

Abstract
We studied the activity of a debranching enzyme (TreX) from Sulfolobus solfataricus on glycogen-mimic substrates, branched maltotetraosyl-β-cyclodextrin (Glc4-β-CD), and natural glycogen to better understand substrate transglycosylation and the effect thereof on glycogen debranching in microorganisms. The validation test of Glc4-β-CD as a glycogen mimic substrate showed that it followed the breakdown process of the well-known yeast and rat liver extract. TreX catalyzed both hydrolysis of α-1,6-glycosidic linkages and transglycosylation at relatively high (>0.5 mM) substrate concentrations. TreX transferred maltotetraosyl moieties from the donor substrate to acceptor molecules, resulting in the formation of two positional isomers of dimaltotetraosyl-α-1,6-β-cyclodextrin [(Glc4)2-β-CD]; these were 61,63- and 61,64-dimaltotetraosyl-α-1,6-β-CD. Use of a modified Michaelis-Menten equation to study substrate transglycosylation revealed that the kcat and Km values for transglycosylation were 1.78 × 103 s−1 and 3.30 mM, respectively, whereas the values for hydrolysis were 2.57 × 103 s−1 and 0.206 mM, respectively. Also, enzyme catalytic efficiency (the kcat/Km ratio) increased as the degree of polymerization of branch chains rose. In the model reaction system of Escherichia coli, glucose-1-phosphate production from glycogen by the glycogen phosphorylase was elevated ∼1.45-fold in the presence of TreX compared to that produced in the absence of TreX. The results suggest that outward shifting of glycogen branch chains via transglycosylation increases the number of exposed chains susceptible to phosphorylase action. We developed a model of the glycogen breakdown process featuring both hydrolysis and transglycosylation catalyzed by the debranching enzyme.
INTRODUCTION
Glycogen is a branched glucose polysaccharide consisting of α-1,4-glucosidic linkages with α-1,6 branches and is the major storage carbohydrate in animals equivalent to starch in plants (1, 2). Glycogen constitutes more than half of the Escherichia coli cell mass when the bacteria are grown under conditions of nitrogen shortage but with an excess carbon source (3–6). Glycogen accumulation commences during the lag phase, is maximal during the stationary phase (∼15 h), and decreases over the next several hours in many bacteria, such as Bacillus subtilis (7, 8). Glycogen biosynthesis in bacteria and archaea is accomplished by production of ADP-glucose and formation of α-1,4 glucosidic chains and α-1,6-linked glucan branches (9), whereas UDP-glucose initiates glycogen biosynthesis in mammalian muscle and liver. Thus, the enzyme is involved in glycogen metabolism in bacteria, and archaea share significant homology and enzymatic mechanisms (9). In contrast, breakdown of glycogen is initiated by debranching enzymes catalyzing hydrolysis of the α-1,6-glucosidic linkages of glycogen, amylopectin, and pullulan. Such enzymes are widely distributed in nature, being found in animals, bacteria (10–14), archaea (15–17), and plants. The glycogen degradation process in animals and yeasts is well known, in which the debranching enzymes exhibit two distinct activities—a glucanotransferase action and amylo-1,6-glucosidase activity (2, 18, 19). However, the glycogen breakdown process is unknown or the mode of action of debranching enzymes is not well understood in bacteria and archaea.
One of the main reasons that debranching enzymes of microorganisms have been poorly studied is the difficulty in identifying intermediate reaction products because the substrate (glycogen) has a complex structure, making it difficult to describe the reaction mechanism in detail. Thus, we prepared maltodextrin-branched cyclodextrin as a glycogen mimic to study branch chain specificity of the debranching enzyme. TreX, a glycogen debranching enzyme cloned from the trehalose biosynthesis gene cluster of Sulfolobus solfataricus P2, catalyzes the debranching reaction of the branch chain of glycogen into maltodextrin. It belongs to the GH13 (glycoside hydrolase 13 in the CAZy classification) family and shows high sequence similarity to isoamylase (20). However, our previous study showed that TreX exhibits both hydrolysis and transglycosylation activities by catalyzing the breakage and transfer of α-1,4-glucan oligosaccharides among chains (20). We also obtained a three-dimensional structure of TreX and identified two distinct active site configurations, in line with the bifunctional nature of the enzyme (21). The bifunctional debranching enzymes from mammalian tissues transfer the maltotetraosyl moiety to the nonreducing end of glucose, elongating the branch chain with an α-1,4-glycosidic linkage that is immediately hydrolyzed by glycogen phosphorylase (GlgP). However, unlike the animal debranching enzyme, TreX transfers the glucan moiety by forming an α-1,6 glycosidic linkage, resulting in translocation of the branch point within the molecule (20). The breakdown processes including the debranching enzyme are likely to have a different mechanism. Therefore, this study aimed to elucidate the mechanism of substrate transglycosylation of the TreX enzyme and its effect on the glycogen breakdown process.
MATERIALS AND METHODS
Materials.
Thin-layer chromatography (TLC) plates were purchased from Whatman International, Ltd. (Maidstone, Kent, United Kingdom), Ni-nitrilotriacetic acid (NTA) Superflow matrices were obtained from Qiagen (Hilden, Germany), and Bacto tryptone and yeast extract were from Difco (Detroit, MI). Other reagents were purchased from Sigma Chemical Co. (St. Louis, MO), Merck (Darmstadt, Germany), Junsei Chemical Co. (Tokyo, Japan), Showa Chemicals, Inc. (Tokyo, Japan), and Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Purification of TreX.
The glycogen debranching enzyme (GDE) of S. solfataricus P2 (TreX) was expressed in E. coli MC1061 [F− araD139 recA13 Δ(araABC-leu)7696 galU galK ΔlacX74 rpsL thi hsdR2 mcrB]. Transformants were cultured in Luria-Bertani (LB) medium (1% [wt/vol] Bacto tryptone, 0.5% [wt/vol] yeast extract, 0.5% [wt/vol] NaCl) containing ampicillin (100 μg ml−1) at 37°C. TreX tagged with six His residues was purified on a 1- by 4-cm Ni-NTA column (Ni-NTA Superflow; Qiagen) as described previously (22), and enzyme purity was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
TreX assays.
TreX activities were measured as described previously (23). Hydrolytic activity was assayed at 75°C in 50 mM sodium acetate buffer (pH 5.5) using 0.5% (wt/vol) amylopectin as the substrate. The reducing power of hydrolysis products was determined using the dinitrosalicylic acid method (24). One unit of TreX was defined as the amount of enzyme releasing 1 μmol equivalent of maltose min−1. Protein concentrations were determined using bovine serum albumin (BSA; Sigma Chemical Co.) as a standard (25).
Synthesis of maltodextrin-branched cyclodextrin and branched maltoheptaose.
