

Abstract
We report the development of SearchDOGS Bacteria, software to automatically detect missing genes in annotated bacterial genomes by combining BLAST searches with comparative genomics. Having successfully applied the approach to yeast genomes, we redeveloped SearchDOGS to function as a standalone, downloadable package, requiring only a set of GenBank annotation files as input. The software automatically generates a homology structure using reciprocal BLAST and a synteny-based method; this is followed by a scan of the entire genome of each species for unannotated genes. Results are provided in a HTML interface, providing coordinates, BLAST results, syntenic location, omega values (Ka/Ks, where Ks is the number of synonymous substitutions per synonymous site and Ka is the number of nonsynonymous substitutions per nonsynonymous site) for protein conservation estimates, and other information for each candidate gene. Using SearchDOGS Bacteria, we identified 155 gene candidates in the Shigella boydii sb227 genome, including 56 candidates of length < 60 codons. SearchDOGS Bacteria has two major advantages over currently available annotation software. First, it outperforms current methods in terms of sensitivity and is highly effective at identifying small or highly diverged genes. Second, as a freely downloadable package, it can be used with unpublished or confidential data.
INTRODUCTION
With the rapidly decreasing cost and increasing speed of genome sequencing, a wealth of genome sequence information is becoming available to the scientific community. As of September 2013, Entrez Genome (http://www.ncbi.nlm.nih.gov) reported 2,664 completely sequenced prokaryotic genomes and another 11,552 in scaffold or contig form. However, the speed at which these new genomes can be accurately annotated is quickly becoming a bottleneck. In the vast majority of cases, protein-coding genes are annotated using automated programs (1), which for the most part can be divided into two classes: “composition-based” gene prediction programs that use characteristics of sequence composition to predict where protein-coding open reading frames exist (2–7) and “sequence similarity” programs that predict protein-coding open reading frames (ORFs) based on the identification of sequence similarity to annotated homologs in other species (8, 9).
However, both these approaches run into difficulties when annotating short or highly diverged genes. Statistical methods such as codon bias have less power in discriminating coding from noncoding DNA for short genes using the “composition-based” approach, and small or highly diverged genes return weak hits to homologs in BLAST searches and therefore cannot be accurately differentiated from ORFs that occur by chance (10). This uncertainty has resulted in overestimation of the number of protein-coding genes in annotated bacterial species (10–12) but also in many bona fide short genes being overlooked (8, 13–16).
One way to overcome this problem is to ascertain the syntenic context of the potential gene. If a potential unannotated gene is found to lie in the same local genomic region as its potential orthologs in other species, then there is a good likelihood that it is a bona fide gene even if it produces relatively weak BLAST hits to its orthologs. Identification of regions of conserved synteny can also be used to detect gene duplications, fusions, and paralogy relations in comparing multiple genomes (17) as well as to make functional-association predictions (18, 19).
We previously described the development of the software SearchDOGS (standing for “Searches against a Database of Orthologous Genomic Segments”), which uses conserved local synteny across species combined with BLASTX sequence similarity searches to identify genes that may have been missed in published annotations due to small size or a high level of divergence (20). Using this approach, we identified 594 previously undetected genes in 11 published Saccharomycetaceae family yeast genomes, including a number of new genes in well-studied model organisms such as Saccharomyces cerevisiae and Eremothecium gossypii. Many of the genes identified are very highly diverged, and 36% are less than 100 amino acids in length. We subsequently adapted the SearchDOGS method for application to 13 species from the “CTG” clade of yeasts, discovering over 1,400 previously unannotated genes (21).
Having applied the method successfully in yeast, we wished to extend the scope of SearchDOGS to allow it to be applicable to any set of suitable species where extensive local synteny can be established. Bacteria are an ideal candidate for the SearchDOGS approach due to their simple genomic architecture, low noncoding DNA content, and the large number of species to choose from for comparison. In this paper, we report the application of SearchDOGS to sets of strains and species from the gammaproteobacteria. This clade includes Gram-negative bacteria, many of which are common human pathogens, and contains the model organism Escherichia coli K-12 (22).
A host of powerful and comprehensive annotation pipelines, such as AGMIAL (23), AGeS (24), MicroScope (25), and NCBI's Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP), have been developed for bacterial genomes in recent years, with others under development. SearchDOGS Bacteria complements these platforms by providing an exceptionally sensitive tool for identifying the genes that prove most tricky for automated annotation programs. In contrast to the yeast implementation of SearchDOGS, which imported information about orthologous and paralogous sequence relationships from the YGOB database, SearchDOGS Bacteria was designed as a standalone piece of software. Thus, the only input data required are GenBank-formatted files of the relevant bacterial genomes. It allows the user to choose a number of annotated genomes to compare and automatically generates ortholog pillars: sets of genes that are orthologous between species. For a given species, it generates a list of candidate loci where a missing gene may exist and where an open reading frame showing sequence homology to orthologous genes has been identified. A detailed HTML output page is generated, providing syntenic location, coordinates, BLASTP results, and omega (Ka/Ks) values as an estimate of protein conservation (26). The aim is to provide the user with sufficient information to accept or reject each candidate with confidence. The software is freely downloadable from http://wolfe.ucd.ie, to be used locally, and thus is suitable for use with confidential or unpublished genomic data. It is written in Perl and runs in a Linux/UNIX environment.
MATERIALS AND METHODS
Overview of SearchDOGS search procedure.
After data input, the first stage of the SearchDOGS Bacteria program is that of automatically developing a set of ortholog pillars analogous to the homology pillars in YGOB. These pillars are used by SearchDOGs as “map coordinates”; for each genomic segment, the ortholog pillars corresponding to the two annotated genes allow the segment to be “mapped” to syntenic regions in other genomes. Ortholog pillars that map to the intergenic region of a genomic segment (i.e., where an ortholog is absent or unannotated in the genome in question but present in related genomes) can also be identified.
To find unannotated genes, each genome is then sliced into overlapping genomic segments containing two genes and the intergenic sequence between them. Each of these genomic segments is tested against the array of ortholog pillars to identify pillars that share synteny with it. The intergenic sequence between the two annotated genes in the genomic segment is then used as a BLASTX query against a small database consisting of the protein sequences of the genes in the syntenic ortholog pillars. For more detail on the search procedure, see Fig. S1 in the supplemental material; further details are also provided by OhÉigeartaigh et al. (20).
Generation of ortholog pillars.
An ortholog pillar represents a set of genes that are orthologous across species. An ortholog pillar is expected to contain a maximum of one gene per species, although for species lacking an ortholog at this locus a pillar “entry” would be lacking. In cases where duplication has occurred and duplicates remain colocated, the duplicate most similar in sequence to its orthologs is placed in the pillar; the other is placed in a new pillar.
Pillars of orthologs are generated using a two-step process. For each genome studied, pairwise BLASTP searches are performed using the protein sequence of each gene as a query against the protein set of the other species. Pillars of reciprocal best BLASTP hits are generated. In this first stage, a reciprocal best BLASTP hit is required for each ortholog against every other ortholog in the pillar, and a cutoff value of 1e-5 is used in the BLASTP searches.
The initial stage of pillar generation is followed by a “Syntenoblast” approach (27) to try to place the remaining genes in pillars (Fig. 1). A second round of BLASTP searches is carried out using a permissive cutoff value of E = 10. For each gene that hits a potential ortholog in another species, the syntenic context of the gene is evaluated. Five “steps out” are performed along the genome of the query and the genome of the hit. If the query and the hit are found to share an ortholog pillar, then they are deemed to be supported by shared synteny and are put in a single ortholog pillar. For this stage, full reciprocity of BLASTP hits with all of the genes in the pillar hit is not required. In cases where there are multiple candidates for a single pillar position, the pillar member is chosen first by the strength of the shared synteny and then by the E value of the BLASTP hit. This process is run in an iterative fashion until each gene has been labeled a singleton or has been placed in an ortholog pillar with synteny and BLASTP support.
FIG 1.
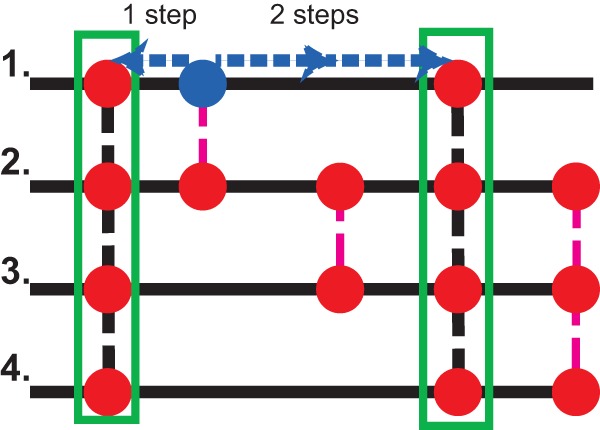
SearchDOGS Bacteria's pillar generation method. A set of initial pillars (highlighted in green) is created using reciprocal best BLASTP results. The orthology of genes producing one-way BLASTP hits is confirmed by automatically searching for shared synteny. In the example shown, the gene from species 1 highlighted in blue has neighboring genes that are in ortholog pillars with neighboring genes of the gene from species 2, confirming that the species 1 gene in blue and the species 2 gene beneath it belong in a single ortholog pillar.
