/labinvest.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ran D, Schubert M, Pietsch L, Taubert I, Wuchter P, Eckstein V, et al. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Experimental hematology. 2009;37:1423–34. doi: 10.1016/j.exphem.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 74.Kuroda T, Hirohashi Y, Torigoe T, Yasuda K, Takahashi A, Asanuma H, et al. ALDH1-High Ovarian Cancer Stem-Like Cells Can Be Isolated from Serous and Clear Cell Adenocarcinoma Cells, and ALDH1 High Expression Is Associated with Poor Prognosis. PloS one. 2013;8:e65158. doi: 10.1371/journal.pone.0065158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohara Y, Oda T, Sugano M, Hashimoto S, Enomoto T, Yamada K, et al. Histological and prognostic importance of CD44(+) /CD24(+) /EpCAM(+) expression in clinical pancreatic cancer. Cancer science. 2013;104:1127–34. doi: 10.1111/cas.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. The Journal of pathology. 2013;229:355–78. doi: 10.1002/path.4086. [DOI] [PubMed] [Google Scholar]
- 77.Neumeister V, Agarwal S, Bordeaux J, Camp RL, Rimm DL. In situ identification of putative cancer stem cells by multiplexing ALDH1, CD44, and cytokeratin identifies breast cancer patients with poor prognosis. The American journal of pathology. 2010;176:2131–8. doi: 10.2353/ajpath.2010.090712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer research. 2008;68:1451–61. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 79.Salnikov AV, Kusumawidjaja G, Rausch V, Bruns H, Gross W, Khamidjanov A, et al. Cancer stem cell marker expression in hepatocellular carcinoma and liver metastases is not sufficient as single prognostic parameter. Cancer letters. 2009;275:185–93. doi: 10.1016/j.canlet.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 80.Shan Y, Huang Y, Xie Y, Tan Y, Chen B-C, Zhou M, et al. Angiogenesis and clinicopathologic characteristics in different hepatocellular carcinoma subtypes defined by EpCAM and α-fetoprotein expression status. Medical oncology (Northwood, London, England) 2011;28:1012–6. doi: 10.1007/s12032-010-9600-6. [DOI] [PubMed] [Google Scholar]
- 81.Wen L, Chen X-Z, Yang K, Chen Z-X, Zhang B, Chen J-P, et al. Prognostic value of cancer stem cell marker CD133 expression in gastric cancer: a systematic review. PloS one. 2013;8:e59154. doi: 10.1371/journal.pone.0059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma Y-C, Yang J-Y, Yan L-N. Relevant markers of cancer stem cells indicate a poor prognosis in hepatocellular carcinoma patients: a meta-analysis. European journal of gastroenterology & hepatology. 2013 doi: 10.1097/MEG.0b013e32836019d8. [DOI] [PubMed] [Google Scholar]
- 83.Cheng W-Y, Ou Yang T-H, Anastassiou D. Development of a prognostic model for breast cancer survival in an open challenge environment. Science translational medicine. 2013;5:181ra50. doi: 10.1126/scitranslmed.3005974. [DOI] [PubMed] [Google Scholar]
- 84.Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clinical chemistry. 2013;59:168–79. doi: 10.1373/clinchem.2012.184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem cells (Dayton, Ohio) 2009;27:980–7. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang TD, Friedland S, Sahbaie P, Soetikno R, Hsiung P-L, Liu JTC, et al. Functional imaging of colonic mucosa with a fibered confocal microscope for real-time in vivo pathology. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2007;5:1300–5. doi: 10.1016/j.cgh.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mangoni M, Livi L, Biti G, Di Cataldo V, Capaccioli N, Castier Y, et al. Stem cell tracking: toward clinical application in oncology? Tumori. 2012;98:535–42. doi: 10.1177/030089161209800501. [DOI] [PubMed] [Google Scholar]
- 88.Shigdar S, Lin J, Li Y, Yang CJ, Wei M, Zhus Y, et al. Cancer stem cell targeting: the next generation of cancer therapy and molecular imaging. Therapeutic delivery. 2012;3:227–44. doi: 10.4155/tde.11.148. [DOI] [PubMed] [Google Scholar]
- 89.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature reviews. Clinical oncology. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 90.Dodge ME, Lum L. Drugging the cancer stem cell compartment: lessons learned from the hedgehog and Wnt signal transduction pathways. Annual review of pharmacology and toxicology. 2011;51:289–310. doi: 10.1146/annurev-pharmtox-010510-100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kiang A, Yu MA, Ongkeko WM. Progress and pitfalls in the identification of cancer stem cell-targeting therapies in head and neck squamous cell carcinoma. Current medicinal chemistry. 2012;19:6056–64. [PubMed] [Google Scholar]