Five milliliters of a solution of concentrated maltosyl transferase from Thermatoga maritima (TmMT) was added to a solution of 5% (wt/vol) maltosyl-β-cyclodextrin (Glc2-β-CD) and 10% (wt/vol) soluble starch in McIlvaine's buffer (pH 6.5), and the reaction proceeded at 70°C for 3 days. The solution was next heated to 100°C for 20 min to inactivate the enzyme and loaded onto a C18 Sep-Pak column to remove small oligosaccharides. The substrate was purified by several-stage preparative high-performance liquid chromatography (HPLC). Dimaltotetraosyl-β-cyclodextrin [(Glc4)2-β-CD] was prepared by a modification of the method of Kang et al. (26). To prepare maltotetraosyl-α-1,6 maltoheptaose, Glc4-β-CD was hydrolyzed with Pyrococcus furiosus amylase (PfTA), and the product was purified on a butyl-Sepharose column (27).
Cloning, expression, and purification of yeast glycogen debranching enzymes.
To clone the GDE of Saccharomyces cerevisiae, four PCR primers were used: forward1 (5′-CGG GCC ATG GGA ATG AAT AGA TCA TTA CTG CTA CGT TTG TCG-3′ [NcoI restriction site underlined]) and reverse1 (TGT CAT AGA TAA GCT TTG TAC AAA CAA TGT AGA [HindIII restriction site underlined]) amplifying bases 1 to 3123 (the large fragment) and forward2 (TCT ACA TTG TTT GTA CAA AGC TTA TCT ATG ACA [HindIII restriction site underlined]) and reverse2 (CGC GAA GCT TTC AGG AAT CAT CTT CGT AGG CAT CCC ATA A [HindIII restriction site underlined]) amplifying bases 3124 to 4608 (the small fragment). These primers were designed with reference to the full genome sequence in GenBank. The large fragment was PCR amplified from genomic DNA of S. cerevisiae S288c. The PCR product was digested with NcoI and HindIII and ligated with identically cut pPRoEX HTa (Invitrogen, Carlsbad, CA). The recombinant plasmid was used as a vector for subsequent cloning of the small fragment, which was amplified using the forward2 and reverse2 primers followed by HindIII digestion. Finally, the small and large fragments were ligated, and the recombinant gene was sequenced (in pPRoEX Hta). The final recombinant plasmid was designated pPRoEXGDB1.
To overexpress GDB1, pPRoEXGDB1 was transformed into E. coli BL21(DE3). A transformant was cultured in 1 liter LB medium (10 g tryptone, 5 g yeast extract, 5 g sodium chloride) at 37°C with shaking (250 rpm), and isopropyl-β-d-thiogalactopyranoside (IPTG) was added to 0.01 mM at an optical density at 600 nm (OD600) of 1.0. After induction, the culture was cooled to 25°C and incubated for 24 h. Cells were harvested by centrifugation (8,000 × g, 20 min), and the pellet was sonicated (VC-600; Sonics & Materials, Danbury, CT) in an ice-cold water bath. After removal of cell debris, GDB1 was purified by Ni2+-nitrilotriacetate (Ni-NTA) column chromatography (Qiagen) following the manufacturer's method.
Overexpression and purification of GlgP.
The glycogen phosphorylase gene (glgP) was amplified by PCR from genomic DNA of E. coli K-12, with the addition of restriction sites (underlined below) required for cloning. The primers were GlgP/XbaI (5′-CGG AAA AAT CTA GAA CCC TTT GGC CCC GTT C-3′) and GlgP/NdeI (5′-GAA ACG CCC ATA TGA ATG CTC CGT TTA CAT ATT C-3′). The PCR product was digested with NdeI and XbaI and ligated into identically cut pTKNd6×H. The resulting construct (pTKNd6×H_GlgP) was transformed into E. coli MC1061, and transformants were cultured on LB medium with kanamycin (50 μg ml−1). GlgP was purified by Ni-NTA chromatography, and enzyme activity on glycogen as the substrate was measured using high-performance anion-exchange chromatography (HPAEC).
Isolation of glycogen from E. coli.
E. coli MC4100 [araD139 deoC1 flbB5301 ptsF25 rbsR relA1 rpsL150 Δ(argF-lac)U169] was cultured in M63 minimal salts (1 liter) supplemented with 0.5% (wt/vol) maltose as a carbon source at 37°C for 20 h with shaking (250 rpm). Cells were harvested by centrifugation (6,000 × g) at 4°C for 20 min, washed with ice-cold water, and resuspended in 5 ml 50 mM sodium acetate (pH 4.5). Glycogen was isolated from this suspension and quantified as described by Park et al. (28).
Reaction of yeast debranching enzyme with branched Glc4βCD.
The purified yeast debranching enzyme (1.5 mU) was added to 1% (wt/vol) solutions of Glc4-β-CD or Glc1-β-CD in 50 mM NaOAc buffer (pH 6.0). Reactions proceeded for 0.5 and 1 h at 37°C, and products were analyzed using TLC and HPAEC.
Preparation of phosphorylase-limited dextrin from glycogen.
Phosphorylase-limited dextrin was prepared as described by Hers et al. (29). Glycogen from E. coli (500 mg) was incubated with 37.5 μg phosphorylase in 20 ml 0.1 M sodium phosphate buffer (pH 6.5) in dialysis tubing and dialyzed against the same buffer. After 24 h, 20 μg phosphorylase a was added to the contents of the bag, and the incubation was continued for an additional 24 h. The reaction was terminated by heating at 100°C for 5 min, and insoluble material was removed by centrifugation. The resulting solution was dialyzed against water and freeze-dried.
Reaction of TreX with branched Glc4-β-CD.
TreX (1.5 mU) was incubated with Glc4-β-CD (0.5 mM) in 50 mM sodium acetate buffer (pH 5.5) in the presence of 10% (vol/vol) dimethyl sulfoxide (DMSO) at 70°C. Samples were taken at 1, 5, 10, 30, 60, and 90 min and heated at 100°C for 10 min. The samples were centrifuged and analyzed by TLC and HPAEC.
Reaction of glycogen phosphorylase with glycogen and phosphorylase-limited dextrin from glycogen, in the presence of TreX.
Glycogen phosphorylase was incubated with 1% (wt/vol) glycogen or phosphorylase-limited dextrin in the presence (0.2 to 1 mU) or absence of TreX in 50 mM sodium phosphate buffer (pH 6.5) at 37°C. Reactions proceeded for 15, 30, and 60 min, and samples were heated at 100°C for 5 min. Insoluble material was removed by centrifugation, and the resulting solutions were analyzed by HPAEC. Glc-1P production was quantified using a standard curve constructed with the aid of authentic material.
Analysis of reaction products. (i) TLC analysis.