Generation of candidate open reading frames.
For each intergenic region that hits one or more genes from a syntenic homology pillar in the BLASTX search, the high-similarity pairs (HSPs) corresponding to the hit with the lowest E value are retained. Open reading frames are obtained using ORFmaker, an inbuilt ORF finder specifically designed for use with SearchDOGS Bacteria (Fig. 2).
FIG 2.

Flowchart illustrating the process by which ORFmaker identifies and accepts a stop-to-stop ORF. (i and ii) If the BLASTX search produces two HSPs of sufficient cumulative length that are in the correct orientation and order, ORFmaker attempts to create a single ORF spanning both by reading across between the HSPs or by correcting a frameshift. If a single HSP of sufficient length is produced by the BLASTX search, this is retained. (iii) The construct is extended outward from each direction until a stop codon is reached, creating a stop-to-stop ORF. The ORF is tested for errors such as the location of the starting methionine and the presence of premature stop codons. (iv) ORFmaker accepts ORFs with a single error, whether it is a frameshift, a premature stop, or a lack of start, to allow for the possibility of a sequence error. ORFs with two or more errors are rejected. In instances where an ORF passing the criteria can be made using a single HSP or a two-HSP construct, the ORF producing the highest BLASTP bit score against its orthologs when translated is retained.
Evidence for protein conservation.
For each candidate ORF, SearchDOGS's results include a calculation of omega values against the ORF's annotated orthologs. Omega (Ka/Ks, where Ks is the number of synonymous substitutions per synonymous site and Ka is the number of nonsynonymous substitutions per nonsynonymous site) value calculations are a standard tool for estimating the level of conservation between two protein-coding genes (28). Genes showing conservation of protein-coding content are expected to contain fewer nonsynonymous changes per site relative to synonymous changes.
For each candidate, the largest standard start-to-stop ORF within the stop-to-stop candidate ORF is used in the omega value calculation, provided no genes with nonconsensus start codons exist within the corresponding ortholog pillar. For cases in which orthologs with nonconsensus start codons do exist, only the length of the ORF from the start of the HSP to the end of the ORF is used; this is due to the difficulty in predicting nonconsensus start codons with certainty.
Pairwise comparisons are performed between each start-to-stop candidate ORF and every gene in the corresponding ortholog pillar using the program yn00 from the PAML package (29). As calculating an accurate standard error measurement for Ka/Ks is problematic, the value and standard error of the difference between Ks and Ka (Ks − Ka) are calculated in order to test the statistical significance of these results. Assuming neutral evolution, a Ks − Ka value of approximately 0 is expected, and a Ks − Ka value significantly greater than 0 indicates constrained protein evolution (26). A 95% confidence interval for Ks − Ka is calculated using the following formula:
| (1) |
where SE(Ks − Ka) is the standard error of (Ks − Ka) and is calculated as follows:
| (2) |
Two problems associated with the Ka/Ks test must be noted: (i) Ka/Ks values are often not statistically significant for genes which are short or have very similar nucleotide sequences due to an insufficiency of informative sequence, and (ii) Ka/Ks values for candidates with a potential nonconsensus start codon are only approximate and may not be entirely accurate if a significant length of ORF upstream of the HSP is excluded from the calculation.
Test set of genomes used in this analysis.
Nine genomes from the gammaproteobacterial clade were downloaded from GenBank (see Table 1). All input files were collected in March 2013. The original annotations for the different genomes differ both in methods used and in level of rigor (details on the GenBank files used and the methods used to annotate these genomes are provided in Table S1 in the supplemental material). The inclusion of the E. coli K-12 MG1655 model organism provides a valuable “gold standard” annotation, useful for identifying hits that correspond to pseudogenic fragments in genomes being tested, as well as pseudogenes that have been misannotated as real genes in the existing annotations for these genomes. The genome annotations included use only the GenBank files listed in table S1; they do not include Refseq edits and have not been updated for more recent reannotations. Plasmids were excluded from the analysis.
TABLE 1.
Genomes used in the SearchDOGS species comparison, including model organism E. coli K-12 MG1655a
| Species | GenBank accession no. | Genome size (MB) | No. of protein-coding genes | Acronym |
|---|---|---|---|---|
| Escherichia coli K-12 substrain MG1655 | U00096.2 | 4.6 | 4,145 | ECK1 |
| Escherichia coli O157:H7 strain Sakai | BA000007.2 | 5.6 | 5,361 | ECO1 |
| Escherichia coli S88 | CU928161.2 | 5.2 | 4,696 | ECS8 |
| Shigella boydii Sb227 | CP000036.1 | 4.9 | 4,136 | SBOY |
| Salmonella enterica subsp. enterica serovar Typhi strain Ty2 | AE014613.1 | 4.8 | 4,323 | SETY |
| Yersinia pestis antiqua | CP000308.1 | 4.9 | 4,167 | YPAN |
| Pseudomonas syringae pv. tomato strain DC3000 | AE016853.1 | 6.5 | 5,482 | PSYR |
| Vibrio cholerae O395 | CP000626.1, CP000627.1 | 4.1 | 3,875 | VCHO |
| Xanthomonas campestris pv. campestris strain ATCC 33913 | AE008922.1 | 5.1 | 4,181 | XCAM |
All species are from the gammaproteobacterial clade.
RESULTS
Generation of results.
For each genome used in the SearchDOGS analysis, every intergenic region is individually tested against the ortholog pillars (pillars that each contain any annotated orthologs of a given locus in each genome in the data set) that share synteny with it (see Materials and Methods). In order to identify weak hits in an orthologous position, all BLASTX hits are considered regardless of E value.
Where a hit occurs, SearchDOGS then tries to identify an intact gene structure corresponding to it. A subroutine called ORFmaker identifies a stop-to-stop ORF (i.e., from one stop codon to the next stop codon in the same frame) corresponding to the HSP. In order to cope with potential sequence errors, ORFmaker's default setting allows the readthrough of a single stop codon or frameshift where HSP evidence exists to indicate the existence of a longer ORF, although such ORFs are flagged as dubious. The set of ORFs predicted in a genome is then filtered based on criteria such as HSP length and position of start and stop codons relative to the HSP (see Materials and Methods). HTML output pages are then produced showing details of the ORFs that pass these criteria (Fig. 3) and providing links to the BLAST results and nucleotide sequences. Conservation of protein sequence across wide evolutionary distances, corresponding to nonsynonymous-to-synonymous rate ratios (Ka/Ks) significantly less than 1, is a strong indicator of the authenticity of a gene (26). SearchDOGS Bacteria integrates PAML software (29) to perform Ka/Ks tests in a pairwise fashion using annotated and potential genes (Fig. 4A). Ka/Ks values significantly lower than 1 are seen as a strong indicator of protein sequence conservation. Individual Ka/Ks values are calculated as well as an average value of Ka/Ks representing the difference between the potential gene and its corresponding ortholog pillar. A 95% confidence interval test of the difference between Ks and Ka (Ks − Ka) is calculated for each pairwise comparison as a measure of statistical significance.
FIG 3.

Identification of a yieP ortholog in S. boydii. (A) SearchDOGS output showing the syntenic neighborhood of the S. boydii hit. The intergenic regions of the S. boydii genomic segments highlighted in red hit proteins in a syntenic ortholog pillar (pillar 5377) in a BLASTX search and contain a candidate gene. Each row names the genes flanking this pillar in each species if their syntenic context is conserved. Dist., distance; nt, nucleotide. (B) Amino acid lengths of proteins coded by predicted (P) and known (K) genes at the yieP locus. (C) Amino acid sequences coded for by the stop-to-stop ORF identified by SearchDOGS in S. boydii (highlighted in yellow) and the annotated genes in the ortholog pillar corresponding to the yieP locus. Start codons are highlighted in green and stop codons in red.
FIG 4.