- 92.Ahmed N, Abubaker K, Findlay J, Quinn M. Cancerous ovarian stem cells: obscure targets for therapy but relevant to chemoresistance. Journal of cellular biochemistry. 2013;114:21–34. doi: 10.1002/jcb.24317. [DOI] [PubMed] [Google Scholar]
- 93.Gangopadhyay S, Nandy A, Hor P, Mukhopadhyay A. Breast cancer stem cells: a novel therapeutic target. Clinical breast cancer. 2013;13:7–15. doi: 10.1016/j.clbc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 94.Economopoulou P, Kaklamani VG, Siziopikou K. The role of cancer stem cells in breast cancer initiation and progression: potential cancer stem cell-directed therapies. The oncologist. 2012;17:1394–401. doi: 10.1634/theoncologist.2012-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang EH, Wicha MS. Colon cancer stem cells: implications for prevention and therapy. Trends in molecular medicine. 2008;14:503–9. doi: 10.1016/j.molmed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Florio T, Barbieri F. The status of the art of human malignant glioma management: the promising role of targeting tumor-initiating cells. Drug discovery today. 2012;17:1103–10. doi: 10.1016/j.drudis.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 97.Liu J, Albrecht AM, Ni X, Yang J, Li M. Glioblastoma tumor initiating cells: therapeutic strategies targeting apoptosis and microRNA pathways. Current molecular medicine. 2013;13:352–7. [PubMed] [Google Scholar]
- 98.Nagano H, Ishii H, Marubashi S, Haraguchi N, Eguchi H, Doki Y, et al. Novel therapeutic target for cancer stem cells in hepatocellular carcinoma. Journal of hepato-biliary-pancreatic sciences. 2012;19:600–5. doi: 10.1007/s00534-012-0543-5. [DOI] [PubMed] [Google Scholar]
- 99.Gottschling S, Schnabel PA, Herth FJF, Herpel E. Are we missing the target? Cancer stem cells and drug resistance in non-small cell lung cancer. Cancer genomics & proteomics. 2012;9:275–86. [PubMed] [Google Scholar]
- 100.Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:4277–84. doi: 10.1158/1078-0432.CCR-11-3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. BioEssays: news and reviews in molecular, cellular and developmental biology. 2009;31:1038–49. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fábián Á, Vereb G, Szöllősi J. The hitchhikers guide to cancer stem cell theory: markers, pathways and therapy. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2013;83:62–71. doi: 10.1002/cyto.a.22206. [DOI] [PubMed] [Google Scholar]
- 103.Korkaya H, Wicha MS. Breast cancer stem cells: we’ve got them surrounded. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:511–3. doi: 10.1158/1078-0432.CCR-12-3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY) 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13820–5. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 107.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer research. 2008;68:3243–50. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature biotechnology. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 110.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 111.Mathews LA, Keller JM, Goodwin BL, Guha R, Shinn P, Mull R, et al. A 1536-well quantitative high-throughput screen to identify compounds targeting cancer stem cells. Journal of biomolecular screening. 2012;17:1231–42. doi: 10.1177/1087057112458152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Parajuli B, Shin S-J, Kwon S-H, Cha S-D, Chung R, Park W-J, et al. Salinomycin induces apoptosis via death receptor-5 up-regulation in cisplatin-resistant ovarian cancer cells. Anticancer research. 2013;33:1457–62. [PubMed] [Google Scholar]
- 114.Koo KH, Kim H, Bae Y-K, Kim K, Park B-K, Lee C-H, et al. Salinomycin induces cell death via inactivation of Stat3 and downregulation of Skp2. Cell death & disease. 2013;4:e693. doi: 10.1038/cddis.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou J, Li P, Xue X, He S, Kuang Y, Zhao H, et al. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicology letters. 2013;222:139–45. doi: 10.1016/j.toxlet.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 116.Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. Journal of biomedicine & biotechnology. 2012;2012:950658. doi: 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y, Zhang T, Korkaya H, Liu S, Lee H-F, Newman B, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:2580–90. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li Y, Zhang T. Targeting cancer stem cells with sulforaphane, a dietary component from broccoli and broccoli sprouts. Future oncology (London, England) 2013;9:1097–103. doi: 10.2217/fon.13.108. [DOI] [PubMed] [Google Scholar]