Silica gel K5F TLC plates (Whatman) were activated for 1 h at 110°C. Samples were spotted onto plates using a pipette, and the plates were placed in a TLC chamber containing n-butanol–ethanol–water at 5:5:3 (vol/vol/vol). Chromatography was conducted at room temperature. Reducing sugars were detected using the naphthol-sulfuric acid (H2SO4) method (30). Each plate was thoroughly dried and developed by rapid dipping into a methanol solution containing 3 g N-(1-naphthyl)-ethylene-diamine and 50 ml concentrated H2SO4 per liter. Each plate was dried and placed in an oven at 110°C for 10 min; purple-black spots appeared on a white background.
(ii) HPAEC.
The reaction mixtures incubated with TreX were boiled for 10 min, centrifuged at 12,000 × g for 10 min, and filtered using a membrane filter kit (pore diameter, 0.45 μm) (Millipore, Billerica, MA). Products were applied to a 0.4- by 25-cm CarboPac PA-1 column (Dionex, Sunnyvale, CA) and analyzed on an HPAEC platform fitted with an ED40 pulsed amperometric detector (Dionex). Sixty-microliter aliquots of sample were injected. Elution was achieved using a NaOAc gradient in 150 mM NaOH (increasing from 60 to 180 mM NaOAc over 0 to 10 min, from 180 to 240 mM over 10 to 16 min, from 240 to 300 mM over 16 to 27 min, from 300 to 360 mM over 27 to 44 min, and from 360 to 372 mM over 44 to 55 min). The flow rate was 1 ml min−1.
(iii) Isolation and identification of transfer products.
TreX (3 mU mg−1 of substrate) was reacted with 0.5% (wt/vol) Glc4-β-CD in 50 mM sodium acetate buffer (pH 5.5) at 70°C for 5 min. The reaction mixture was next heated at 100°C for 10 min and passed through a 0.45-μm-pore Millex-HV filter (Millipore) prior to two-step HPLC (Jaigel W-251 column, 2 × 50 cm; W-252 column, 2 × 50 cm) (Japan Analytical Industry, Tokyo, Japan) using distilled water to elute bound materials (flow rate, 3.5 ml min−1). Fractions were analyzed by HPAEC. To identify positional isomers of transfer products, (Glc4)2-β-CD was converted into dimaltosyl-β-cyclodextrin [(Glc2)2-β-CD] by reaction with β-amylase (30 U) at 37°C for 12 h, followed by heating at 100°C for 5 min and filtration through a 0.45-μm-pore diameter membrane to improve isomer separation by HPLC. An 1100 HPLC system (Agilent Technologies, Inc., Waldbronn, Germany) fitted with a Hypercarb column (150- by 4.6-mm internal diameter) (Thermo-Scientific, Loughborough, United Kingdom) and an evaporative light scattering detector (ELSD 2000; Alltech Associates, Inc., Deerfield, IL) was used in the analysis. Elution was performed at 60°C using an isocratic solvent system of acetonitrile and water (20:80 [vol/vol]) at a flow rate of 1 ml min−1.
(iv) Mathematical characterization of the TreX reaction with Glc4-β-CD.
The kinetics of substrate transglycosylation and hydrolysis were calculated using the minimal kinetic scheme for substrate transglycosylation proposed previously by Kawai et al. (31) and Gusakov et al. (32).
Substrate transglycosylation by TreX was considered to proceed as shown in equations 1 and 2 below.
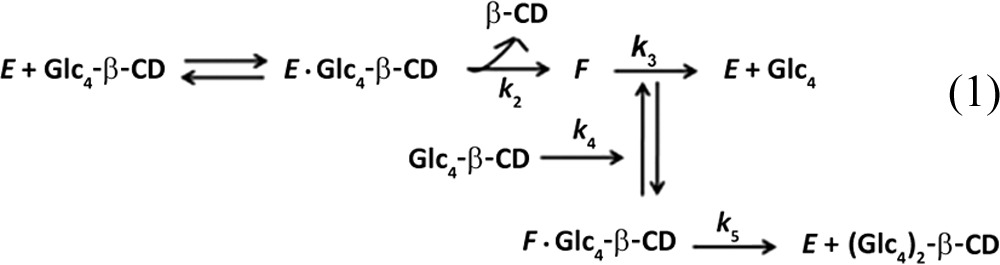 |
| (2) |
Equation 1 yields the rate equations
| (3) |
| (4) |
where Km and kcat are parameters for the hydrolysis reaction, Km2 and kcat2 are the parameters for the transglycosylation reaction, and E and S represent the enzyme and the substrate, respectively.
To determine the transglycosylation kinetic parameters, the hydrolytic parameters Km and kcat were measured in reactions with Glc4-β-CD as the substrate at 0.05 to 0.3 mM. The transglycosylation kinetic parameters were determined at higher substrate concentrations (0.5 to 2 mM). Additionally, the first-order reaction rate for hydrolysis of (Glc4)2-β-CD was separately determined after isolation of (Glc4)2-β-CD.
RESULTS
The glycogen mimic substrate (Glc4-β-CD) is suitable for characterization of the glycogen breakdown process.
To validate the suitability of the glycogen mimic substrate for a glycogen study, Glc4-β-CD was tested with a mouse liver extract and yeast debranching enzymes whose reaction modes are well known. TLC (Fig. 1) and HPAEC (data not shown) yielded information on the reaction products of mouse liver and yeast debranching enzymes acting on Glc4-β-CD (Fig. 1). When the glycogen debranching enzyme of yeast (GDE) was incubated with Glc4-β-CD, Glc1-β-CD (product no. 3 in Fig. 1A) and a series of transfer products—Glc7-β-CD to Glc2-β-CD (no. 6, 7, 8, and 9 in Fig. 1A)—were obtained. The maltotriosyl moiety of Glc4-β-CD was transferred to another molecule of Glc4-β-CD and also to other materials (Fig. 1). Thus, the GDE of yeast elongated branch chain length via the formation of α-1,4-glucosidic linkages in the absence of glycogen phosphorylase. One product, Glc1-β-CD (no. 3), was further hydrolyzed by the yeast GDE to glucose (product 1) and β-CD (product 2). In addition, Glc3-β-CD (product 4) was produced by GDE action; glucose was released from Glc4-β-CD. However, no (Glc4)2-β-CD was produced when yeast GDE reacted with Glc4-β-CD. HPAEC and analysis of Rf values by TLC also identified Glc7-β-CD and Glc1-β-CD. Formation of Glc7-β-CD was confirmed by incubating the putative product with isoamylase; Glc7 and β-CD were produced. These results show that glycogen degradation by the yeast debranching enzyme can be followed with Glc4-β-CD as a substrate analog.
FIG 1.