Insertion in the genomic sequence of S. boydii fadB. (A) By reading through a frameshift, SearchDOGS's ORFmaker program was able to create a full-length ORF (highlighted in red) at the locus corresponding to the highly conserved fadB gene. (B) Screenshot of the SearchDOGS Ka/Ks output for the reconstructed fadB candidate against the annotated FadB orthologs. Omega (Ka/Ks) values are highlighted in red. Tests to determined 95% confidence intervals carried out on the value of Ks − Ka indicate that, in all cases except the S. boydii/V. cholerae pairwise comparison, Ks − Ka is greater than 0 with statistical significance, indicating protein sequence conservation. (C) ClustalW alignment of the nucleotide sequence surrounding an apparent expansion of a 7-bp repeat in S. boydii fadB. E. coli K-12, E. coli S88, E. coli O157:H7, and S. boydii are shown, and the repeat sequence is highlighted (83). The entire length of the insertion is 28 bp and thus causes a frameshift at this location in S. boydii fadB.
Gammaproteobacterial species used in the study.
To test the software, we chose 9 species from the gammaproteobacterial clade (Table 1). These included the E. coli K-12 strain MG1655 model organism, some closely related strains (E. coli, Shigella), and some species of increasing evolutionary distance (Vibrio, Pseudomonas, Xanthomonas). The genomes range in size from 4.1 Mb and 3,875 protein-coding genes (Vibrio cholerae) to 6.5 Mb and 5,482 protein-coding genes (Pseudomonas syringae). Other than Vibrio cholerae, which has two chromosomes, all consist of a single circular chromosome. All 9 genomes were obtained from GenBank (see Materials and Methods). SearchDOGS Bacteria was successfully able to generate an extensive ortholog pillar structure for these species (Table 2). Using these pillars, we predicted candidate genes that remained unannotated in each genome within this data set.
TABLE 2.
Breakdown of ortholog pillar structure for each species in the comparison set, where pillars containing 9 orthologs represent loci with an ortholog present in every species and single ortholog pillars represent species-specific singletons relative to the other species in the seta
| No. of genes in pillar | No. or % of genes in: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli K-12 | E. coli O157:H7 | E. coli S88 | S. boydii | S. enterica | Y. pestis | P. syringae | V. cholerae | X. campestris | |
| 9 | 832 | 832 | 832 | 832 | 832 | 832 | 832 | 832 | 832 |
| 8 | 479 | 479 | 477 | 458 | 473 | 459 | 398 | 374 | 235 |
| 7 | 629 | 625 | 631 | 589 | 611 | 553 | 217 | 423 | 146 |
| 6 | 545 | 540 | 543 | 498 | 465 | 375 | 157 | 110 | 97 |
| 5 | 540 | 558 | 546 | 475 | 447 | 138 | 121 | 90 | 70 |
| 4 | 449 | 470 | 463 | 293 | 228 | 135 | 129 | 89 | 108 |
| 3 | 286 | 393 | 346 | 168 | 208 | 186 | 303 | 197 | 238 |
| 2 | 200 | 469 | 403 | 134 | 343 | 378 | 889 | 409 | 643 |
| 1 | 185 | 995 | 455 | 689 | 716 | 1,111 | 2,436 | 1,351 | 1,812 |
| Total no. of genes in species | 4,145 | 5,361 | 4,696 | 4,136 | 4,323 | 4,167 | 5,482 | 3,875 | 4,181 |
| % of genes in pillars containing 5+ orthologs | 73 | 57 | 65 | 69 | 65 | 57 | 32 | 47 | 33 |
| No. of candidate genes predicted per species | 270 | 203 | 170 | 480 | 233 | 86 | 90 | 36 | 114 |
For example, E. coli K-12 has 185 singletons and 832 genes with orthologs in all species. Species with a greater percentage of genes in higher-number pillars are mapped more successfully against the species set. This is reflected in the identification of more candidate ORFs in species with few singletons (E. coli, Shigella) as opposed to species in which a large percentage of the genome lacks identifiable orthologs (Xanthomonas, Pseudomonas, Vibrio).
Missing genes in the Shigella boydii genome annotation.
We analyzed in detail the set of results for Shigella boydii strain Sb227 (30) (see Tables S2 and S3 in the supplemental material). While Shigella has historically been treated as a species different from E. coli, the two genera are actually part of the same, diverse genus (31). SearchDOGS generated an initial candidate list of 480 additional ORFs in this species, including some very short predictions that are unlikely to be real genes. Table S2 contains 122 candidate unannotated genes from this initial list in order of decreasing length. In this analysis, candidates under 90% of the median length of their orthologs were excluded and are not listed, although some of these truncated genes may well be functional; this is a user-adjustable parameter. In most cases, the “low-hanging fruit”—large, intact genes with strong sequence identity to genes in related species—were already correctly identified in the initial annotations. However, we identified 7 gene candidates of length > 200 codons, each conserved across a number of species and showing protein sequence conservation, as well as 59 other candidates of length 60 to 200 codons.
For example, E. coli K-12 yieP, coding for a 230-amino-acid predicted transcriptional regulator (32), has a well-conserved ortholog annotated in each of the E. coli strains included (33, 34) as well as in Salmonella enterica and Yersinia pestis (35, 36). We identified a 231-codon candidate in S. boydii in a conserved location (Fig. 3A) showing very high similarity in length and sequence to the annotated orthologs (Fig. 3B and C). Two additional stains of Shigella flexneri examined were found to have an annotated ortholog of yieP.
We identified 56 candidates of length < 60 codons (see Table S2 in the supplemental material). Of these, 36 correspond to unannotated homologs of short genes in E. coli K-12 and 15 correspond to homologs of genes annotated in other species and predicted to exist in E. coli K-12 (i.e., E. coli K-12 also contains a suitable intact ORF). A further 5 are predicted to exist in S. boydii but not in E. coli K-12.
Of the S. boydii candidates we identified, 31 contained short (20 bp or fewer) overlaps with an adjacent annotated gene. This may have led to the rejection of these ORFs by ab initio gene-finding programs despite homology and conserved protein sequence with genes in related species (37, 38). It is likely that some of these annotated neighbors that overlap conserved, unannotated genes are spurious or possess incorrectly annotated start coordinates (39, 40). We also identified many instances of candidates with nonconsensus start codons (9% of annotated genes in E. coli K-12 are currently annotated as having a TTG or GTG start [32]), and there are rare instances of bacterial genes starting with TTC, CTG, and ATC (32, 38, 41).
In 33 cases, a very highly conserved protein-coding gene of a length identical to the length of its homologs could be produced by allowing readthrough of a single stop codon or correction of a single frameshift (see Table S3 in the supplemental material). The possibility must be considered that these are not genuine truncation events but are instead the result of a sequencing or assembly error, particularly in the case of very long genes or genes encoding proteins that appear to be highly conserved in many related species. An example of an unexpected stop codon occurs in the S. boydii orthologs of E. coli nemA. This gene codes for N-ethylmaleimide reductase, a member of the “Old Yellow Enzyme” family (42). It plays a role in the beta-oxidation of fatty acids by being involved in reductive degradation of toxic nitrous compounds (43, 44) and is regulated by nemR. The nemA-nemR operon is present in every species in this study except S. boydii, where nemR is present but nemA is annotated as a pseudogene truncated by an in-frame stop codon at position 101 of 367 (30). By reading through the stop codon, SearchDOGS was able to produce a full-length NemA protein showing very high sequence similarity to its annotated orthologs. The pairwise Ka/Ks ratios against the included orthologs in Yersinia pestis and the E. coli strains range from 0.07 to 0.19. An intact nemA gene is also present in several strains of Shigella flexneri that we examined (30), indicating that the CAG-to-TAG transition leading to the premature stop codon in S. boydii may be due to a point sequence error. It is also possible that a readthrough of a genuine stop codon may occur. E. coli K-12 gene fdhF, coding for a subunit of a formate dehydrogenase complex involved in anaerobic respiration, has an annotated homolog in each species studied except Pseudomonas syringae and Shigella boydii. The UGA stop codon at position 140 in the E. coli K-12 protein sequence is translated in vivo as selenocysteine under anaerobic conditions (45), allowing readthrough of the entire protein, and is indicated as a “U” in the protein sequences of the E. coli strains. However, the in frame stop codon has led to the rejection of the S. boydii homolog in the original annotation (30) despite protein sequence conservation over the entire 716-amino-acid length of the protein. It appears that an A-C transversion has resulted in a TGC codon coding for a cysteine at that location in Y. pestis, V. cholerae, and Xanthomonas campestris (36, 46, 47).
Identification of short bacterial proteins using SearchDOGS.
