

Abstract
Members of the trimeric autotransporter adhesin (TAA) family play a crucial role in adhesion of Gram-negative pathogens to host cells. Moreover, these proteins are multifunctional virulence factors involved in several other biological traits, including invasion into host cells and evasion of the host immune system. In cystic fibrosis epidemic Burkholderia cenocepacia strain J2315, we identified a unique TAA (BCAM0224)-encoding gene, previously described as being implicated in virulence. Here, we characterized this multifunctional protein, trying to establish its role in B. cenocepacia pathogenicity. We show that BCAM0224 occurs on the bacterial surface and adopts a trimeric conformation. Furthermore, we demonstrated that BCAM0224 is needed for earlier stages of biofilm formation and is required for swarming motility. In addition, BCAM0224 plays an important role in evasion of the human innate immune system, providing resistance against the bactericidal activity of serum via the complement classical pathway. Finally, BCAM0224 mediates bacterial adhesion to and invasion of cultured human bronchial epithelial cells. Together, these data reveal the high versatility of the BCAM0224 protein as a virulence factor in the pathogenic bacterium B. cenocepacia.
INTRODUCTION
For most bacterial pathogens, the crucial sequential steps in the establishment of infection are first the attachment and then the colonization of the host cells. Attachment involves binding to either specific host cell receptors or extracellular matrix (ECM) proteins. Associated with such processes, pili or fimbrial adhesins have been the main factors studied in Gram-negative bacteria. In recent years, trimeric autotransporter adhesins (TAAs), a group of nonfimbrial adhesins, have emerged as key determinants of the pathogen's success (1). TAAs share an organization consisting of a conserved membrane-embedded C-terminal domain forming a trimeric beta-barrel pore and providing the transition of the passenger domains (stalk and head[s]) to the bacterial cell surface (1). YadA from Yersinia enterocolitica is the prototype TAA (2). Besides YadA, several other TAAs have been identified, all described to act as adhesins, promoting adherence to host cells and/or ECM proteins (3). Moreover, these proteins proved to be multifunctional virulence factors involved in other biological traits, including biofilm formation (4), hemagglutination (5), serum resistance (6, 7), and adhesion to and invasion of host cells (8, 9).
The complement system is the first line of innate immune defense against intruders. The activation of this system triggers a cascade of protein deposition on the bacterial surface, resulting in the assembly of the membrane attack complex and opsonization of the pathogen, followed by phagocytosis. Several bacterial pathogens are known to be resistant to the innate immune system, being able to escape bactericidal killing by the complement system (10). A list of TAAs have been reported to be part of the complement evasion mechanisms of many bacteria, such as YadA (11), NhhA from Neisseria meningitides (12), UspA1 and UspA2 from Moraxella catarrhalis (7), DsrA from Haemophilus ducreyi (6), Eibs from Escherichia coli (13, 14), and Omp100 from Aggregatibacter actinomycetemcomitans (15).
The Burkholderia cepacia complex (Bcc) consists of 17 closely related species that emerged as opportunistic human pathogens, particularly in patients with cystic fibrosis (CF) (16). Although all Bcc species can cause respiratory infections, B. cenocepacia and B. multivorans are the species most commonly recovered from sputum of CF patients (16). They have the ability to spread from patient to patient and are associated with unpredictable clinical outcomes ranging from asymptomatic carriage to fatal acute pneumonia (cepacia syndrome) (17). Within the species B. cenocepacia, several epidemic lineages have been described and were responsible for various outbreaks in large geographic regions. Among these, B. cenocepacia CF isolate J2315, belonging to the epidemic lineage Edinburgh-Toronto (ET-12) (18, 19), is one of the best-studied Bcc pathogens. Despite the identification of a considerable number of virulence factors associated with this pathogen, many aspects of infection, namely, bacterial adhesion to and invasion of host cells and complement escape, remain to be elucidated. In an attempt to contribute to the elucidation of such events, we aimed to investigate the role of newly identified B. cenocepacia J2315 TAA-encoding genes.
The use of computational searches revealed the existence of seven TAA-encoding genes in the genome of epidemic B. cenocepacia strain J2315 (20). Among these, three TAA-encoding genes (BCAM0219, BCAM0223, and BCAM0224) are clustered in a 30-kb region together with neighboring two-component systems, which may regulate their virulence-associated functions (20, 21). In a previous study, we reported that the BCAM0224 gene was detected exclusively in epidemic B. cenocepacia strain J2315. We also found that a B. cenocepacia BCAM0224 mutant exhibited attenuated virulence in a Galleria mellonella infection model (22). For the neighboring TAA gene BCAM0223, we showed that its disruption in B. cenocepacia caused marked decreases in hemagglutination, properties of adherence to vitronectin, serum resistance, adhesion to epithelial cells, and virulence (21). Furthermore, in a recent study, atomic force microscopy (AFM) was used to measure the homophilic trans-interaction forces between trimeric forms of BCAM0224 (23). In order to gain new insights into the mode of action of the B. cenocepacia TAA virulence-associate factors, this study provides a more complete understanding of how the TAA-encoding gene BCAM0224 contributes to bacterial virulence, highlighting its multifaceted roles during host infection.
MATERIALS AND METHODS
Bacterial isolates and culture conditions.
B. cenocepacia clinical isolate K56-2, an ET-12 isolate, was kindly provided by J. J. LiPuma (University of Michigan). The B. cenocepacia K56-2 BCAM0224::Tp mutant and Escherichia coli strain BL21(DE3) expressing the BCAM0224 gene were described in a previous study (22). Bacteria were cultured in Luria-Bertani (LB) broth (Conda, Pronadisa) at 37°C with orbital agitation at 250 rpm. When appropriate, medium was supplemented with 150 mg/liter of ampicillin (for E. coli), 150 mg/liter of trimethoprim, or 100 mg/liter chloramphenicol (for the B. cenocepacia mutant). For functional studies, the parental and mutant B. cenocepacia strains were grown in 24-well plates for 17 h under conditions of limited oxygen supply in LB broth supplemented with 300 mM NaCl and 10 mM H2O2. A limited oxygen supply (6 to 12%) was achieved by growing cultures in incubation jars containing an atmosphere generator for microaerophilic conditions (GENbox; bioMérieux).
Cell lines and cell culture.
Two human bronchial epithelial cell lines were used: 16HBE14o− cells, which are normal bronchial cells, and CFBE41o− cells, which are homozygous for the delta F508 mutation in a cyclic AMP (cAMP)-activated chloride channel (CF transmembrane conductance regulator), leading to decreased chloride ion transport and impaired water transport across the cell membrane, corresponding to a CF airway. Both cell lines were immortalized and characterized (24, 25). Cells were maintained in fibronectin-collagen I-coated flasks in minimum essential medium with Earle's salt (MEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Lonza), 0.292 g/liter l-glutamine (Sigma-Aldrich), and penicillin-streptomycin (100 U/ml; Alfagene) in a humidified atmosphere at 37°C with 5% CO2.
3D structure prediction.
Three-dimensional (3D) structure prediction of the full-length monomer of BCAM0224 was performed by using the I-TASSER fold recognition method (http://zhanglab.ccmb.med.umich.edu/I-TASSER) (26) and visualized with Swiss-PdbViewer V.4.0. The predicted 3D structure of the C-terminal region (residues 822 to 910) was obtained with Swiss Model Workspace (http://swissmodel.expasy.org/workspace/), based on the crystal structure of the Hia outer membrane (OM) domain (PDB accession number 2GR7). To predict the trimerization of the C-terminal region (anchor) of BCAM0224, we used Cluspro 2.0 software (http://cluspro.bu.edu/home.php), an automated docking and discrimination method for the prediction of protein complexes (27).