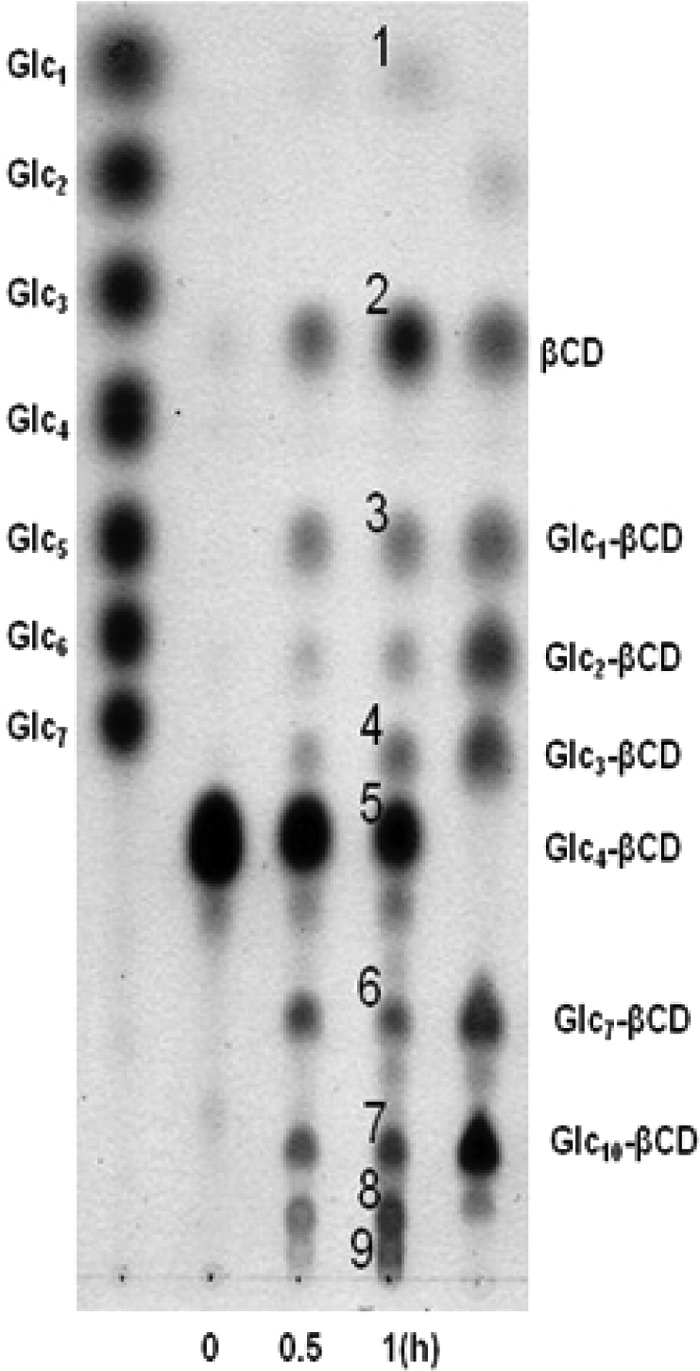
Thin-layer chromatography analysis of products from reaction mixtures containing yeast glycogen debranching enzyme. Product numbers: 1, glucose; 2, β-CD; 3, glucose-α-1,6-β-CD; 4, maltotriosyl-α-1,6-β-CD; 5, maltotetraosyl-α-1,6-β-CD; 6, maltoheptaosyl-α-1,6-β-CD; 7, maltodecaosyl-α-1,6-β-CD; 8, maltotridecaosyl-α-1,6-β-CD; 9, maltotetradecaosyl-α-1,6-β-CD.
TreX has both hydrolytic and transglycosylation activity at high substrate concentrations (>0.5 mM).
The enzyme was incubated with Glc4-β-CD (0.5 mM) in 50 mM sodium acetate buffer (pH 5.5) in the presence of 10% (vol/vol) DMSO at 70°C. Samples were taken at various time intervals and heated at 100°C for 10 min prior to analysis by TLC and HPAEC. Unlike what was noted when the debranching enzymes of yeast and mouse liver extract were studied, TreX did not generate α-1,4 hydrolysis products such as Glc3-, Glc2-, or Glc1-β-CD. TreX reacted with Glc4-β-CD to initially form the transglycosylation product (Glc4)2-β-CD. These concentrations then decreased gradually, whereas β-CD and maltotetraose concentrations increased, as the reaction time was extended (Fig. 2, 3, and 4). When the Glc4-β-CD concentration was 0.05 mM, the Glc4-β-CD level decreased gradually, whereas the β-CD and maltotetraose concentrations increased with the reaction time (Fig. 2B to 4B). As the positional isomers of the (Glc4)2-β-CD transfer products were not separated on the Hypercarb column, they were first converted to (Glc2)2-β-CD by β-amylase (Fig. 5A and B) and analyzed by HPLC using the Hypercarb column. As shown in Fig. 5C, comparison of the chromatographic profile with that of (Glc2)2-β-CD reference materials (21) indicated that the transfer product was a mixture of 61,63- and 61,64-(Glc4)2-β-CD at a molar ratio close to unity. Additionally, the formation of 61,64-(Glc4)2-β-CD observed in the present study and the synthesis of 61,64-(Glc2)2-β-CD reported by Kang et al. (26) support the idea that the transfer products produced from branch chains varying widely in size are similar.
FIG 2.
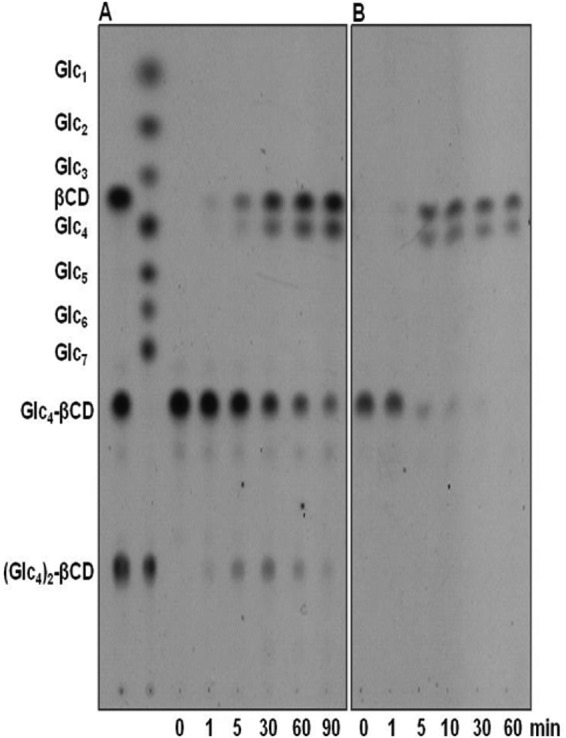
Thin-layer chromatography analysis of the reaction products yielded by TreX acting on 0.5 mM (A) and 0.05 mM (B) Glc4-β-CD. Glc1 to Glc7, standard maltooligosaccharides from glucose to maltoheptaose; β-CD, β-cyclodextrin; Glc4-β-CD, 6-O-α-maltotetraosyl-β-cyclodextrin; (Glc4)2-β-CD, dimaltotetraosyl-β-cyclodextrin.