Very small genes are notoriously difficult to accurately identify and annotate by experimental, ab initio, and homology-based approaches (8, 48–51). Using the February 2013 release of the E. coli K-12 MG1655 genome (GenBank accession number U00096.2) as a gold standard and the same set of test genomes (Table 1), we tested the ability of SearchDOGS to identify unannotated homologs of short genes showing both conserved sequence similarity and synteny with their annotated counterparts. Among the 114 proteins less than 60 amino acids in length annotated in E. coli K-12, we found that 66 (58%) were not correctly annotated across all of the other 8 genomes in our test data set (see Table S4 in the supplemental material). Many of these are small toxic or membrane-associated proteins recently added to the E. coli K-12 annotation (52, 53), but we also identified genes coding for the 50S ribosomal subunit L36 protein in E. coli S88 and Yersinia pestis, as well as the gene coding for ribosome-associated protein Sra in E. coli S88. The sensitivity of the SearchDOGS method allowed us to accurately predict the location of homologs of the smallest leader peptide genes annotated in E. coli K-12, such as a homolog of the 17-codon hisL gene in Yersinia pestis and the 18-codon mgtL gene in E. coli O157:H7, E. coli S88, S. boydii, and S. enterica.
Potential missing genes in the E. coli K-12 MG1655 annotation.
As a model organism, the E. coli K-12 MG1655 genome is annotated to a very high standard and is frequently updated (32). However, we identified a number of unannotated ORFs showing retention in each of the E. coli/Shigella strains and a high degree of conservation between the E. coli strains, S. boydii, and, often, S. enterica (see Table S5 in the supplemental material). Again, the majority of the candidates are short ORFs, ORFs featuring short coding sequence overlaps, and ORFs beginning with nonstandard start codons. Due to how closely the E. coli and Shigella strains are related, it is likely that some of these ORFs are noncoding and happen to be retained by chance, but many are likely to be genuine and warrant study.
One such example is Shigella boydii SBO_4385, an 86-codon gene encoding a hypothetical protein which has orthologs of codons of length 89 annotated in E. coli S88 and E. coli O157:H7 (30, 33, 34). An 86-codon ORF corresponding to a protein of nearly identical sequence was found by SearchDOGS at an orthologous position in E. coli K-12 MG1655 and shows statistically significant Ka/Ks values of 0.31 and 0.28 against the other E. coli orthologs. A 4-bp overlap between the E. coli K-12 ortholog and the neighboring gene yjiL may have led to it being overlooked.
Conservation in many species can be a good indicator that an ORF represents a bona fide gene. A 41-codon gene annotated in S. enterica, t4378 (protein identification no. AAO71829), hits nearly identical unannotated ORFs in E. coli K-12, E. coli O157:H7, E. coli S88, and S. boydii, each beginning with the GTG nonconsensus start codon (35) (Fig. 5A). A 70-codon gene (ECs2526) annotated in E. coli O157:H7 hits highly similar unannotated ORFs in E. coli K-12, E. coli S88, S. boydii, S. enterica, and Y. pestis (33) (Fig. 5B). In five species, the ORF overlaps by 13 bp with the neighboring gene.
FIG 5.
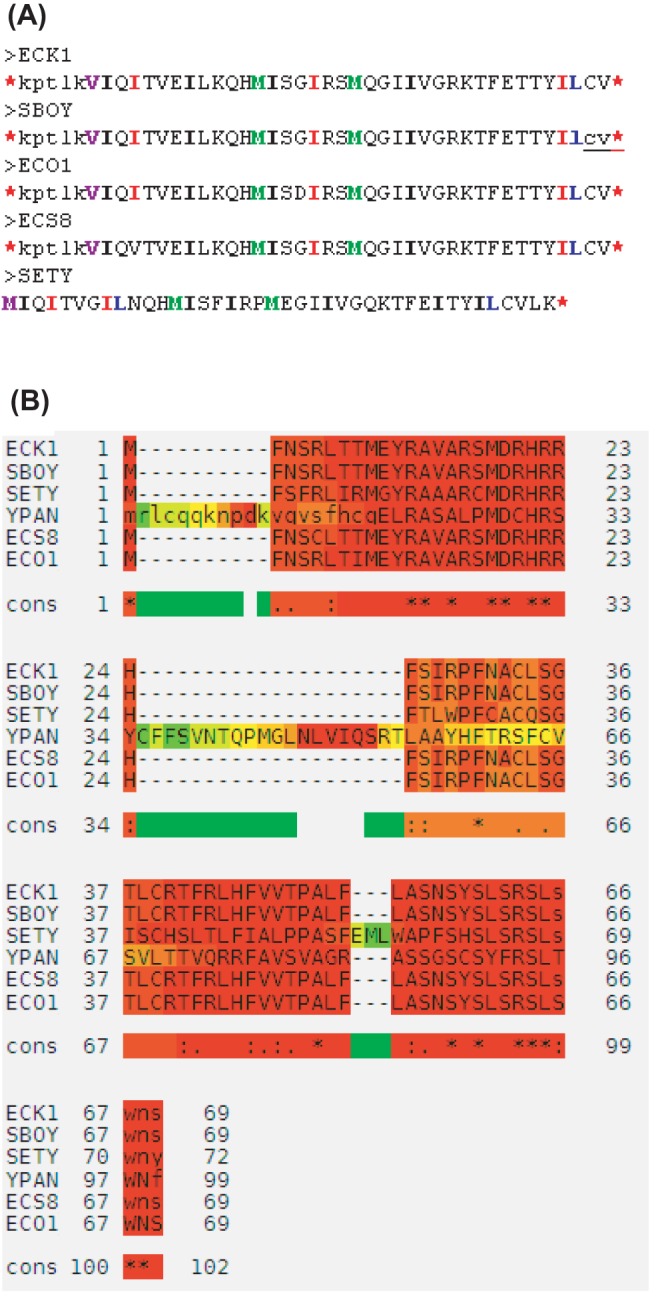
Further examples of candidate genes identified by SearchDOGS. (A) Salmonella enterica subsp. enterica serovar Typhi strain Ty2 gene t4378 coding for hypothetical protein AAO71829.1 hits ORFs coding for proteins with near-identical protein sequences at a syntenic location in E. coli K-12, E. coli O157:H7, E. coli S88, and S. boydii. (B) TCOFFEE protein sequence alignment corresponding to an annotated gene in E. coli O157:H7 (bottom sequence) and highly conserved unannotated ORFs at a syntenic location in E. coli K-12, E. coli S88, S. boydii, S. enterica, and Y. pestis (84). SearchDOGS also hits a more highly diverged ORF with sequence similarity in P. syringae (not shown).
Improving the annotation of the E. coli S88 genome.
The genome of E. coli S88 (34) was annotated by initially using the sequence composition-based prediction software AMIGene. Results were then refined using MaGe, a sophisticated annotation pipeline that employs the estimation of synteny conservation both to identify genes and to resolve gene duplicate/fusion/paralog relationships (17). In order to test whether SearchDOGS improves on the sensitivity of this method, we performed a run testing the E. coli S88 annotation against the same version of the E. coli K-12 MG1655 genome that was available to Touchon et al. (34) at the time that they annotated the S88 genome. This version of the E. coli K-12 MG1655 genome dates from February 2006 and was obtained from GenBank (accession number U00096.2; version GI:48994873). It lacks several newly discovered genes (32). Using this input, SearchDOGS identified 14 likely gene candidates, 5 of which are 60 codons or fewer in length. It also identified 7 cases in which frameshift correction or stop codon readthrough creates a highly conserved full-length gene in strain S88 (Table 3).
TABLE 3.