Generation of an anti-BCAM0224 antibody.
A fragment of the BCAM0224 gene, encoding 284 amino acids (aa) of the passenger domain (aa 383 to 666), was amplified by using B. cenocepacia K56-2 genomic DNA as the template and primers FBCAM0224383 (5′-TTTCATATGTCGCAACTGCACGGTGTGT-3′) and RBCAM0224666 (5′-AAACTCGAGGGTCACGCTGTCGAACGACAA-3′), containing NdeI and XhoI restriction sites (underlined), respectively. The fragment was cloned into plasmid pET23a (Novagen) to generate pDM5 and introduced by electroporation into E. coli BL21(DE3). To overexpress the BCAM0224383–666 polypeptide with a 6×His tag at its C terminus, 1 liter of LB broth was inoculated at an initial optical density at 640 nm (OD640) of 0.1 by using a culture grown overnight. The cells were grown to an OD640 of 0.6 and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Sigma-Aldrich) for 3 h at 37°C with orbital agitation at 250 rpm. BCAM0224383–666-His protein was purified by affinity chromatography on a Histrap column prepacked with high-performance Ni2+ Sepharose (GE Healthcare), and purity was verified by 12% acrylamide sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Polyclonal antibodies against BCAM0224 were produced by immunization (Agro-Bio, France) of two rabbits with recombinant BCAM0224383–666-His. The resulting immunized serum was then purified by affinity chromatography (Agro-Bio, France), and purified anti-BCAM0224383–666-His was used in all experiments.
Bacterial membrane extracts.
Bacterial cultures of B. cenocepacia K56-2 and the BCAM0224::Tp mutant were grown in 200 ml of supplemented LB broth under microaerophilic conditions, as mentioned above. After 17 h of growth, bacteria were harvested by centrifugation at 9,000 × g for 10 min at 4°C. Bacterial pellets were washed with 50 mM Tris-HCl (pH 7.4) and resuspended in 5 ml of the same buffer containing a protease inhibitor mixture (Roche), 1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma), 50 μg/ml DNase (Qiagen), and 50 μg/ml RNase (Sigma). The suspension was sonicated 8 times for 30 s at 40 W (Branson). Unbroken cells and debris were removed by centrifugation at 10,000 × g for 15 min at 4°C, and the supernatant was ultracentrifuged at 100,000 × g for 1 h at 4°C (SW41Ti rotor; Beckman) to pellet the cell envelopes. The total membranes were resuspended in 50 mM Tris-HCl (pH 7.4) buffer, and total protein quantification was performed by using a protein assay kit (Bio-Rad Laboratories).
Western blot analysis.
For Western analysis, a volume of the membrane extracts corresponding to 100 μg of the total protein was dissolved in sample buffer (Laemmli buffer with 5% [vol/vol] 2-β-mercaptoethanol and 5% [vol/vol] bromophenol blue), boiled for 10 min, and separated by 6% SDS-PAGE. Some samples were denatured by using 6 and 8 M urea (Sigma) in the sample buffer. Proteins were transferred onto nitrocellulose membranes (Bio-Rad) by using a Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% (wt/vol) nonfat dry milk in phosphate-buffered saline (PBS) containing 0.5% (vol/vol) Tween 20 (PBS-T) for 1 h, incubated with purified anti-BCAM0224 antibody (diluted 1:5,000), and washed three times for 5 min with PBS-T. Membranes were then incubated with a secondary antibody (diluted 1:2,000), goat anti-rabbit serum conjugated to horseradish peroxidase (Santa Cruz), for 1 h. Proteins were detected by the addition of ECL reagent (Pierce) as a substrate and exposed to autoradiographic film (Amersham).
Confocal laser scanning microscopy assays.
Confocal laser scanning microscopy analyses were performed as previously described (21). Briefly, bacteria were stained by using plasmid pIN29, which encodes the red fluorescent protein DsRed (28). The plasmid was electroporated into B. cenocepacia K56-2 and the BCAM0224::Tp mutant. For surface localization experiments, B. cenocepacia K56-2 and the BCAM0224::Tp mutant were grown in supplemented LB broth under microaerophilic conditions, as mentioned above. After 17 h of growth, 200 μl of bacterial suspension at an OD640 of ∼1.5 was harvested by centrifugation and washed with PBS. For bacterial adhesion, the procedure was carried out as described above, but glass coverslips were placed into 24-well plates before cell seeding. At the end of the adhesion experiment, cells were washed three times with PBS.
Immunostaining of samples from both experiments was done by using the same steps. Samples were fixed with 3.7% paraformaldehyde (20 min), quenched with 50 mM NH4Cl (10 min), immersed in 0.2% Triton X-100 (5 min), and saturated with 5% bovine serum albumin (BSA) (30 min). Samples were incubated with primary mouse E-cadherin antibody (Santa Cruz) (1:100 dilution) for adhesion experiments and anti-BCAM0224 antibody (1:50 dilution) for localization experiments, followed by secondary polyclonal goat anti-mouse serum coupled to Alexa 488 (Santa Cruz) (1:500 dilution). Finally, the coverslips or bacterial suspensions were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories).
All samples were examined on a Leica TCS SP5 (Leica Microsystems CMS GmbH, Mannheim, Germany) inverted microscope (model no. DMI6000) with a 63× water (1.2-numerical-aperture) apochromatic objective (29).
Complementation of the B. cenocepacia K56-2 BCAM0224::Tp mutant.
The complete coding sequence of the BCAM0224 gene was obtained from plasmid pDM3 (22) by digestion with NdeI and XbaI restriction enzymes. This DNA fragment was ligated into plasmid pIN177 (tac promoter; Cmr) (A. Vergunst and D. O'Callagan, unpublished data) before transformation into E. coli DH5α and selection with 100 mg/liter chloramphenicol. The resulting plasmid, pDM6, was introduced into the BCAM0224::Tp mutant by electroporation, being replicative in Burkholderia.
Biofilm formation assays.
Biofilm formation on polystyrene surfaces was measured by using the crystal violet method as follows: bacterial cultures were grown in 3 ml LB broth supplemented with 300 mM NaCl and 10 mM H2O2 (initial OD640 of 0.2) in glass tubes for 17 h at 37°C at 60 rpm under microaerophilic conditions. In each case, cells were harvested, washed, and diluted in LB broth to an OD640 of 0.05, and 200 μl of these cell suspensions was inoculated into wells of a 96-well polystyrene microtiter plate. Plates were statically incubated at 37°C for 24 or 48 h. Quantification was achieved by removing the culture medium and unattached bacterial cells. Wells were rinsed three times with deionized water, and adherent bacteria were stained with 200 μl of a 1% (wt/vol) crystal violet solution for 15 min at room temperature. The solution was removed, and three gentle rinses with deionized water were performed. Finally, the dye was solubilized with 95% ethanol, and the absorbance was measured at 595 nm in a microplate reader (Versamax; Molecular Devices). Results are mean values of at least 20 replicates from 3 independent experiments.
Motility assays.
Swimming plates (0.3% Bacto agar [Difco], 10 g/liter tryptone, and 5 g/liter NaCl) and swarming plates (0.5% Bacto agar and LB medium supplemented with 5 g/liter glucose) were prepared by using square plates. For estimations of motility, bacterial cultures were grown under defined conditions. The culture OD640 was adjusted to match a density of 1, and one 3-μl drop of each culture was inoculated onto agar surface and incubated at 37°C for 48 h, followed by halo diameter determinations.
Serum bactericidal assay.