FIG 3.

Time course HPAEC analysis of the reaction products of TreX acting on 0.5 mM (A) and 0.05 mM (B) Glc4-β-CD. 1, maltotetraosyl-1,6-β-CD; 2, β-CD; 3, mixture of isomers of dimaltotetraosyl-1,6-β-CD; 4, maltotetraose.
FIG 4.
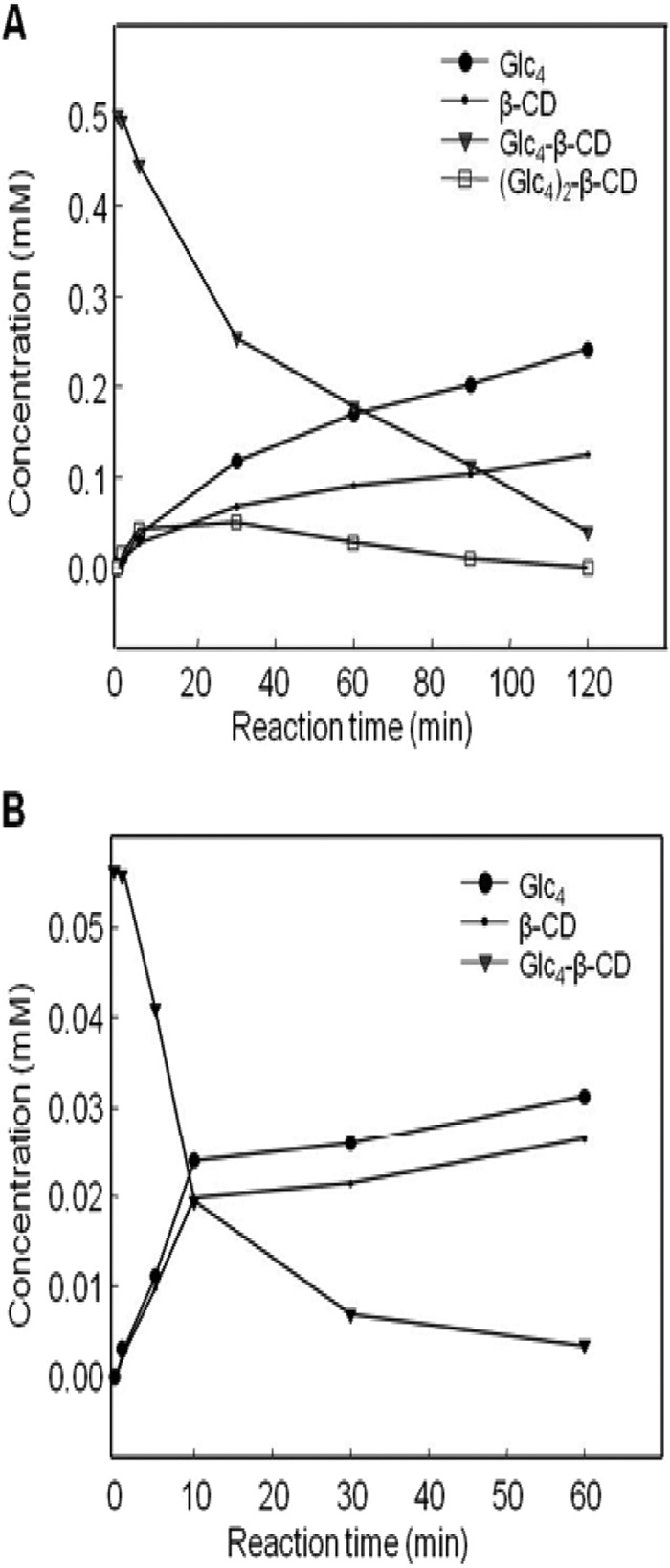
Time course analysis of TreX products produced when the enzyme acted on 0.5 mM (A) and 0.05 mM (B) Glc4-β-CD. ●, maltotetraose; •, β-CD; ▼, maltotetraosyl-1,6-β-CD; □, dimaltotetraosyl-β-CD.
FIG 5.
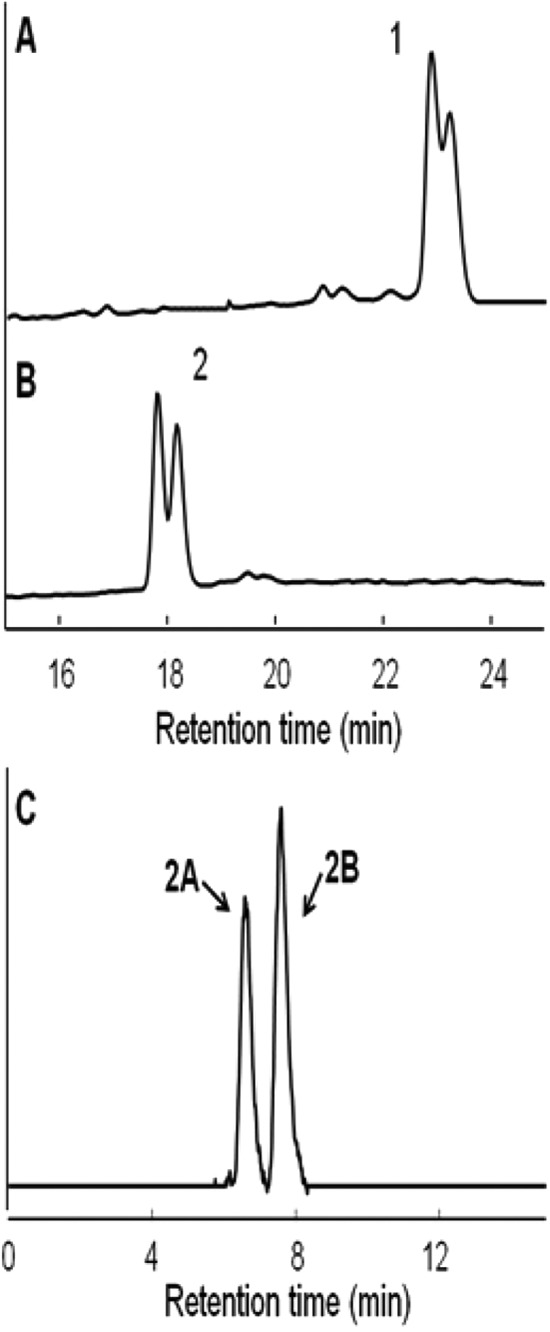
HPAEC analysis (A and B) of transfer products prior to β-amylase treatment (A) and after β-amylase treatment (B). The positional isomers of (B) were analyzed by HPLC using a Hypercarb column (C). Products: 1, a mixture of 61,63- and 61,64-dimaltotetraosyl-β-CD; 2, a mixture of 61,63- and 61,64-dimaltosyl-β-CD; 2A, 61,63-dimaltosyl-β-CD; 2B, 61,64-dimaltosyl-β-CD.