List of loci at which orthologs of E. coli K-12 MG1655 genes have been annotated in E. coli S88 using the February 2006 annotation of E. coli K-12 MG1655a
| Neighboring genes in E. coli S88 (protein ID) | Length (no. of codons) | E. coli K-12 ortholog name | E. coli K-12 protein product (32) |
|---|---|---|---|
| CAR05156.1 | 253 | yhjH | EAL domain-containing protein involved in flagellar function |
| CAR05160.1 | |||
| CAR03076.1 | 181 | infC | Protein chain initiation factor IF3 |
| CAR03078.1 | |||
| CAR02425.1 | 125 | yceQ | Predicted protein |
| CAR02426.1 | |||
| CAR01639.1 | 110 | ykgJ | Predicted ferredoxin |
| CAR01640.1 | |||
| CAR03844.1 | 92 | ypdI | Predicted lipoprotein involved in colanic acid biosynthesis |
| CAR03845.1 | |||
| CAR03119.1 | 91 | ynjH | Predicted protein |
| CAR03120.1 | |||
| CAR04458.1 | 84 | yqgD | Predicted inner membrane protein |
| CAR04459.1 | |||
| CAR02336.1 | 77 | ymcE | Cold shock gene |
| CAR02337.1 | |||
| CAR02668.1 | 72 | hokD | Qin prophage; small toxic polypeptide |
| CAR02669.1 | |||
| CAR01645.1 | 47 | ykgO | rplJ (L36) paralog |
| CAR01646.1 | |||
| CAR06223.1 | 47 | yjjY | Predicted protein |
| CAR06224.1 | |||
| CAR02507.1 | 47 | ylcG | DLP12 prophage; predicted protein |
| CAR02508.1 | |||
| CAR04903.1 | 38 | rpmJ | 50S ribosomal subunit protein L36 |
| CAR04904.1 | |||
| CAR02096.1 | 38 | ybgT | Conserved protein |
| CAR02097.1 | |||
| Frameshift correction/stop readthrough allowed | |||
| CAR02369.1 | 808 | putP | Proline:sodium symporter |
| CAR02372.1 | |||
| CAR05021.1 | 533 | rtcR | Sigma 54-dependent transcriptional regulator of rtcBA expression |
| CAR05024.1 | |||
| CAR02124.1 | 478 | ybhI | Predicted transporter |
| CAR02127.1 | |||
| CAR03673.1 | 444 | yfaV | Predicted transporter |
| CAR03676.1 | |||
| CAR01981.1 | 387 | ybdL | Methionine aminotransferase, PLP dependent |
| CAR01984.1 | |||
| CAR02773.1 | 351 | ycjQ | Predicted oxidoreductase; Zn dependent and NAD(P) binding |
| CAR02776.1 | |||
| CAR02219.1 | 172 | ybjP | Predicted lipoprotein |
| CAR02222.1 |
Cases in which a single frameshift correction/stop codon readthrough produces a full-length gene candidate are also presented. ID, identification number. PLP, pyridoxal phosphate.
Automated annotation programs can miss or misannotate certain genes due to unusual start codons or nucleotide composition in these genes. The essential translation initiation factor IF3 is encoded by infC, one of only two E. coli genes known to start with the rare start codon ATT (54). IF3 plays a role in the fidelity of translation initiation and has been shown to regulate the frequency of initiation from noncanonical start codons (55, 56). In this fashion, it functions in a negative-feedback loop, repressing its own translation when in abundance (54, 57). We identified a full-length ORF corresponding to infC in E. coli S88. A truncated infC, beginning at a downstream GTG, is annotated as a pseudogene in the E. coli S88 genome, presumably because MaGe does not consider ATT to be a possible start codon. An incorrect start was also annotated for this locus in the existing S. boydii, Y. pestis, V. cholerae, P. syringae, and X. campestris annotations; these genes all appear to begin with an ATT start codon, although they are annotated with different start codons. This highlights the need for manual curation or more rigorous comparative genomic approaches to correctly annotate loci with unusual features.
Identification of a possible gene fusion in Xanthomonas campestris.
We identified a possible case of gene fusion within a biosynthetic operon in Xanthomonas campestris. The Xanthomonas campestris gene cobC (which is called cobD in Salmonella [58]) codes for a 327-amino-acid enzyme in the cobalamin (vitamin B12) biosynthesis pathway. This gene has no annotated homolog in X. campestris pathovar campestris strain ATCC 33913 but is annotated in two other X. campestris pathovars, campestris strain B100 and vesicatoria strain 85-10, where it overlaps by 4 bases with the downstream cobQ (59, 60). The apparent gene fusion occurs in X. campestris pathovar campestris strain ATCC 33913. Here, a single base pair insertion near the end of the cobC ORF has led to the loss of the stop codon and has also brought it into frame with cobQ, creating what appears to be an 817-codon CobCQ fusion protein (Fig. 6). De novo cobalamin production from uroporphyrinogen III is a complex pathway involving 30 or so enzymes; however, some bacteria are able to produce cobalamin from pathway intermediates. The Salmonella and Pseudomonas strains can produce cobalamin de novo (61), whereas E. coli and Shigella strains can produce cobalamin only from the intermediate molecule cobinamide and are missing the early genes in the pathway (a set of genes known as the CobI genes) (62). Xanthomonas is widely reported to produce cobalamin and is used in industrial production (63) but also appears to lack the full pathway (61). However, it appears to have fully retained the 9 genes of the cobA-cobS operon and thus appears capable of producing cobalamin from the intermediate hydrogenobyrinic acid (64). The cobalamin pathways differ extensively even between de novo cobalamin-producing bacteria (64), so it is possible that Xanthomonas can perform other steps of the pathway using nonhomologous proteins.
FIG 6.
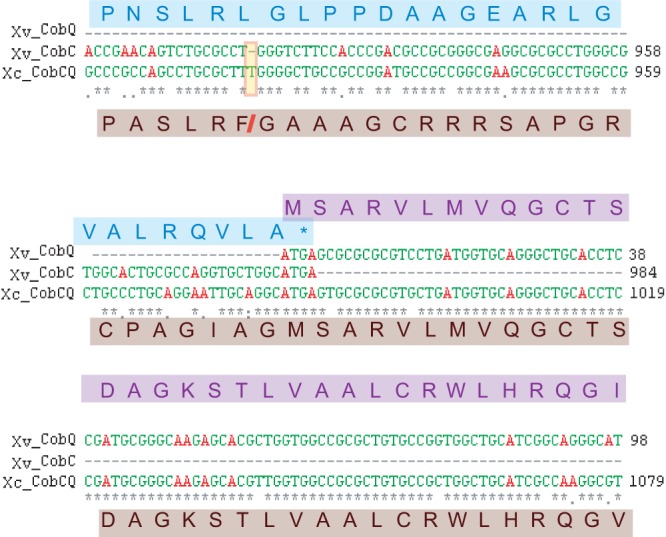
Possible gene fusion in the X. campestris cobalamin synthesis pathway. Nucleotide alignment of the point of overlap between the adjacent Xanthomonas campestris pv. vesicatoria genes cobC and cobQ (Xv_CobC, Xv_CobQ) against the Xanthomonas campestris pv. campestris strain ATCC 33913 fused cobCQ ORF (Xv_CobCQ). Protein sequences are shown with Xv_CobQ above in blue, Xv_CobC in purple, and the Xv_CobCQ fusion below in brown. A single base pair insertion toward the end of the X. campestris pv. campestris cobC gene (highlighted in yellow) has abolished the stop codon at the end of this ORF and has brought it into frame with the downstream CobQ, creating a gene fusion.
Identification of pseudogenes.
A significant problem associated with homology-based automated annotation methods is the difficulty in differentiating bona fide unannotated genes from pseudogenes that share both sequence similarity and location with their intact orthologs in other species. SearchDOGS tries to overcome this by providing as much information as possible in order for the user to make an informed choice on whether to accept, reject, or further study a candidate. In theory, low Ka/Ks ratios between a candidate gene and its orthologs across a range of evolutionary distances provide strong evidence of protein conservation (26). However, even a Ka/Ks ratio < 1 does not guarantee the existence of a functional gene, because recently formed pseudogenes still bear the hallmarks of sequence constraint to code for protein (65). Furthermore, bona fide unannotated genes often do not return statistically significant values in these tests if they are short or are too highly similar in nucleotide sequence to their orthologs. One pragmatic approach is to ignore any potential candidates that are shorter than 80% of the median length of annotated orthologs in closely related species (66); however, there is no guarantee that such a truncation will render a gene's protein product nonfunctional.
For illustration, ilvG is a recently formed pseudogene in E. coli K-12 MG1655. The functional gene codes for a catalyst of 2-acetolactate synthesis from pyruvate (33) and exists as a 549-to-574-codon gene in E. coli S88, E. coli O157:H7, S. boydii, S. enterica, Y. pestis, V. cholera, and X. campestris (30, 33–36, 46, 47). In E. coli K-12 MG1655, a frameshift at codon 328 (Ecogene [67]) has resulted in a truncated 329-codon pseudogene which was identified as a candidate gene by SearchDOGS. Ka/Ks ratios between the annotated orthologs and the hypothetical translation of the pseudogene are low (0.04 to 0.18 for the statistically significant values), and there is very high protein sequence similarity between the annotated orthologs and the pseudogene. It appears that ilvG is the victim of a recent pseudogenization event in E. coli K-12 and has not degenerated enough to lose the evolutionary characteristics of a bona fide protein-coding gene.