- 119.Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low concentrations of metformin selectively inhibit CD133+ cell proliferation in pancreatic cancer and have anticancer action. PloS one. 2013;8:e63969. doi: 10.1371/journal.pone.0063969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Würth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell cycle (Georgetown, Tex) 2013;12:145–56. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:972–7. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (Clinical research ed) 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:2593–600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
- 124.Kourelis TV, Siegel RD. Metformin and cancer: new applications for an old drug. Medical oncology (Northwood, London, England) 2012;29:1314–27. doi: 10.1007/s12032-011-9846-7. [DOI] [PubMed] [Google Scholar]
- 125.Marangoni E, Lecomte N, Durand L, de Pinieux G, Decaudin D, Chomienne C, et al. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. British journal of cancer. 2009;100:918–22. doi: 10.1038/sj.bjc.6604953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kondratyev M, Kreso A, Hallett RM, Girgis-Gabardo A, Barcelon ME, Ilieva D, et al. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. 2012;31:93–103. doi: 10.1038/onc.2011.212. [DOI] [PubMed] [Google Scholar]
- 127.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2784–9. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–94. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iyengar R. Why we need quantitative dynamic models. Science signaling. 2009;2:eg3. doi: 10.1126/scisignal.264eg3. [DOI] [PubMed] [Google Scholar]
- 130.Guardavaccaro D, Clevers H. Wnt/β-catenin and MAPK signaling: allies and enemies in different battlefields. Science signaling. 2012;5:pe15. doi: 10.1126/scisignal.2002921. [DOI] [PubMed] [Google Scholar]
- 131.Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, et al. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Science signaling. 2012;5:ra3. doi: 10.1126/scisignal.2002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jeong W-J, Yoon J, Park J-C, Lee S-H, Lee S-H, Kaduwal S, et al. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Science signaling. 2012;5:ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 133.Millat T, Bullinger E, Rohwer J, Wolkenhauer O. Approximations and their consequences for dynamic modelling of signal transduction pathways. Mathematical biosciences. 2007;207:40–57. doi: 10.1016/j.mbs.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 134.Neves SR, Tsokas P, Sarkar A, Grace EA, Rangamani P, Taubenfeld SM, et al. Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell. 2008;133:666–80. doi: 10.1016/j.cell.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science (New York, NY) 1999;283:381–7. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 136.Lipshtat A, Jayaraman G, He JC, Iyengar R. Design of versatile biochemical switches that respond to amplitude, duration, and spatial cues. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1247–52. doi: 10.1073/pnas.0908647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shinar G, Feinberg M. Structural sources of robustness in biochemical reaction networks. Science (New York, NY) 2010;327:1389–91. doi: 10.1126/science.1183372. [DOI] [PubMed] [Google Scholar]
- 138.Sasagawa S, Ozaki Y, Fujita K, Kuroda S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nature cell biology Nature Publishing Group. 2005;7:365–73. doi: 10.1038/ncb1233. [DOI] [PubMed] [Google Scholar]
- 139.Sung M-H, Simon R. In silico simulation of inhibitor drug effects on nuclear factor-kappaB pathway dynamics. Molecular pharmacology. 2004;66:70–5. doi: 10.1124/mol.66.1.70. [DOI] [PubMed] [Google Scholar]
- 140.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. 2012;11:873–86. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- 142.Stuelten CH, Mertins SD, Busch JI, Gowens M, Scudiero DA, Burkett MW, et al. Complex display of putative tumor stem cell markers in the NCI60 tumor cell line panel. Stem cells (Dayton, Ohio) 2010;28:649–60. doi: 10.1002/stem.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pastrana E, Silva-Vargas V, Doetsch F. Eyes Wide Open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–98. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Research. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 145.Greve B, Kelsch R, Spaniol K, Eich HT, Götte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2012;81:284–93. doi: 10.1002/cyto.a.22022. [DOI] [PubMed] [Google Scholar]