Kinetics of hydrolysis and transglycosylation of Glc4-β-CD by TreX.
The enzyme was added to Glc4-β-CD solutions of various concentrations (0.05 to 2 mM). As transglycosylation was observed at substrate concentrations of >0.5 mM (Fig. 2 and 3), a kinetic analysis was conducted using two ranges of substrate concentration. The Km and kcat of hydrolysis were assessed at substrate concentrations of 0.05 to 0.3 mM, and the Km2 and kcat2 of transglycosylation were determined at 0.5 to 2 mM. Data plotting yielded the initial velocities of Glc4 production at various substrate concentrations. A Lineweaver-Burk plot (data not shown) showed that the Km and kcat of hydrolysis were 0.206 ± 0.003 mM and (2.57 ± 0.334) × 103 s−1, respectively, and the kcat/Km ratio was (12.42 ± 0.162) × 103 mM−1· s−1. To determine the kinetic parameters of transglycosylation, TreX was reacted with Glc4-β-CD at 0.5 to 2 mM. The formation of β-CD was quantified by HPAEC to obtain initial reaction velocities. Equation 4 was used to estimate the transglycosylation parameters Km2 = 3.39 ± 0.057 mM, kcat2 = (1.78 ± 0.03) × 103 s−1, and kcat2/Km2 = (0.54 ± 0.01) × 103 mM−1·s−1, which indicated that transglycosylation occurred principally at high substrate concentrations. The branched glycosyl chain of the Glc4 substrate was transferred to a hydroxyl group of an acceptor molecule other than water, and the extent of hydrolysis was reduced. Thus, transglycosylation may influence the overall reaction rate. As the reaction proceeds, hydrolysis began to dominate over transglycosylation, leading to the production of Glc4 and β-CD from the TreX transglycosylation products (Fig. 4).
TreX has high specific activity for a wide range of branch chain lengths.
Glycogen contains branch chains of various lengths. To explore TreX activities on various branch chains, Glc2- to Glc10-β-CDs were incubated with TreX, and the products were analyzed by HPAEC and TLC. Unexpectedly, TreX was active on all branch chains studied and the kcat/Km ratio increased as the branch chain degree of polymerization (DP) rose (Fig. 6).
FIG 6.
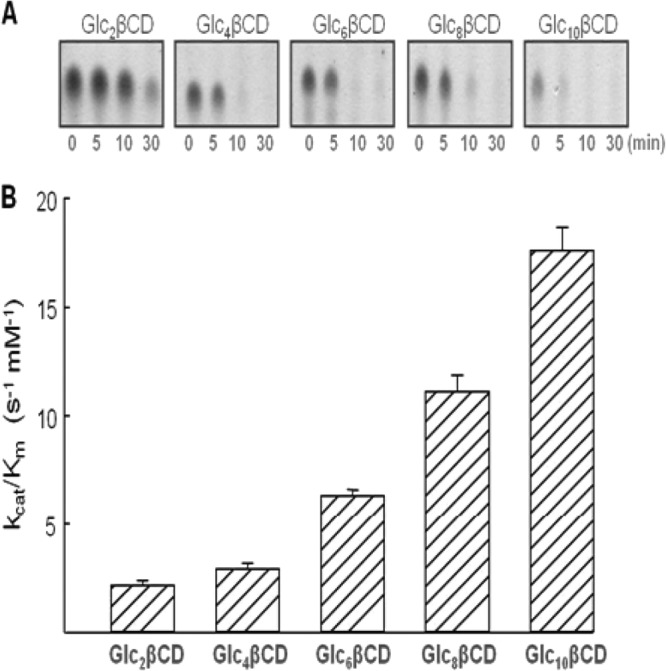
TreX activities toward various branched cyclodextrins: Glc2-β-CD (6-O-α-maltosyl-β-cyclodextrin), Glc4-β-CD (6-O-α-maltotetraosyl-β-cyclodextrin), Glc6-β-CD (6-O-α-maltohexaosyl-β-cyclodextrin), and Glc8-β-CD (6-O-α-maltooctaosyl-β-cyclodextrin). (A) TLC analysis of the substrate concentration at various reaction times; (B) values of the kcat/Km ratio.
Action of TreX on branched maltotetraosyl-α-1,6-maltoheptaose.
Unlike cyclodextrin, glycogen is a linear glucan. To confirm that TreX was active on linear glucans, the enzyme was incubated with maltotetraosyl-α1,6-maltoheptaose, yielding three major products after a 10-min reaction. The substrate was hydrolyzed to maltoheptaose and maltotetraose. In parallel, the maltotetraose moiety was transferred to another substrate molecule, producing dimaltotetraosyl maltoheptaose, which was in turn further hydrolyzed to maltotetraose and maltoheptaose (data not shown). This supports the suggestion that TreX exerts α-1,6-transfer activity on branched glucans. The reaction did not yield either maltooctaose (Glc8) or maltoundecaose (Glc11), indicating that the maltotetraosyl moiety was not transferred via formation of an α-1,4 linkage but rather an α-1,6 linkage.
Transglycosylation enhances the rate of glycogen dephosphorylation.
To explore the effect of transglycosylation on glycogen degradation in a model system, glycogen (E. coli MC4100) was incubated with glycogen phosphorylase in the presence or absence of TreX. The latter enzyme significantly increased the rate of Glc-1P formation (5.48 × 10−3 mmol·liter−1·min−1 compared to the control value of 3.91 × 10−3 mmol·liter−1·min−1). Glc-1P was formed more rapidly when glycogen was present at 1.0 mM than that at 0.5 mM (Table 1), which was in line with the observation that transglycosylation occurred preferentially at higher concentrations of Glc4-β-CD (Fig. 2 and 3). In contrast, TreX activity on limited dextrin did not vary with substrate concentration (Table 1). A slight increase in the rate in the absence of TreX was likely due to the incomplete limited dextrin preparation. These results indicate that shifting branch chains to the outer layers of glycogen was achieved via transglycosylation, thus, increasing the number of nonreducing glucose moieties available to phosphorylase (Table 1).
TABLE 1.