We identified one interesting example in which an apparent expansion of a 7-bp sequence appears to have led to the pseudogenization of a highly conserved gene in Shigella boydii. Degradation of fatty acids under aerobic conditions in E. coli is carried out by the FadBA enzyme complex, a tetramer made up of proteins encoded by fadB and fadA (68, 69), as part of the fadR regulon (70). fadB and fadA are highly conserved throughout both Gram-positive and Gram-negative bacteria (71), and both genes are present in all of the 8 species in our study set, with the exception of S. boydii, where fadB is annotated as a truncated pseudogene containing a frameshift (30). By correcting the frameshift, SearchDOGS was able to automatically predict a full-length (738-amino-acid) FadB protein showing very high sequence similarity to its annotated orthologs (Fig. 4B). This full-length construct shows Ka/Ks values ranging from 0.02 to 0.19 compared to its orthologs (Fig. 4A). Closer examination of the S. boydii fadB nucleotide sequence shows that the frameshift is caused by a 28-bp insertion consisting of four tandem repeats of a 7-bp repeat (Fig. 4C). This 7-bp repeat matches a 7-bp segment that is imperfectly repeated in E. coli K-12 (6 of 7 bases match); in S. boydii, 2 copies have expanded to 6. As well as being intact in all the E. coli strains studied, fadB is also intact in several strains of Shigella flexneri that we examined (72, 73). Without experimental evidence, it is unclear whether the fadB truncation in S. boydii is due to an error in sequencing or assembly or to a genuine pseudogenization event.
Obligate and facultative pathogens have been shown to harbor a relatively high number of pseudogenes for reasons that may be due to ongoing genome reduction as an adaptation to the host environment (74). The inclusion of E. coli K-12 MG1655, a reference sequence with a high degree of annotation accuracy (32), allowed us to identify many cases of what appear to be pseudogenes incorrectly annotated as real genes in the genomes studied. Table S6 in the supplemental material includes a list of likely pseudogenes identified in the genomes studied based on a high degree of similarity in length and sequence to known E. coli K-12 pseudogenes. These genes are automated predictions, and most represent a truncated form of an ortholog present in one of the other species. (Table S6 is likely to represent only a subset of all incorrectly annotated pseudogenes in these genomes. It does not include those automatic gene predictions that appear to be truncated orthologs of full-length genes in other species where there is no E. coli K-12 feature at the locus to compare against.)
DISCUSSION
A crucial requirement of the SearchDOGS method is the accurate and unambiguous ascertainment of orthology of genomic segments in multiple species. For this to be possible for a given set of species, several conditions must be met. Species included cannot have diverged too far from each other; otherwise, rearrangements and gene gain or loss will obscure attempts to identify conserved synteny. In the SearchDOGS analysis, the five species with the most candidate hits were also the species with the highest numbers of genes in ortholog pillars of 5 or more members, i.e., the species that could be mapped most effectively against the majority of other species (Table 2) However, if a set of species with too narrow a divergence range is chosen, it is often difficult to get a clear signal of protein sequence conservation (65).
In addition to a suitable evolutionary range, several other factors must be considered when choosing a comparison species set. SearchDOGS identifies only unannotated genes for which an ortholog exists and is annotated in another species. Hence, we suggest that at least one closely related species with a very high quality of annotation should be used for reference. Gene content in a genome is also affected by lifestyle and environment. Furthermore, genes may be annotated in certain strains that have been missed in others due to different annotation methods, differing degrees of sequencing and annotation rigor, or studies carried out under different ranges of conditions.
One of the greatest challenges when using sequence homology-based techniques to predict genes is differentiating bona fide genes from pseudogenes. A pseudogene can be incorrectly annotated as a functional gene for two reasons. First, a genuine gene can hit a pseudogene showing sequence similarity to the query. If the pseudogenization event is sufficiently recent, the remaining gene fragment may still bear many of the hallmarks of a genuine gene, including protein conservation (65). Second, it is likely that many pseudogenes in sequenced genomes are currently incorrectly annotated as if they are functional. This is evidenced by the number of “genes” in other strains and species that we identified that are identical in sequence to known E. coli K-12 pseudogene features (see Table S6 in the supplemental material). As many automatic annotation procedures, including SearchDOGS, rely heavily on sequence similarity to a reference set of annotated features, these “false genes” in the reference set can lead to the spurious misannotation of other pseudogenes, spreading false annotations through databases if unchecked (53). Brown and Sjolander estimated in 2006 that only 3% of those proteins in the UniProt database not labeled as “hypothetical” or “unknown” had experimental support (75), the remainder having been inferred by bioinformatics means, and this percentage is surely considerably lower by now.
A third problem lies in correctly differentiating genuine frameshift mutations (creating pseudogenes) from sequencing errors. Current next-generation sequencing methods such as Roche/454 and Illumina have a higher background rate of errors than previous methods such as Sanger sequencing (76–79), particularly in genomes sequenced at a low level of coverage, and these errors mainly take the form of single nucleotide insertions and deletions (indels). SearchDOGS Bacteria is designed to correct a single frameshift, if HSP evidence indicates that a sensible and conserved full-length protein can thereby be created. However, as with other gene disruption events, a gene in which a genuine frameshift mutation has occurred recently has many of the same characteristics, such as a low Ka/Ks ratio, as a genuine gene containing a sequencing error.
For each species in the input set, SearchDOGS generates a list of automatic predictions based on sequence similarity and conserved genomic location. Our aim was to include as much information as possible in order for the user to make an informed decision as to whether to reject a candidate gene or accept it for further study. Users are encouraged to look at the length, sequence similarity, and level of protein conservation of a candidate relative to its annotated orthologs. However, full proof that a candidate gene genuinely codes for a functional protein requires detection of the protein translation product, for example, by mass spectrometry. It should be noted that as a result of the expert input needed in both selecting the genomes to be analyzed and evaluating the candidate genes identified, SearchDOGS is not suitable for automated, large-scale analyses. This is particularly relevant when defining bona fide genes that are to be used in downstream analyses by the scientific community. However, we believe that it provides a powerful tool for targeted analyses.
As the tools of transcriptome sequencing (RNA-seq) and proteomics come of age (80–82) and bioinformatics methods become ever more sophisticated, these approaches combined should result in fast, accurate, and complete genome annotations to complement the accelerating pace of genome sequencing.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Science Foundation Ireland (07/IN1/B911) and the European Research Council (advanced grant 268893).
We are grateful to Estelle Proux-Wéra for extensive discussion and advice.
Footnotes
Published ahead of print 21 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01368-13.