A serum bactericidal assay was performed as previously described (21). Briefly, bacterial cultures were grown under microaerophilic conditions, as mentioned above. After 17 h of growth, they were diluted in PBS to a final concentration of 106 CFU/ml. Subsequently, 10 μl (104 CFU) of the cell suspension was added to 30 μl of normal human serum (NHS) (Sigma-Aldrich) and 60 μl of PBS, resulting in a final serum concentration of 30%. Tubes were incubated at 37°C for 1 h and then placed on ice. After this, samples were serially diluted and plated. To determine the initial bacterial load, we used a tube with NHS replaced by PBS. As a control, we performed all experiments in parallel using heat-inactivated NHS (hNHS) at 56°C for 30 min. The percentage of survival was determined as the quantity of bacteria that survived relative to the initial bacterial load.
To test the involvement of the various complement pathways, we used factor B-depleted NHS (B−NHS) (Quidel), which has the alternative complement activation pathway blocked. To block both the classical and lectin pathways, we used PBS containing 10 mM MgCl2 and 10 mM EGTA to equilibrate NHS (LC−NHS) for 15 min on ice prior to the addition of bacteria. Finally, to selectively block the classical pathway, a C1q-depleted serum (C−NHS) (Quidel) was used.
Adhesion and invasion of epithelial cells.
Adhesion and invasion experiments were carried out on 16HBE14o− (non-CF) and CFBE41o− (CF) bronchial epithelial cells as described previously (21), with some modifications. Cells were seeded in 24-well plates (Orange Scientific) 1 day prior to infection at 5 × 105 cells per well in supplemented MEM. Bacterial strains were grown as described above and used to infect cells at a multiplicity of infection (MOI) of 50:1. After infection, plates were centrifuged at 700 × g for 5 min. For adhesion assays, the infected monolayers were incubated for 30 min at 37°C in an atmosphere containing 5% CO2. Each well was washed three times with PBS, and cells were lysed by applying lysis buffer (10 mM EDTA, 0.25% Triton X-100) for 30 min at room temperature. The adherent bacteria were quantified by plating serial dilutions of the cell lysates onto LB agar plates and incubating the plates for 24 h at 37°C. For invasion assays, the infected monolayers were incubated for 2 h to allow bacterial entry and washed three times with PBS, MEM containing amikacin and ceftazidime (2 mg/ml each) was added, and the mixture was incubated for another 2 h. Supernatants were then plated to confirm the effectiveness of the antibiotic treatment, and cell monolayers were washed three times with PBS. The number of intracellular bacteria was determined by lysing cells with lysis buffer (30 min at room temperature) and plating serial dilutions onto LB agar plates. Results are expressed as a ratio of the wild-type cells and corrected with the initial bacterial dose applied.
We also tested the ability of the antibody (anti-BCAM0224) to inhibit adhesion to and invasion of epithelial cells by wild-type B. cenocepacia K56-2 and the BCAM0223::Tp mutant. Bacterial suspensions were incubated with the purified anti-BCAM0224 rabbit polyclonal antibody (diluted 1:10) for 1 h at room temperature. Samples were washed with PBS and used to infect cell monolayers according to the adhesion and invasion assays described above. Control experiments including an irrelevant rabbit serum (Sigma) were used as a control.
Statistical analysis.
Data are expressed as mean values of a minimum of three independent experiments ± standard deviations (SD). Statistical analysis was carried out by using Statistica 7 software. One-way analysis of variance (ANOVA) and Bonferroni's multiple-comparison test were performed to determine statistically significant differences. Tukey's test for unequal group sample sizes was used for biofilm comparisons. A P value of <0.05 was considered statistically significant.
RESULTS
Predicted structure of the TAA BCAM0224.
In a previous work (22), we identified the BCAM0224 gene, encoding a protein with similarity to the prototype member of the TAA family, the Yersinia adhesin YadA. In an attempt to gain insight into its structure, we have built 3D models for BCAM0224 domains using the Web-based I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) (26). In Fig. 1A, we show the top 3D models for each BCAM0224 domain, with C scores ranging from −0.65 to 1.52. As expected, the C-terminal region (anchor domain) yielded the highest C score. It comprises four antiparallel β-sheets connected by short turns and one extended α-helix and has structural homology to the outer membrane (OM) translocator domain of the Haemophilus influenzae Hia trimeric autotransporter. Furthermore, the surface-exposed part of the BCAM0224 protein contains two heads and one stalk, composed primarily of antiparallel beta-sheets and connecting loops with a small percentage of alpha-helices. The two head models (models 1 and 2) show relatively good alignments with the corresponding parts of YadA from Yersinia and BoaA from Burkholderia pseudomallei, respectively (Fig. 1A).
FIG 1.

BCAM0224 structure prediction. (A) The top 3D models of the BCAM0224 protein domains predicted by I-TASSER (based on the C score). The C score values were estimated based on Z scores obtained for the various templates used in the I-TASSER simulations. (B) Comparative alignment of the C-terminal 83-amino-acid sequence of BCAM0224 with the membrane anchor domain of TAAs (http://toolkit.tuebingen.mpg.de/dataa). The secondary-structure elements that are conserved between the two sequences are boxed (four β-sheets, one hairpin loop, and one α-helix). The arrow highlights the glycine residue (residue 876) homologous to the conserved Gly-389 in YadA. (C) Computer modeling of the putative 3D structure for the membrane anchor region of BCAM0224. (i) Monomeric structure obtained with Swiss Model Workspace, based on the structure of the Hia outer membrane translocator domain; (ii) trimeric structure obtained by Cluspro 2.0 software; (iii) upper view of the trimeric structure.
Further analysis of the C-terminal region of BCAM0224 containing 83 aa shows the secondary-structure features of the membrane-anchoring domain of TAAs (http://toolkit.tuebingen.mpg.de/dataa) (30). A strictly conserved glycine residue (Gly-876 in BCAM0224), which was proven to be important for both the translocation and the stability of YadA (31), is highlighted (Fig. 1B). Computer modeling using Swiss Model Workspace yielded the 3D structure of this C-terminal region, based on the structure of the Hia OM translocator domain. Using Cluspro 2.0 software, we predicted the trimerization of this region, showing the formation of a 12-strand β-barrel that could form a pore in the OM and facilitate the translocation of the N-terminal part of the protein, in analogy to other members of the trimeric family (Fig. 1C). We also observed that the conserved Gly-876 residue faces the pore lumen in this model (not shown), which is in accordance with other TAAs such as YadA and Hia (31).
BCAM0224 forms stable trimers at the cell surface.
The structural prediction obtained for the C-terminal part of BCAM0224 suggested that this protein forms trimers in the bacterial membrane. To verify this assumption, membrane proteins of wild-type, BCAM0224 mutant, and complemented-mutant strains were isolated and subjected to immunoblotting using an anti-BCAM0224 antibody. The Western blot showed the presence of two main products in membrane extracts from wild-type and complemented BCAM0224-deficient cells, one of approximately 110 kDa and another of >300 kDa, which were absent in BCAM0224 mutant extracts (Fig. 2A). The 110-kDa product is above the expected BCAM0224 molecular mass (86 kDa), which could be due to posttranslational modifications or altered migration as a consequence of the protein profile. The higher-mass product is about three times the size of the lower-mass product, corresponding to trimeric forms that are distinctive of TAAs. The complete disintegration of the trimers occurred with the higher concentration of urea (8 M) used (Fig. 2A).
FIG 2.