Effect of TreX on formation of glucose-1-phosphatea
| Substrate | TreX concn (mU) | Glucose-1-phosphate concn (mM) at: |
Rate constant (mmol liter−1·min−1) × 10−3 | ||
|---|---|---|---|---|---|
| 15 min | 30 min | 60 min | |||
| Limited dextrin, 10 mg/ml | 0.0 | 0.030 ± 0.002 | 0.052 ± 0.004 | 0.071 ± 0.058 | 1.15 ± 0.09 |
| 0.2 | 0.030 ± 0.002 | 0.049 ± 0.003 | 0.067 ± 0.042 | 1.08 ± 0.07 | |
| 0.5 | 0.028 ± 0.002 | 0.048 ± 0.003 | 0.064 ± 0.040 | 1.04 ± 0.06 | |
| Glycogen | |||||
| 1 mM | 0.0 | 0.086 ± 0.006 | 0.137 ± 0.011 | 0.242 ± 0.019 | 3.91 ± 0.31 |
| 0.2 | 0.092 ± 0.003 | 0.180 ± 0.012 | 0.331 ± 0.022 | 5.48 ± 0.36 | |
| 0.5 | 0.092 ± 0.001 | 0.171 ± 0.002 | 0.323 ± 0.005 | 5.71 ± 0.39 | |
| 0.5 mM | 0.0 | 0.045 ± 0.004 | 0.062 ± 0.05 | 0.132 ± 0.012 | 2.13 ± 0.19 |
| 0.2 | 0.045 ± 0.004 | 0.078 ± 0.08 | 0.148 ± 0.016 | 2.44 ± 0.13 | |
| 0.5 | 0.045 ± 0.012 | 0.075 ± 0.20 | 0.145 ± 0.039 | 2.38 ± 0.16 | |
Glycogen or phosphorylase-limited dextrin was incubated with TreX (0.2 to 0.5 mU) in the presence of 60 mU of glycogen phosphorylase at 37°C for 15 to 60 min.
DISCUSSION
Branched cyclodextrins as substrate mimics were used previously to characterize yeast debranching enzymes (33). However, transfer products were only indirectly identified by further hydrolysis with pullulanase, in which dibranched products cannot be detected using such a method. Kang et al. (26) synthesized di-O-α-maltosyl-β-cyclodextrin [(Glc2)2-β-CD] from 6-O-α-maltosyl-β-cyclodextrin via transglycosylation catalyzed by TreX. This finding suggested that TreX transfers the maltosyl residue of Glc2-α-1,6-β-CD with formation of an α-1,6-glycosidic linkage. TreX also yielded (Glc4)2-β-CD containing an α-1,6-glucosidic linkage (Fig. 7). Similarly, 63-maltotetraosyl maltoheptaose, similar to a glycogen branch, produced the transfer product of (Glc4)2-maltoheptaose (data not shown). Thus, transglycosylation occurs via formation of an α-1,6-glycosidic linkage when either branched linear maltodextrin or Glc4-β-CD serves as the substrate (data not shown). Additionally, transglycosylation of branch chains can occur between two molecules or within a molecule. Therefore, glycogen consisting of a large number of branch chains in a molecule possibly includes a transglycosylation step in which the same glycogen molecule has both acceptor and donor sites. The branch chains occupy ∼62% of the space within a glycogen molecule, indicating an extremely high density between branch chains in glycogen (34). Therefore, we assume that the local concentrations of donors and acceptors within an individual glycogen molecule may be sufficiently high to trigger transglycosylation. This observation indicates that TreX exerts transfer activity, forming α-1,6-glycosidic linkages in the outer layers of glycogen. In addition, the extent of transglycosylation is probably much higher when authentic glycogen is the substrate, rather than artificial branched β-CDs. Tabata et al. observed that the transferase activity of yeast debranching enzyme acting on limited dextrin was higher than that when a substrate mimic, Glc4-β-CD, was employed (33).
FIG 7.

Schematic diagram of hydrolysis and transglycosylation of maltotetraosyl β-cyclodextrin (Glc4-β-CD) catalyzed by TreX.
The substrate specificity to various branch chain lengths of glycogen was determined in detail to determine which branch chain could be efficiently employed during glycogen breakdown. Possible transglycosylation reactions of microbial debranching enzymes require more intensive study, as the enzymes of archaea and bacteria have received little attention compared to those of animals and yeasts. A previous study indicated that TreX accepts a wide spectrum of substrates, the chains of which have DP values of 3 to 6 (23). In the present study, we further analyzed the effects of branch chain length using a wide range of substrates. Unexpectedly, TreX was active on branched chains of DP2 to DP10; transfer products were evident with all tested substrates. This result agrees with the suggestion of Woo et al. (21) that subunit tetramerization generates a channel-like cavity that allows long branch chains to be accommodated. An isoamylase from Pseudomonas spp. had the highest specific activity toward a branch chain of DP7, but debranching of the DP2 chain was minimal (data not shown). The debranching enzyme from Nostoc punctiforme exhibits a catalytic preference for longer maltooligosaccharides (DP, >8) (11). In contrast, GlgX, a debranching enzyme from E. coli, is active mainly against DP4 chains (5, 10), and AmyX from B. subtilis is specific for DP3 to -6 chains (7). A substrate with a branch chain length of DP4 can be considered a suitable substrate for kinetic analysis because such a substrate is within the range of preferable chain length for TreX. Moreover, a limited dextrin containing a branch chain of DP4 is produced via the action of glycogen phosphorylase and is also conveniently studied in relation to mammalian debranching enzyme. The average branch chain lengths of glycogen in E. coli (28), S. solfataricus (35), and B. subtilis (36) are DP7 to -11, and the structures are expected to be similar to that of mammalian glycogen (DP13). Meléndez-Hevia et al. (34) and Meléndez et al. (37) developed a mathematical model of the mammalian glycogen molecule. An optimized glycogen structure features chains that are an average of 13 glucose residues in length. The chains are exposed to the action of phosphorylase because they are sufficiently flexible to allow binding to the enzyme active site. Moreover, the presence of a large number of such chains ensures that a phosphorylase active site rendered vacant by loss of a chain can be immediately filled by another adjacent chain (34).
Branch chains on the glycogen surface are accessible by phosphorylase. The three-dimensional structure of mammalian glycogen phosphorylase features a narrow 30-Å crevice that binds glycogen, accommodating 4 to 5 glucosyl residues (38, 39). Similarly, archaeal phosphorylase acting on external chains releases glucose, thus allowing the enzyme to act on internal chains on the same surface. A consequence of transglycosylation of branch chains is an increase in chain density on the outer glycogen layer. Internal chains are shifted outward, increasing the number of nonreducing ends available to phosphorylase (Fig. 8). The kinetic data (Table 1) revealed that such outward transfer provides many points of attack for phosphorylase, increasing glucose release. As shown in Fig. 2A, transglycosylation occurs preferentially at high substrate concentrations (∼0.5 mM Glc4-β-CD). Branch chains occupy ∼62% of the space within a glycogen molecule, indicating an extremely high density between branch chains in glycogen (34). Therefore, we can assume that the local concentrations of branch chains in the outer tier of the glycogen molecule may be sufficiently high to enable transglycosylation.
FIG 8.