REFERENCES
- 1.Stothard P, Wishart DS. 2006. Automated bacterial genome analysis and annotation. Curr. Opin. Microbiol. 9:505–510. 10.1016/j.mib.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 2.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641. 10.1093/nar/27.23.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frishman D, Mironov A, Mewes HW, Gelfand M. 1998. Combining diverse evidence for gene recognition in completely sequenced bacterial genomes. Nucleic Acids Res. 26:2941–2947. 10.1093/nar/26.12.2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsen TS, Krogh A. 2003. EasyGene—a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics 4:21. 10.1186/1471-2105-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besemer J, Borodovsky M. 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33:W451–W454. 10.1093/nar/gki487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bocs S, Cruveiller S, Vallenet D, Nuel G, Médigue C. 2003. AMIGene: Annotation of MIcrobial Genes. Nucleic Acids Res. 31:3723–3726. 10.1093/nar/gkg590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mir K, Neuhaus K, Scherer S, Bossert M, Schober S. 2012. Predicting statistical properties of open reading frames in bacterial genomes. PLoS One 7:e45103. 10.1371/journal.pone.0045103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samayoa J, Yildiz F, Karplus K. 5 May 2011. Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics; 10.1093/bioinformatics/btr275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39:W29–W37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skovgaard M, Jensen LJ, Brunak S, Ussery D, Krogh A. 2001. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 17:425–428. 10.1016/S0168-9525(01)02372-1 [DOI] [PubMed] [Google Scholar]
- 11.Nielsen P, Krogh A. 2005. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics 21:4322–4329. 10.1093/bioinformatics/bti701 [DOI] [PubMed] [Google Scholar]
- 12.Yu JF, Xiao K, Jiang DK, Guo J, Wang JH, Sun X. 2011. An integrative method for identifying the over-annotated protein-coding genes in microbial genomes. DNA Res. 18:435–449. 10.1093/dnares/dsr030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemm MR, Paul BJ, Miranda-Rios J, Zhang A, Soltanzad N, Storz G. 2010. Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J. Bacteriol. 192:46–58. 10.1128/JB.00872-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kucerova E, Clifton SW, Xia XQ, Long F, Porwollik S, Fulton L, Fronick C, Minx P, Kyung K, Warren W, Fulton R, Feng D, Wollam A, Shah N, Bhonagiri V, Nash WE, Hallsworth-Pepin K, Wilson RK, McClelland M, Forsythe SJ. 2010. Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS One 5:e9556. 10.1371/journal.pone.0009556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren AS, Archuleta J, Feng WC, Setubal JC. 2010. Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics 11:131. 10.1186/1471-2105-11-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood DE, Lin H, Levy-Moonshine A, Swaminathan R, Chang YC, Anton BP, Osmani L, Steffen M, Kasif S, Salzberg SL. 2012. Thousands of missed genes found in bacterial genomes and their analysis with COMBREX. Biol. Direct 7:37. 10.1186/1745-6150-7-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G, Scarpelli C, Medigue C. 2006. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 34:53–65. 10.1093/nar/gkj406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedberg I. 2006. Automated protein function prediction—the genomic challenge. Brief. Bioinform. 7:225–242. 10.1093/bib/bbl004 [DOI] [PubMed] [Google Scholar]
- 19.Enault F, Suhre K, Claverie JM. 2005. Phydbac “Gene Function Predictor”: a gene annotation tool based on genomic context analysis. BMC Bioinformatics 6:247. 10.1186/1471-2105-6-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.OhÉigeartaigh SS, Armisén D, Byrne KP, Wolfe KH. 2011. Systematic discovery of unannotated genes in 11 yeast species using a database of orthologous genomic segments. BMC Genomics 12:377. 10.1186/1471-2164-12-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maguire SL, ÓhÉigeartaigh SS, Byrne KP, Schröder MS, O'Gaora P, Wolfe KH, Butler G. 2013. Comparative genome analysis and gene finding in Candida species using CGOB. Mol. Biol. Evol. 30:1281–1291. 10.1093/molbev/mst042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 60:708–720. 10.1007/s00248-010-9717-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryson K, Loux V, Bossy R, Nicolas P, Chaillou S, van de Guchte M, Penaud S, Maguin E, Hoebeke M, Bessieres P, Gibrat JF. 2006. AGMIAL: implementing an annotation strategy for prokaryote genomes as a distributed system. Nucleic Acids Res. 34:3533–3545. 10.1093/nar/gkl471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar K, Desai V, Cheng L, Khitrov M, Grover D, Satya RV, Yu C, Zavaljevski N, Reifman J. 2011. AGeS: a software system for microbial genome sequence annotation. PLoS One 6:e17469. 10.1371/journal.pone.0017469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fevre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith AA, Weiman M, Medigue C. 2013. MicroScope—an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41:D636–D647. 10.1093/nar/gks1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Bielawski JP. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15:496–503. 10.1016/S0169-5347(00)01994-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrne KP, Wolfe KH. 2005. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 15:1456–1461. 10.1101/gr.3672305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li WH, Wu CI, Luo CC. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 2:150–174 [DOI] [PubMed] [Google Scholar]
- 29.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24:1586–1591. 10.1093/molbev/msm088 [DOI] [PubMed] [Google Scholar]
- 30.Yang F, Yang J, Zhang X, Chen L, Jiang Y, Yan Y, Tang X, Wang J, Xiong Z, Dong J, Xue Y, Zhu Y, Xu X, Sun L, Chen S, Nie H, Peng J, Xu J, Wang Y, Yuan Z, Wen Y, Yao Z, Shen Y, Qiang B, Hou Y, Yu J, Jin Q. 2005. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 33:6445–6458. 10.1093/nar/gki954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lan R, Reeves PR. 2002. Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect. 4:1125–1132. 10.1016/S1286-4579(02)01637-4 [DOI] [PubMed] [Google Scholar]
- 32.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G, III, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL. 2006. Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 34:1–9. 10.1093/nar/gkj405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, Tanaka M, Tobe T, Iida T, Takami H, Honda T, Sasakawa C, Ogasawara N, Yasunaga T, Kuhara S, Shiba T, Hattori M, Shinagawa H. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8:11–22. 10.1093/dnares/8.1.11 [DOI] [PubMed] [Google Scholar]
- 34.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, Karoui ME, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguenec C, Lescat M, Mangenot S, Martinez-Jehanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Ruf CS, Schneider D, Tourret J, Vacherie B, Vallenet D, Medigue C, Rocha EP, Denamur E. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5:e1000344. 10.1371/journal.pgen.1000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng W, Liou SR, Plunkett G, III, Mayhew GF, Rose DJ, Burland V, Kodoyianni V, Schwartz DC, Blattner FR. 2003. Comparative genomics of Salmonella enterica serovar Typhi strains Ty2 and CT18. J. Bacteriol. 185:2330–2337. 10.1128/JB.185.7.2330-2337.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chain PS, Hu P, Malfatti SA, Radnedge L, Larimer F, Vergez LM, Worsham P, Chu MC, Andersen GL. 2006. Complete genome sequence of Yersinia pestis strains Antiqua and Nepal516: evidence of gene reduction in an emerging pathogen. J. Bacteriol. 188:4453–4463. 10.1128/JB.00124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aggarwal G, Worthey EA, McDonagh PD, Myler PJ. 2003. Importing statistical measures into Artemis enhances gene identification in the Leishmania genome project. BMC Bioinformatics 4:23. 10.1186/1471-2105-4-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poptsova MS, Gogarten JP. 2010. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology 156:1909–1917. 10.1099/mic.0.033811-0 [DOI] [PubMed] [Google Scholar]
- 39.Pallejà A, Harrington ED, Bork P. 2008. Large gene overlaps in prokaryotic genomes: result of functional constraints or mispredictions? BMC Genomics 9:335. 10.1186/1471-2164-9-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakke P, Carney N, Deloache W, Gearing M, Ingvorsen K, Lotz M, McNair J, Penumetcha P, Simpson S, Voss L, Win M, Heyer LJ, Campbell AM. 2009. Evaluation of three automated genome annotations for Halorhabdus utahensis. PLoS One 4:e6291. 10.1371/journal.pone.0006291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu HQ, Hu GQ, Ouyang ZQ, Wang J, She ZS. 2004. Accuracy improvement for identifying translation initiation sites in microbial genomes. Bioinformatics 20:3308–3317. 10.1093/bioinformatics/bth390 [DOI] [PubMed] [Google Scholar]
- 42.Williams RE, Bruce NC. 2002. ‘New uses for an Old Enzyme'—the Old Yellow Enzyme family of flavoenzymes. Microbiology 148:1607–1614 [DOI] [PubMed] [Google Scholar]
- 43.Miura K, Tomioka Y, Suzuki H, Yonezawa M, Hishinuma T, Mizugaki M. 1997. Molecular cloning of the nemA gene encoding N-ethylmaleimide reductase from Escherichia coli. Biol. Pharm. Bull. 20:110–112. 10.1248/bpb.20.110 [DOI] [PubMed] [Google Scholar]