Subcellular localization and trimerization of BCAM0224. (A) Denatured Western blot of B. cenocepacia membrane extracts, probed with a polyclonal antibody specific for BCAM0224. The monomeric and trimeric forms of BCAM0224 are indicated. (B) Fluorescence confocal microscopy images of B. cenocepacia K56-2 and the BCAM0224::Tp mutant using the anti-BCAM0224 antibody. Bacteria were labeled with DsRed (red) or DAPI (blue), and BCAM0224 was visualized with Alexa Fluor 488 (green). The membrane-associated fluorescence signal of anti-BCAM0224-Alexa 488 corresponds to 88% ± 2% of total cellular Alexa 488 fluorescence after immunostaining.
To demonstrate the surface localization of BCAM0224, we performed immunofluorescence confocal microscopy (Fig. 2B) using the anti-BCAM0224 antibody. B. cenocepacia K56-2 bacteria harboring a plasmid expressing a red protein or labeled with DAPI showed reactivity to the specific antibody (green) only at the cell surface (88% ± 2% of total fluorescence after immunostaining). No reaction was observed with BCAM0224 mutant cells (Fig. 2B).
BCAM0224 mutation reduces biofilm formation and motility of B. cenocepacia K56-2.
Bacterial TAAs are often associated with the trait of biofilm formation (32, 33). We therefore examined the role of BCAM0224 mutation in biofilm formation by B. cenocepacia K56-2. The results presented in Fig. 3A show that compared to the wild type, the BCAM0224::Tp mutant exhibits a significant decrease in biofilm formation on polystyrene surfaces after 24 h and 48 h of incubation (P < 0.01). Complementation of the B. cenocepacia K56-2 BCAM0224::Tp mutant with the BCAM0224 gene in trans (BCAM0224::Tp pDM6) restored the biofilm formation phenotype of the B. cenocepacia K56-2 strain (Fig. 3A), indicating that the reduction in biofilm formation was due to the lack of the BCAM0224 gene.
FIG 3.

(A) Biofilm formation in polystyrene microtiter plates by wild-type B. cenocepacia K56-2 and the BCAM0224::Tp mutant at 24 and 48 h. Complementation of the mutant was achieved by using plasmid pIN177-BCAM0224 (pDM6) and the empty plasmid as a control. Biofilm growth was quantified by the solubilization of crystal violet-stained cells with ethanol. Under the experimental conditions used, the strains had similar growth rates. The BCAM0224::Tp mutant showed a decreased capacity to form biofilm compared to the wild-type strain (**, P < 0.01) by Tukey's multiple-comparison test for unequal group sample sizes. (B) Swarming and swimming motilities after 48 h of incubation of wild-type B. cenocepacia K56-2, the BCAM0224::Tp mutant, the BCAM0224::Tp mutant with empty vector pIN177, and the BCAM0224::Tp mutant complemented with pIN177-BCAM0224 (pDM6). The graph translates halo diameter measurements from three independent experiments. The mutant has motility that is significantly affected compared to that of the wild-type strain by Bonferroni's multiple test (**, P < 0.01 for swarming; *, P < 0.05 for swimming).
At different stages of biofilm formation, motility is implied in diverse ways (34). This prompted us to evaluate whether the BCAM0224::Tp mutant displayed repressed swarming and swimming phenotypes. Indeed, the lack of BCAM0224 significantly reduced the swarming capacity and only attenuated swimming motility (P < 0.01 and P < 0.05, respectively) (Fig. 3B). Complementation of the BCAM0224::Tp mutant with the wild-type BCAM0224 gene partially restored the motility phenotype (Fig. 3B).
BCAM0224 is required for B. cenocepacia K56-2 serum resistance.
To determine whether BCAM0224 is involved in the capacity of B. cenocepacia K56-2 to resist complement killing, we conducted a comparison of serum killing assays between the wild type and the mutant, using 30% normal human serum (NHS). As shown in Fig. 4A, after 1 h of incubation at 37°C, the BCAM0224::Tp mutant was found to be sensitive to serum (15% survival), whereas the parental strain was completely resistant (97% survival). Both strains survived uniformly in heat-inactivated NHS (hNHS), used as a control. Restoration of the serum resistance phenotype in the complemented mutant (BCAM0224::Tp pDM6) confirms that the serum sensitivity of the mutant was due to the lack of the BCAM0224 gene product (Fig. 4A). Additionally, the BCAM0224::Tp mutant carrying empty vector pIN177 was used as a control to account for any potential effects of the plasmid per se on serum resistance. The observed effect may be the result of the use of a extra antibiotic (chloramphenicol) necessary for plasmid maintenance.
FIG 4.

Serum resistance. (A) Serum bactericidal assay with 30% normal human serum (NHS) incubated with B. cenocepacia K56-2 and the BCAM0224::Tp mutant showing impaired sensitivity of the mutant to serum killing (***, P < 0.001; *, P < 0.05). Complementation in trans of the mutant was achieved by using plasmid pIN177-BCAM0224 (pDM6). The BCAM0224::Tp mutant carrying the empty vector pIN177 was used as a control. Heat-inactivated NHS (hNHS) was used as the control for the experiment. (B, top) Determination of the pathway involved in serum killing of the BCAM0224 mutant, performed with various sera, including hNHS, NHS, NHS plus 10 mM MgCl2 and 10 mM EGTA (LC−NHS) (lectin and classical pathways blocked), factor B-depleted human serum (B−NHS) (alternative pathway blocked), and C1q-depleted serum (C−NHS) (classical pathway blocked). B. cenocepacia K56-2 showed resistance to all sera, whereas the BCAM0224::Tp mutant was equivalently sensitive to NHS and B−NHS, showing that the mutant is killed via the classical pathway. All the results are from three independent experiments; bars indicate SD. (Bottom) Schematic representation of the three complement pathways illustrating blockage by the sera used. MASP, mannose-associated serine protease.
To investigate further how BCAM0224 contributes to the evasion of B. cenocepacia K56-2 of complement-dependent killing by NHS, we selectively blocked the complement activation pathways (Fig. 4B). First, we blocked both the classical and lectin pathways with NHS containing MgCl2 and the chelating agent EGTA to remove Ca2+ (LC−NHS) (35). Ca2+ is essential for complement activation, and thereby, only the alternative pathway remains active. Under such conditions, the wild type and the BCAM0224::Tp mutant survived equally (about 100%) (Fig. 4B). Second, we used factor B-depleted serum (B−NHS), which selectively blocks the alternative pathway. Contrary to the above-described findings, the use of B−NHS serum restored its killing activity against the BCAM0224::Tp mutant (23% survival; P < 0.001) but had little effect on the wild-type strain (92% survival) (Fig. 4B). Third, to validate the results and distinguish between the classical and lectin pathways, we conducted assays using C1q-depleted serum (C−NHS) (selectively blocks the classical pathway). No or minimal killing was observed for both strains (Fig. 4B). Altogether, our results suggest that the killing of the serum-sensitive BCAM0224 mutant is mediated by the classical pathway.
BCAM0224 contributes to adhesion to and invasion of human bronchial epithelial cells.
We further investigated the possible role of BCAM0224 in adhesion and invasion of two human bronchial epithelial cell lines (16HBE14o− and CFBE41o−), which have non-cystic fibrosis (CF) and CF phenotypes, respectively (24, 25). Analysis by confocal microscopy revealed that lower levels of the BCAM0224::Tp mutant adhered to the cells (Fig. 5A). Moreover, cell monolayers were infected with B. cenocepacia K56-2 and the BCAM0224::Tp mutant, and cell-associated bacteria were detected by plating of cell lysates. For both cell lines, the BCAM0224::Tp mutant showed decreased adhesion levels compared to the wild type, being more pronounced in non-CF 16HBE14o− cells. Complementation with BCAM0224 in trans partially restored the wild-type phenotype (Fig. 5B). To determine whether the presence of the empty vector alone affects the levels of adhesion, the BCAM0224::Tp mutant carrying plasmid pIN177 was used as a control.
FIG 5.

Cellular adhesion. (A) Fluorescence confocal microscopy images of B. cenocepacia K56-2 and the BCAM0224::Tp mutant adhering to 16HBE14o− (non-CF) and CFBE41o− (CF) cells. Images correspond to 3D projections obtained from 0.4-μm confocal slices of a monolayer of cells. Eukaryotic cell plasma membranes and nuclei were visualized by using Alexa 488 (green) and DAPI (blue) fluorophores, respectively; bacteria were labeled with DsRed (red). (B, top) Adherence to 16HBE14o− (non-CF) and CFBE41o− (CF) epithelial cell lines by the BCAM0224::Tp mutant, expressed as a ratio of B. cenocepacia K56-2 adherence (***, P < 0.001). Complementation with BCAM0224 in trans using plasmid pDM6 partially restores the wild-type phenotype. The BCAM0224::Tp mutant harboring empty vector pIN177 was used as a control (**, P < 0.01; *, P < 0.05, respectively). (Bottom) Inhibition of epithelial cell adhesion was achieved by preincubation of B. cenocepacia K56-2 and the BCAM0223::Tp mutant with anti-BCAM0224 antibody (open and striped bars, respectively) (***, P < 0.01; **, P < 0.01; *, P < 0.05). No inhibitory activity was observed with an irrelevant rabbit serum used as a negative control. (C) Adherence to 16HBE14o− (non-CF) and CFBE41o− (CF) cells using E. coli strain BL21 expressing BCAM0224, expressed as a ratio of E. coli cells harboring the empty plasmid (***, P < 0.001; **, P < 0.01).
The capacity of the anti-BCAM0224 antibody to influence the adhesion of B. cenocepacia K56-2 was also analyzed. As shown in Fig. 5B, preincubation of B. cenocepacia K56-2 with anti-BCAM0224 resulted in a significant decrease in adherence (P < 0.001), similar to that observed for the BCAM0224::Tp mutant. We also used the adhesion-deficient B. cenocepacia BCAM0223::Tp mutant, as previously reported (21), as a control. The phenotype of the BCAM0223::Tp mutant treated with anti-BCAM0224 antibody is additive, displaying substantially reduced levels of binding to host cells (P < 0.01 for 16HBE14o− and P < 0.05 for CFBE41o− cells) (Fig. 5B). Furthermore, we used a nonpathogenic and noninvasive E. coli strain expressing BCAM0224 to evaluate the adhesion capacity of this protein. The adhesion assay, performed with both cell lines, showed a significant fold increase for E. coli expressing BCAM0224 in contrast to the empty vector (Fig. 5C), being more marked in non-CF 16HBE14o− cells (P < 0.001).
Some TAAs have the ability not only to adhere to epithelial cells but also to invade them. To test this tenet, CF and non-CF cell monolayers were infected with wild-type and mutant B. cenocepacia strains, and their invasiveness was evaluated by using a modified gentamicin invasion assay. Antibiotics were used to promote extracellular bacterial death. The quantification of intracellular bacteria after 4 h of infection revealed a reduction of invasion by the BCAM0224::Tp mutant compared to the parental strain (Fig. 6A) for both cell lines (P < 0.001 for 16HBE14o− cells and P < 0.01 for CFBE41o− cells). Furthermore, as shown in Fig. 6A, CF cells had a decreased level of invasion compared to the wild-type cell line. Next, we demonstrated that the BCAM0224 gene partially complemented the mutation on the chromosome (Fig. 6A). Finally, we also used B. cenocepacia K56-2 preincubated with anti-BCAM0224 antibody to reinforce the possible involvement of BCAM0224 in cellular invasion. The results show a significant decrease of invasion for bacteria treated with the anti-BCAM0224 antibody in both CF and non-CF cell monolayers (P < 0.01 and P < 0.001, respectively) (Fig. 6B). As for adhesion, we used the BCAM0223::Tp mutant treated with anti-BCAM0224 antibody as a control. This combination acts synergistically to decrease the ability of the mutant strain to invade host cells (P < 0.05 for 16HBE14o− cells) (Fig. 6B). Taken together, our results indicate that BCAM0224 mediates bacterial adhesion to and invasion of the 16HBE14o− (non-CF) and CFBE41o− (CF) epithelial cell lines.
FIG 6.
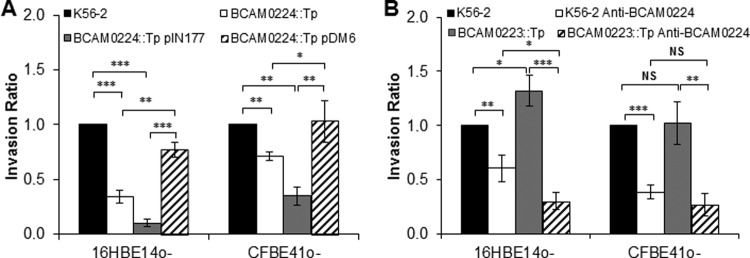
Role of BCAM0224 in cell invasion. (A) Invasion 4 h after infection of the 16HBE14o− (non-CF) and CFBE41o− (CF) epithelial cell lines by the BCAM0224::Tp mutant, expressed as a ratio of B. cenocepacia K56-2 cells (***, P < 0.001; **, P < 0.01). The invasion results for BCAM0224::Tp mutant complementation using plasmid pDM6 and the empty plasmid are shown for both cell lines (***, P < 0.001; **, P < 0.01; *, P < 0.05). (B) Inhibition of epithelial cell invasion was achieved by preincubation of B. cenocepacia K56-2 and the BCAM0223::Tp mutant with anti-BCAM0224 antibody (**, P < 0.01; ***, P < 0.001). No inhibitory activity was observed with an irrelevant rabbit serum, used as a negative control. Results are expressed as a ratio of B. cenocepacia K56-2 values (NS, no statistical significance). All results are from three independent experiments; bars indicate SD.
DISCUSSION
In a previous study, we identified a gene, BCAM0224, encoding a trimeric autotransporter adhesin (TAA), which appears to be restricted to epidemic-associated B. cenocepacia strains of the ET-12 lineage (22). Transcriptional analysis revealed that BCAM0224 is located within a bicistronic operon composed of 9 genes, which comprises, among others, two additional TAA-encoding genes (BCAM0219 and BCAM0223) (21). In an attempt to characterize the functions of the TAA-encoding genes in B. cenocepacia, we have generated mutations and analyzed the associated phenotypes. Based on this, we previously proposed that the BCAM0224 protein is involved in virulence, since the BCAM0224 mutant was less virulent than the wild-type strain in an insect infection model (22). In addition, disruption of the BCAM0223 gene impairs the ability of the mutant strain to adhere to epithelial cells, decreases serum resistance, and reduces virulence (21). Finally, in a recent report, we used atomic force microscopy (AFM) to study association constants and conformational properties of the BCAM0224 protein. We have determined homophilic trans-interactions (BCAM0224-BCAM0224) as well as heterophilic interactions (BCAM0224 with collagen). Both interactions displayed relatively low binding affinity, which could be important for epithelium colonization (23).
In the present study, we are continuing to characterize the role of the BCAM0224 protein, aiming to assess its contribution to the overall pathogenicity of B. cenocepacia. The BCAM0224::Tp mutant described previously (22) was used in this study. To investigate whether the insertion of a trimethoprim resistance cassette within the BCAM0224 gene may have polar effects on the expression of the neighboring gene BCAM0223, we performed real-time reverse transcription-PCR. The results obtained showed that the BCAM0224 mutation does not affect the downstream gene in the operon (data not shown).