- 44.Umezawa Y, Shimada T, Kori A, Yamada K, Ishihama A. 2008. The uncharacterized transcription factor YdhM is the regulator of the nemA gene, encoding N-ethylmaleimide reductase. J. Bacteriol. 190:5890–5897. 10.1128/JB.00459-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen GT, Axley MJ, Hacia J, Inouye M. 1992. Overproduction of a selenocysteine-containing polypeptide in Escherichia coli: the fdhF gene product. Mol. Microbiol. 6:781–785. 10.1111/j.1365-2958.1992.tb01528.x [DOI] [PubMed] [Google Scholar]
- 46.da Silva AC, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, Monteiro-Vitorello CB, Van Sluys MA, Almeida NF, Alves LM, do Amaral AM, Bertolini MC, Camargo LE, Camarotte G, Cannavan F, Cardozo J, Chambergo F, Ciapina LP, Cicarelli RM, Coutinho LL, Cursino-Santos JR, El-Dorry H, Faria JB, Ferreira AJ, Ferreira RC, Ferro MI, Formighieri EF, Franco MC, Greggio CC, Gruber A, Katsuyama AM, Kishi LT, Leite RP, Lemos EG, Lemos MV, Locali EC, Machado MA, Madeira AM, Martinez-Rossi NM, Martins EC, Meidanis J, Menck CF, Miyaki CY, Moon DH, Moreira LM, Novo MT, Okura VK, Oliveira MC, Oliveira VR, et al. 2002. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417:459–463. 10.1038/417459a [DOI] [PubMed] [Google Scholar]
- 47.Feng L, Reeves PR, Lan R, Ren Y, Gao C, Zhou Z, Cheng J, Wang W, Wang J, Qian W, Li D, Wang L. 2008. A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS One 3:e4053. 10.1371/journal.pone.0004053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H, Chan WS, Li Z, Wang D, Liu S, Zhou Y. 2011. Small open reading frames: current prediction techniques and future prospect. Curr. Protein Pept. Sci. 12:503–507. 10.2174/138920311796957667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basrai MA, Hieter P, Boeke JD. 1997. Small open reading frames: beautiful needles in the haystack. Genome Res. 7:768–771 [DOI] [PubMed] [Google Scholar]
- 50.Ochman H. 2002. Distinguishing the ORFs from the ELFs: short bacterial genes and the annotation of genomes. Trends Genet. 18:335–337. 10.1016/S0168-9525(02)02668-9 [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Tschaplinski TJ, Hurst GB, Jawdy S, Abraham PE, Lankford PK, Adams RM, Shah MB, Hettich RL, Lindquist E, Kalluri UC, Gunter LE, Pennacchio C, Tuskan GA. 2011. Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Res. 21:634–641. 10.1101/gr.109280.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fozo EM, Hemm MR, Storz G. 2008. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72:579–589. 10.1128/MMBR.00025-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. 2008. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 70:1487–1501. 10.1111/j.1365-2958.2008.06495.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Binns N, Masters M. 2002. Expression of the Escherichia coli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol. Microbiol. 44:1287–1298. 10.1046/j.1365-2958.2002.02945.x [DOI] [PubMed] [Google Scholar]
- 55.Meinnel T, Sacerdot C, Graffe M, Blanquet S, Springer M. 1999. Discrimination by Escherichia coli initiation factor IF3 against initiation on non-canonical codons relies on complementarity rules. J. Mol. Biol. 290:825–837. 10.1006/jmbi.1999.2881 [DOI] [PubMed] [Google Scholar]
- 56.Maar D, Liveris D, Sussman JK, Ringquist S, Moll I, Heredia N, Kil A, Blasi U, Schwartz I, Simons RW. 2008. A single mutation in the IF3 N-terminal domain perturbs the fidelity of translation initiation at three levels. J. Mol. Biol. 383:937–944. 10.1016/j.jmb.2008.09.012 [DOI] [PubMed] [Google Scholar]
- 57.Butler JS, Springer M, Dondon J, Graffe M, Grunberg-Manago M. 1986. Escherichia coli protein synthesis initiation factor IF3 controls its own gene expression at the translational level in vivo. J. Mol. Biol. 192:767–780. 10.1016/0022-2836(86)90027-6 [DOI] [PubMed] [Google Scholar]
- 58.Brushaber KR, O'Toole GA, Escalante-Semerena JC. 1998. CobD, a novel enzyme with L-threonine-O-3-phosphate decarboxylase activity, is responsible for the synthesis of (R)-1-amino-2-propanol O-2-phosphate, a proposed new intermediate in cobalamin biosynthesis in Salmonella typhimurium LT2. J. Biol. Chem. 273:2684–2691. 10.1074/jbc.273.5.2684 [DOI] [PubMed] [Google Scholar]
- 59.Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Buttner D, Caldana C, Gaigalat L, Goesmann A, Kay S, Kirchner O, Lanz C, Linke B, McHardy AC, Meyer F, Mittenhuber G, Nies DH, Niesbach-Klosgen U, Patschkowski T, Ruckert C, Rupp O, Schneiker S, Schuster SC, Vorholter FJ, Weber E, Puhler A, Bonas U, Bartels D, Kaiser O. 2005. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 187:7254–7266. 10.1128/JB.187.21.7254-7266.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vorhölter FJ, Schneiker S, Goesmann A, Krause L, Bekel T, Kaiser O, Linke B, Patschkowski T, Rückert C, Schmid J, Sidhu VK, Sieber V, Tauch A, Watt SA, Weisshaar B, Becker A, Niehaus K, Pühler A. 2008. The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J. Biotechnol. 134:33–45. 10.1016/j.jbiotec.2007.12.013 [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. 2009. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 10:78. 10.1186/1471-2164-10-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawrence JG, Roth JR. 1995. The cobalamin (coenzyme B12) biosynthetic genes of Escherichia coli. J. Bacteriol. 177:6371–6380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martens JH, Barg H, Warren MJ, Jahn D. 2002. Microbial production of vitamin B12. Appl. Microbiol. Biotechnol. 58:275–285. 10.1007/s00253-001-0902-7 [DOI] [PubMed] [Google Scholar]
- 64.Raux E, Lanois A, Levillayer F, Warren MJ, Brody E, Rambach A, Thermes C. 1996. Salmonella typhimurium cobalamin (vitamin B12) biosynthetic genes: functional studies in S. typhimurium and Escherichia coli. J. Bacteriol. 178:753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ochman H, Davalos LM. 2006. The nature and dynamics of bacterial genomes. Science 311:1730–1733. 10.1126/science.1119966 [DOI] [PubMed] [Google Scholar]
- 66.Lerat E, Ochman H. 2004. Psi-Phi: exploring the outer limits of bacterial pseudogenes. Genome Res. 14:2273–2278. 10.1101/gr.2925604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou J, Rudd KE. 2013. EcoGene 3.0. Nucleic Acids Res. 41:D613–D624. 10.1093/nar/gks1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Binstock JF, Pramanik A, Schulz H. 1977. Isolation of a multi-enzyme complex of fatty acid oxidation from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 74:492–495. 10.1073/pnas.74.2.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pramanik A, Pawar S, Antonian E, Schulz H. 1979. Five different enzymatic activities are associated with the multienzyme complex of fatty acid oxidation from Escherichia coli. J. Bacteriol. 137:469–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cho BK, Knight EM, Palsson BO. 2006. Transcriptional regulation of the fad regulon genes of Escherichia coli by ArcA. Microbiology 152:2207–2219. 10.1099/mic.0.28912-0 [DOI] [PubMed] [Google Scholar]
- 71.Fujita Y, Matsuoka H, Hirooka K. 2007. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 66:829–839. 10.1111/j.1365-2958.2007.05947.x [DOI] [PubMed] [Google Scholar]
- 72.Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, Zhang X, Zhang J, Yang G, Wu H, Qu D, Dong J, Sun L, Xue Y, Zhao A, Gao Y, Zhu J, Kan B, Ding K, Chen S, Cheng H, Yao Z, He B, Chen R, Ma D, Qiang B, Wen Y, Hou Y, Yu J. 2002. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 30:4432–4441. 10.1093/nar/gkf566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nie H, Yang F, Zhang X, Yang J, Chen L, Wang J, Xiong Z, Peng J, Sun L, Dong J, Xue Y, Xu X, Chen S, Yao Z, Shen Y, Jin Q. 2006. Complete genome sequence of Shigella flexneri 5b and comparison with Shigella flexneri 2a. BMC Genomics 7:173. 10.1186/1471-2164-7-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17:589–596. 10.1016/S0168-9525(01)02447-7 [DOI] [PubMed] [Google Scholar]
- 75.Brown D, Sjolander K. 2006. Functional classification using phylogenomic inference. PLoS Comput. Biol. 2:e77. 10.1371/journal.pcbi.0020077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kircher M, Kelso J. 2010. High-throughput DNA sequencing—concepts and limitations. Bioessays 32:524–536. 10.1002/bies.200900181 [DOI] [PubMed] [Google Scholar]
- 77.Farrer RA, Kemen E, Jones JD, Studholme DJ. 2009. De novo assembly of the Pseudomonas syringae pv. syringae B728a genome using Illumina/Solexa short sequence reads. FEMS Microbiol. Lett. 291:103–111. 10.1111/j.1574-6968.2008.01441.x [DOI] [PubMed] [Google Scholar]
- 78.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quinlan AR, Stewart DA, Stromberg MP, Marth GT. 2008. Pyrobayes: an improved base caller for SNP discovery in pyrosequences. Nat. Methods 5:179–181. 10.1038/nmeth.1172 [DOI] [PubMed] [Google Scholar]
- 80.Pinto AC, Melo-Barbosa HP, Miyoshi A, Silva A, Azevedo V. 2011. Application of RNA-seq to reveal the transcript profile in bacteria. Genet. Mol. Res. 10:1707–1718. 10.4238/vol10-3gmr1554 [DOI] [PubMed] [Google Scholar]
- 81.Altelaar AF, Munoz J, Heck AJ. 2013. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14:35–48. 10.1038/nrg3356 [DOI] [PubMed] [Google Scholar]
- 82.Ansong C, Purvine SO, Adkins JN, Lipton MS, Smith RD. 2008. Proteogenomics: needs and roles to be filled by proteomics in genome annotation. Brief. Funct. Genomic. Proteomic. 7:50–62. 10.1093/bfgp/eln010 [DOI] [PubMed] [Google Scholar]
- 83.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 84.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.