Although the structure of a full-length TAA has not yet been solved, structures for individual domains have been determined (36). In this study, the BCAM0224 3D structures of various domains were modeled by using I-TASSER, an automated structure prediction tool. Although the primary sequences between the query BCAM0224 and the templates were significantly different, I-TASSER is effective at predicting the structures. When displayed on the cell surface, BCAM0224 appears to comprise one apical YadA-like head domain followed by an extended stalk and finally a second head. Overall, approximately 63% of the surface-exposed structure is made up of stacks of beta-sheets (Fig. 1). It is noteworthy that the structure of the stalk is unique among TAAs in lacking the common coiled-coil structural motif. In contrast, the hydrophobic C-terminal domain (anchor domain) represents the most highly conserved domain within the TAA proteins. Finally, in good agreement with bioinformatic predictions, we detected the presence of BCAM0224 assembled into trimers in the membrane of wild-type and BCAM0224-complemented strains of B. cenocepacia K56-2 (Fig. 2A). These trimeric structures were stable under standard denaturing treatment conditions (heating and SDS) but dissociated completely in the presence of chaotropes (e.g., 8 M urea), a common feature of TAAs (37). By confocal microscopy, we were able to confirm the localization of BCAM0224 at the bacterial surface (Fig. 2B).
Being an adhesin, we analyzed the possible involvement of BCAM0224 in biofilm formation. In fact, the BCAM0224::Tp mutant showed a decreased capacity to form biofilm on a polystyrene surface (Fig. 3), suggesting some involvement of BCAM0224 in biofilm formation. This phenotype has been implicated in the persistence of pathogens in their host (34) and is also associated with many TAA proteins (32, 33). Additionally, it is recognized that motility influences biofilm formation at different stages (34). Indeed, at the initial stages of biofilm formation, motility is required and crucial for biofilm architecture (34). We saw that mutation of the BCAM0224 gene abolished flagellum-based motility (swarming), thereby contributing to the observed impaired biofilm formation of the BCAM0224::Tp mutant. It should be noted that the crystal violet-based assay likely represents only a few early events in biofilm formation (38). Therefore, we hypothesize that BCAM0224 expression in response to particular environmental conditions might be involved in fine-tuning the balance between surface motility and biofilm.
Several bacterial pathogens are known to be resistant to the innate immune system, being able to escape bactericidal killing by complement. B. cenocepacia K56-2 is resistant to the bactericidal effect of serum complement (39), and we showed in our previous work that the TAA-encoding BCAM0223 gene is involved in this capacity (21). In fact, TAAs are known to play a major role in serum resistance, and some bacteria possess more than one TAA involved in this process, like M. catarrhalis and E. coli (6, 7, 13, 14). In the present study, we demonstrated that the BCAM0224 TAA is also involved in normal human serum resistance. The BCAM0224 mutant was serum sensitive, and complementation in trans restored serum resistance although not completely (Fig. 4). This could be the result of an excessive level of BCAM0224 in the complemented strain, which could be harmful to the host and thereby may explain the partial complementation of the phenotypes (data not shown). Furthermore, we conclude that the classical complement pathway is essential for the killing of the BCAM0224 mutant. The mechanism of action of BCAM0224 in the classical pathway is not yet known; however, it is possible that it uses of one of the typical strategies used by other pathogens, such as the recruitment or mimicking of complement regulators/inhibitors and the modulation or inhibition of complement proteins by direct interactions (40). Interestingly, BCAM0224 promotes adhesion to vitronectin (data not shown), a known inhibitor of the complement cascade acting to limit the self-reactivity of the innate immune response (41, 42). The ability to bind vitronectin suggests that BCAM0224 could use this property to inhibit complement-mediated attack. On the other hand, BCAM0224 vitronectin binding confers bacterial camouflage with nonimmunogenic proteins. Several TAAs have been described as the key points for vitronectin-dependent serum resistance in bacterial pathogens. Indeed, vitronectin binding to TAAs was shown to contribute to serum resistance in other pathogens, such as Hsf from H. influenzae type b (41), DsrA from H. ducreyi (6), and UspA2 from M. catarrhalis (43).
Attachment to lung epithelial cells is a preliminary step in lung colonization by Bcc species. Sajjan and coauthors (44) demonstrated the role of cable pili in association with the 22-kDa adhesin in the binding of piliated B. cenocepacia to lung explants from CF patients. Furthermore, we recently showed that BCAM0223 (neighbor of the BCAM0224 gene) is also involved in host cell adhesion (21). Here, we provide evidence that BCAM0224 is another important adhesin involved in B. cenocepacia (ET-12 lineage) adhesion to CF (CFBE41o−) and non-CF (16HBE14o−) cultured human epithelial cells (Fig. 5). We found that the inhibitory effects of both BCAM0223 and BCAM0224 on the adherence of B. cenocepacia to host cells appeared to have a more profound effect on cells with a non-CF phenotype (Fig. 5) (21). These observations suggest that, at least in vitro and under the experimental conditions used, CF cells may have BCAM0223 and BCAM0224 adherence receptors on the surface at a lower level.
Another important step of Bcc infection is the capacity to invade epithelial cells. Several reports have shown that B. cenocepacia J2315 is able to invade human respiratory epithelial cells in culture and that this process is facilitated by flagellar and lipase activities (45–47). Entry into epithelial cells is a multifactorial action of pathogens that enables infection by facilitating the evasion of immune responses, penetration of host epithelial barriers, and migration through host tissues. Ultimately, this results in the pathogen becoming blood borne, an outcome of systemic infection. Our findings indicate that BCAM0224 is involved in the invasion capacity of B. cenocepacia isolates of the ET-12 lineage (Fig. 6A) in both cell lines. However, the lack of flagellum-based motility of the BCAM0224 mutant (Fig. 3B) could be involved in the decreased invasion of this mutant. More studies will be needed to understand the correlations of BCAM0224 and motility. The combination of multivalent interactions with host cells, such as adhesion and invasion, observed for BCAM0224 was also described for other TAAs, such as ApiA from Actinobacillus actinomycetemcomitans (48), HadA and NadA from N. meningitides (8, 49), and YadBC from Yersinia pestis (50).
In this study, we observed that the use of an anti-BCAM0224 antibody inhibits the ability of wild-type B. cenocepacia strain K56-2 to adhere to and invade host cells in vitro. We also tested the BCAM0223::Tp mutant (21) for the inhibitory effect of an anti-BCAM0224 antibody, resulting in a synergistic defect in cell adhesion and invasion. This finding indicates the possible use of anti-BCAM0224 antibody for passive immunization in antiadhesion therapies. In fact, adhesin-based vaccines have emerged as a new attractive approach to treat bacterial diseases (51), and a recent study points to anti-TAA antibody as a valuable vaccine candidate against Acinetobacter baumannii infections (52). Research is under way in our laboratory to pursue this possibility.
In conclusion, we characterized in detail the multifunctional TAA BCAM0224 from B. cenocepacia and showed that it affects earlier stages of biofilm formation and motility. This protein also plays an important role in the evasion of the host immune system, providing bactericidal serum resistance via the classical pathway. Moreover, it seems to be important for epithelial cell adhesion and invasion. Altogether, these results underlie the high versatility of the BCAM0224 virulence factor, which plays multiple roles in the pathogenesis of B. cenocepacia. By comparing the multifaceted activity of the BCAM0224 TAA with the multifunctionality associated with the disruption of the TAA gene BCAM0223 (21), we speculate that they can work together, cooperating for an established trait.
ACKNOWLEDGMENTS
This work was supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal (grants PTDC/EBB-BIO/100326/2008 and PTDC/BIA-MIC/118386/2010), and by a postdoctoral fellowship to D.M.-H.
We express our gratitude to A. Vergunst and D. O'Callaghan from INSERM, Université de Montpellier, France, for kindly providing plasmids pIN29 and pIN177 and for all their help. We gratefully acknowledge M. Prieto from the Centro de Química Física Molecular, Instituto Superior Técnico, Portugal, for the confocal microscope. We acknowledge Conceição Amado from the Department of Mathematics, Instituto Superior Técnico, for assistance in the statistical analysis of data. We thank Dieter Gruenert, Pacific Medical Center Research Institute, San Francisco, CA, for providing us with the 16HBE14o− and CFBE41o− cell lines. We also acknowledge John LiPuma from the University of Michigan, who kindly provided a Burkholderia strain.
Footnotes
Published ahead of print 21 March 2014
REFERENCES
- 1.Cotter SE, Surana NK, St Geme JW., III 2005. Trimeric autotransporters: a distinct subfamily of autotransporter proteins. Trends Microbiol. 13:199–205. 10.1016/j.tim.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 2.Linke D, Riess T, Autenrieth IB, Lupas A, Kempf VAJ. 2006. Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol. 14:264–270. 10.1016/j.tim.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 3.Łyskowski A, Leo JC, Goldman A. 2011. Structure and biology of trimeric autotransporter adhesins. Adv. Exp. Med. Biol. 715:143–158. 10.1007/978-94-007-0940-9_9 [DOI] [PubMed] [Google Scholar]
- 4.Pearson MM, Laurence CA, Guinn SE, Hansen EJ. 2006. Biofilm formation by Moraxella catarrhalis in vitro: roles of the UspA1 adhesin and the Hag hemagglutinin. Infect. Immun. 74:1588–1596. 10.1128/IAI.74.3.1588-1596.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fitzgerald M, Mulcahy R, Murphy S, Keane C, Coakley D, Scott T. 1997. A 200 kDa protein is associated with haemagglutinating isolates of Moraxella (Branhamella) catarrhalis. FEMS Immunol. Med. Microbiol. 18:209–216. 10.1111/j.1574-695X.1997.tb01047.x [DOI] [PubMed] [Google Scholar]
- 6.Elkins C, Morrow KJ, Jr, Olsen B. 2000. Serum resistance in Haemophilus ducreyi requires outer membrane protein DsrA. Infect. Immun. 68:1608–1619. 10.1128/IAI.68.3.1608-1619.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aebi C, Lafontaine ER, Cope LD, Latimer JL, Lumbley SL, McCracken GHJ, Hansen EJ. 1998. Phenotypic effect of isogenic uspA1 and uspA2 mutations on Moraxella catarrhalis 035E. Infect. Immun. 66:3113–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serruto D, Spadafina T, Scarselli M, Bambini S, Comanducci M, Höhle S, Kilian M, Veiga E, Cossart P, Oggioni MR, Savino S, Ferlenghi I, Taddei AR, Rappuoli R, Pizza M, Masignani V, Aricò B. 2009. HadA is an atypical new multifunctional trimeric coiled-coil adhesin of Haemophilus influenzae biogroup aegyptius, which promotes entry into host cells. Cell. Microbiol. 11:1044–1063. 10.1111/j.1462-5822.2009.01306.x [DOI] [PubMed] [Google Scholar]
- 9.Heise T, Dersch P. 2006. Identification of a domain in Yersinia virulence factor YadA that is crucial for extracellular matrix-specific cell adhesion and uptake. Proc. Natl. Acad. Sci. U. S. A. 103:3375–3380. 10.1073/pnas.0507749103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kline KA, Fälker S, Dahlberg S, Normark S, Henriques-Normark B. 2009. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5:580–592. 10.1016/j.chom.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 11.Pilz D, Vocke T, Heesemann J, Brade V. 1992. Mechanism of YadA-mediated serum resistance of Yersinia enterocolitica serotype O3. Infect. Immun. 60:189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sjolinder H, Eriksson J, Maudsdotter L, Aro H, Jonsson A-B. 2008. Meningococcal outer membrane protein NhhA is essential for colonization and disease by preventing phagocytosis and complement attack. Infect. Immun. 76:5412–5420. 10.1128/IAI.00478-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandt CH, Hill CW. 2000. Four different genes responsible for nonimmune immunoglobulin-binding activities within a single strain of Escherichia coli. Infect. Immun. 68:2205–2214. 10.1128/IAI.68.4.2205-2214.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandt CH, Hill CW. 2001. Nonimmune binding of human immunoglobulin A (IgA) and IgG Fc by distinct sequence segments of the EibF cell surface protein of Escherichia coli. Infect. Immun. 69:7293–7303. 10.1128/IAI.69.12.7293-7203.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asakawa R, Komatsuzawa H, Kawai T, Yamada S, Goncalves RB, Izumi S, Fujiwara T, Nakano Y, Suzuki N, Uchida Y, Ouhara K, Shiba H, Taubman MA, Kurihara H, Sugai M. 2003. Outer membrane protein 100, a versatile virulence factor of Actinobacillus actinomycetemcomitans. Mol. Microbiol. 50:1125–1139. 10.1046/j.1365-2958.2003.03748.x [DOI] [PubMed] [Google Scholar]
- 16.Drevinek P, Mahenthiralingam E. 2010. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 16:821–830. 10.1111/j.1469-0691.2010.03237.x [DOI] [PubMed] [Google Scholar]
- 17.Mahenthiralingam E, Urban TA, Goldberg JB. 2005. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3:144–156. 10.1038/nrmicro1085 [DOI] [PubMed] [Google Scholar]
- 18.Johnson WM, Tyler SD, Rozee KR. 1994. Linkage analysis of geographic and clinical clusters in Pseudomonas cepacia infections by multilocus enzyme electrophoresis and ribotyping. J. Clin. Microbiol. 32:924–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ledson MJ, Gallagher MJ, Jackson M, Hart CA, Walshaw MJ. 2002. Outcome of Burkholderia cepacia colonisation in an adult cystic fibrosis centre. Thorax 57:142–145. 10.1136/thorax.57.2.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mil-Homens D, Fialho AM. 2011. Trimeric autotransporter adhesins in members of the Burkholderia cepacia complex: a multifunctional family of proteins implicated in virulence. Front. Cell. Infect. Microbiol. 1:13. 10.3389/fcimb.2011.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mil-Homens D, Fialho AM. 2012. A BCAM0223 mutant of Burkholderia cenocepacia is deficient in hemagglutination, serum resistance, adhesion to epithelial cells and virulence. PLoS One 7:e41747. 10.1371/journal.pone.0041747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mil-Homens D, Rocha EPC, Fialho AM. 2010. Genome-wide analysis of DNA repeats in Burkholderia cenocepacia J2315 identifies a novel adhesin-like gene unique to epidemic-associated strains of the ET-12 lineage. Microbiology 156:1084–1096. 10.1099/mic.0.032623-0 [DOI] [PubMed] [Google Scholar]
- 23.El-Kirat-Chatel S, Mil-Homens D, Beaussart A, Fialho AM, Dufrêne YF. 2013. Single-molecule atomic force microscopy unravels the binding mechanism of a Burkholderia cenocepacia trimeric autotransporter adhesin. Mol. Microbiol. 89:649–659. 10.1111/mmi.12301 [DOI] [PubMed] [Google Scholar]
- 24.Bruscia E, Sangiuolo F, Sinibaldi P, Goncz K, Novelli G, Gruenert D. 2002. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 9:683–685. 10.1038/sj.gt.3301741 [DOI] [PubMed] [Google Scholar]
- 25.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. 1994. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10:38–47. 10.1165/ajrcmb.10.1.7507342 [DOI] [PubMed] [Google Scholar]
- 26.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. 2004. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. 10.1093/bioinformatics/btg371 [DOI] [PubMed] [Google Scholar]
- 28.Vergunst AC, Meijer AH, Renshaw SA, O'Callaghan D. 2010. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect. Immun. 78:1495–1508. 10.1128/IAI.00743-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto SN, Silva LC, de Almeida RFM, Prieto M. 2008. Membrane domain formation, interdigitation, and morphological alterations induced by the very long chain asymmetric C24:1 ceramide. Biophys. J. 95:2867–2879. 10.1529/biophysj.108.129858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szczesny P, Lupas A. 2008. Domain annotation of trimeric autotransporter adhesions—daTAA. Bioinformatics 24:1251–1256. 10.1093/bioinformatics/btn118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grosskinsky U, Schütz M, Fritz M, Schmid Y, Lamparter MC, Szczesny P, Lupas AN, Autenrieth IB, Linke D. 2007. A conserved glycine residue of trimeric autotransporter domains plays a key role in Yersinia adhesin A autotransport. J. Bacteriol. 189:9011–9019. 10.1128/JB.00985-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Totsika M, Wells TJ, Beloin C, Valle J, Allsopp LP, King NP, Ghigo J-M, Schembri MA. 2012. Molecular characterization of the EhaG and UpaG trimeric autotransporter proteins from pathogenic Escherichia coli. Appl. Environ. Microbiol. 78:2179–2189. 10.1128/AEM.06680-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghunathan D, Wells TJ, Morris FC, Shaw RK, Bobat S, Peters SE, Paterson GK, Jensen KT, Leyton DL, Blair JMA, Browning DF, Pravin J, Flores-Langarica A, Hitchcock JR, Moraes CTP, Piazza RMF, Maskell DJ, Webber MA, May RC, MacLennan CA, Piddock LJ, Cunningham AF, Henderson IR. 2011. SadA, a trimeric autotransporter from Salmonella enterica serovar Typhimurium, can promote biofilm formation and provides limited protection against infection. Infect. Immun. 79:4342–4352. 10.1128/IAI.05592-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verstraeten N, Braeken K, Debkumari B, Fauvart M, Fransaer J, Vermant J, Michiels J. 2008. Living on a surface: swarming and biofilm formation. Trends Microbiol. 16:496–506. 10.1016/j.tim.2008.07.004 [DOI] [PubMed] [Google Scholar]
- 35.James K. 1982. Complement: activation, consequences, and control. Am. J. Med. Technol. 48:735–742 [PubMed] [Google Scholar]
- 36.Hartmann MD, Grin I, Dunin-Horkawicz S, Deiss S, Linke D, Lupas AN, Hernandez Alvarez B. 2012. Complete fiber structures of complex trimeric autotransporter adhesins conserved in enterobacteria. Proc. Natl. Acad. Sci. U. S. A. 109:20907–20912. 10.1073/pnas.1211872110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoiczyk E, Roggenkamp A, Reichenbecher M, Lupas A, Heesemann J. 2000. Structure and sequence analysis of Yersinia YadA and Moraxella UspAs reveal a novel class of adhesins. EMBO J. 19:5989–5999. 10.1093/emboj/19.22.5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461. 10.1046/j.1365-2958.1998.00797.x [DOI] [PubMed] [Google Scholar]
- 39.Ortega X, Hunt TA, Loutet S, Vinion-Dubiel AD, Datta A, Choudhury B, Goldberg JB, Carlson R, Valvano MA. 2005. Reconstitution of O-specific lipopolysaccharide expression in Burkholderia cenocepacia strain J2315, which is associated with transmissible infections in patients with cystic fibrosis. J. Bacteriol. 187:1324–1333. 10.1128/JB.187.4.1324-1333.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat. Rev. Microbiol. 6:132–142. 10.1038/nrmicro1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preissner KT, Seiffert D. 1998. Role of vitronectin and its receptors in haemostasis and vascular remodeling. Thromb. Res. 89:1–21. 10.1016/S0049-3848(97)00298-3 [DOI] [PubMed] [Google Scholar]
- 42.Hallström T, Trajkovska E, Forsgren A, Riesbeck K. 2006. Haemophilus influenzae surface fibrils contribute to serum resistance by interacting with vitronectin. J. Immunol. 177:430–436 http://www.jimmunol.org/content/177/1/430.long [DOI] [PubMed] [Google Scholar]
- 43.Attia AS, Ram S, Rice PA, Hansen EJ. 2006. Binding of vitronectin by the Moraxella catarrhalis UspA2 protein interferes with late stages of the complement cascade. Infect. Immun. 74:1597–1611. 10.1128/IAI.74.3.1597-1611.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sajjan U, Sylvester FA, Forstner JF. 2000. Cable-piliated Burkholderia cepacia binds to cytokeratin 13 of epithelial cells. Infect. Immun. 68:1787–1795. 10.1128/IAI.68.4.1787-1795.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin DW, Mohr CD. 2000. Invasion and intracellular survival of Burkholderia cepacia. Infect. Immun. 68:24–29. 10.1128/IAI.68.1.24-29.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullen T, Markey K, Murphy P, McClean S, Callaghan M. 2007. Role of lipase in Burkholderia cepacia complex (Bcc) invasion of lung epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 26:869–877. 10.1007/s10096-007-0385-2 [DOI] [PubMed] [Google Scholar]
- 47.Tomich M, Herfst CA, Golden JW, Mohr CD. 2002. Role of flagella in host cell invasion by Burkholderia cepacia. Infect. Immun. 70:1799–1806. 10.1128/IAI.70.4.1799-1806.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Matevski D, Aspiras M, Ellen RP, Lépine G. 2004. Two epithelial cell invasion-related loci of the oral pathogen Actinobacillus actinomycetemcomitans. Oral Microbiol. Immunol. 19:16–25. 10.1046/j.0902-0055.2003.00102.x [DOI] [PubMed] [Google Scholar]
- 49.Capecchi B, Adu-Bobie J, Di Marcello F, Ciucchi L, Masignani V, Taddei A, Rappuoli R, Pizza M, Aricò B. 2005. Neisseria meningitidis NadA is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol. Microbiol. 55:687–698. 10.1111/j.1365-2958.2004.04423.x [DOI] [PubMed] [Google Scholar]
- 50.Forman S, Wulff CR, Myers-Morales T, Cowan C, Perry RD, Straley SC. 2008. yadBC of Yersinia pestis, a new virulence determinant for bubonic plague. Infect. Immun. 76:578–587. 10.1128/IAI.00219-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cozens D, Read RC. 2012. Anti-adhesion methods as novel therapeutics for bacterial infections. Expert Rev. Anti Infect. Ther. 10:1457–1468. 10.1586/eri.12.145 [DOI] [PubMed] [Google Scholar]
- 52.Bentancor LV, Routray A, Bozkurt-Guzel C, Camacho-Peiro A, Pier GB, Maira-Litrán T. 2012. Evaluation of the trimeric autotransporter Ata as a vaccine candidate against Acinetobacter baumannii infections. Infect. Immun. 80:3381–3388. 10.1128/IAI.06096-11 [DOI] [PMC free article] [PubMed] [Google Scholar]