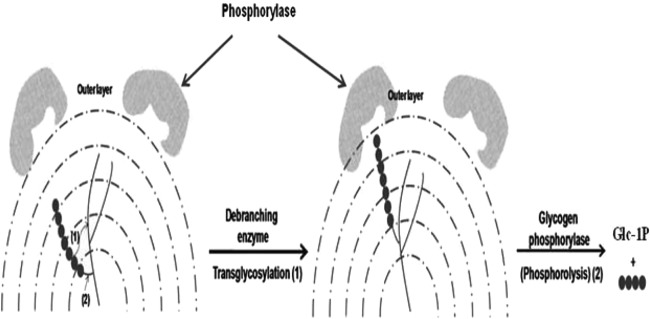
A model of outward shifting of glycogen branch chains during the debranching process, catalyzed by transglycosylation.
Glycogen metabolism is highly interconnected with a wide variety of cellular processes, including stringent response, Mg2+ availability, and osmotic pressure (40). Therefore, glycogen as a reserve macromolecule has little effect on the internal osmotic pressure of the cell (2). Maintaining osmotic pressure during the glycogen degradation is important. Instead of hydrolysis of the branch chain, in this model, transglycosylation may maintain the level of osmotic pressure and fluid viscosity, as the release of linear branch chains (DPs of >5 to 12) by hydrolysis may cause a drastic increase in osmotic pressure and viscosity of cellular fluid, whereas Glc-1P from the transferred branch chains can be immediately metabolized into the glycolytic pathway without a significant change in osmotic pressure. A mutant enzyme that has high hydrolysis activity with low transglycosylation activity or cloning of a novel debranching enzyme from another microbial source having low transglycosylation selectivity is required for further study. Moreover, transglycosylation inhibits hydrolysis (31). Depending on the environment of a given microorganism, debranching (hydrolysis) of glycogen may be controlled by product inhibition and may be further hindered by transglycosylation side reactions. Until recently, the physiological function of the microbial debranching enzyme has been considered to be principally hydrolysis. However, we suggest that transglycosylation may also be involved in glycogen degradation. Similarly, trehalose biosynthesis in Sulfolobus may proceed via transglycosylation catalyzed by TreX in association with the actions of enzymes encoded by treZY (15, 16), and glucan chains shifted to the outer layer by TreX may facilitate the actions of maltooligosyl trehalose synthase (TreY) and maltooligosyl trehalose hydrolase (TreZ). We have shown that just as mammalian debranching enzymes exert dual functions, transglycosylation in archaea may assist in glycogen breakdown by forming and shifting α-1,6-glucosidic linkages by chain transglycosylation to the outer layers of glycogen. However, in the present study, we used kinetic data to explore the possible contribution of transglycosylation to glycogen breakdown in a model system in which TreX from S. solfataricus was introduced to glycogen and glycogen phosphorylase from E. coli. Therefore, a full set of enzymes involved in the glycogen metabolism of a strain of bacteria or archaea should be introduced, and their actions should be analyzed in concert.
ACKNOWLEDGMENTS
This study was supported by the Basic Research Program of the National Research Foundation (grant no. 2012R1A1A2005012) and the Next Generation BioGreen21 Program (SSAC, grant no. PJ009086), Rural Development Administration, Republic of Korea.
Footnotes
Published ahead of print 7 March 2014
REFERENCES
- 1.Ball SG, Morell MK. 2003. From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Annu. Rev. Plant Biol. 54:207–233. 10.1146/annurev.arplant.54.031902.134927 [DOI] [PubMed] [Google Scholar]
- 2.Wilson WA, Roach PJ, Montero M, Baroja-Fernández E, Munoz FJ, Eydallin G, Viale AM, Pozueta-Romero J. 2010. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 34:952–985. 10.1111/j.1574-6976.2010.00220.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Preiss J. 1984. Bacterial glycogen synthesis and its regulation. Annu. Rev. Microbiol. 38:419–458. 10.1146/annurev.mi.38.100184.002223 [DOI] [PubMed] [Google Scholar]
- 4.Preiss J, Romeo T. 1989. Physiology, biochemistry and genetics of bacterial glycogen synthesis. Adv. Microb. Physiol. 30:183–238 [DOI] [PubMed] [Google Scholar]
- 5.Dauvillée D, Kinderf IS, Li Z, Kosar-Hashemi B, Samuel MS, Rampling L, Ball S, Morell MK. 2005. Role of the Escherichia coli glgX gene in glycogen metabolism. J. Bacteriol. 187:1465–1473. 10.1128/JB.187.4.1465-1473.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dippel R, Bergmiller T, Bohm A, Boos W. 2005. The maltodextrin system of Escherichia coli: glycogen-derived endogenous induction and osmoregulation. J. Bacteriol. 187:8332–8339. 10.1128/JB.187.24.8332-8339.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JT. 2008. Roles of glycoside hydrolase family 13 enzymes in maltodextrin/glycogen metabolism in Bacillus subtilis and Escherichia coli. Ph.D. thesis Seoul National University, Seoul, South Korea [Google Scholar]
- 8.Kiel JAKW, Boels JM, Geldman G, Venema G. 1994. Glycogen in Bacillus subtilis: molecular characterization of an operon encoding enzymes involved in glycogen biosynthesis and degradation. Mol. Microbiol. 11:203–218. 10.1111/j.1365-2958.1994.tb00301.x [DOI] [PubMed] [Google Scholar]
- 9.Sheng F, Jia X, Yep A, Preiss J, Geiger H. 2009. The crystal structures of the open and catalytically competent closed conformation of Escherichia coli glycogen synthase. J. Biol. Chem. 284:17796–17807. 10.1074/jbc.M809804200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song HN, Jung TY, Park JT, Park BC, Myung PK, Boos W, Woo EJ, Park KH. 2010. Structural rationale for the short branched substrate specificity of the glycogen debranching enzyme GlgX. Proteins 78:1847–1855. 10.1002/prot.22697 [DOI] [PubMed] [Google Scholar]
- 11.Dumbrepatil AB, Choi JH, Park JT, Kim MJ, Kim TJ, Woo EJ, Park KH. 2010. Structural features of the Nostoc punctiforme debranching enzyme reveal the basis of its mechanism and substrate specificity. Proteins 78:348–356. 10.1002/prot.22548 [DOI] [PubMed] [Google Scholar]
- 12.Yokobayashi K, Misaki A, Harada T. 1970. Purification and properties of Pseudomonas isoamylase. Biochim. Biophys. Acta 212:458–469. 10.1016/0005-2744(70)90252-4 [DOI] [PubMed] [Google Scholar]
- 13.Asha R, Niyonzima FN, Sunil SM. 2013. Purification and properties of pullulanase from Bacillus halodurans. Int. Res. J. Biol. Sci. 2:35–43 http://www.isca.in/IJBS/Archive/v2i3/7.ISCA-IRJBS-2013-005.pdf [Google Scholar]
- 14.Maruta K, Kubota M, Fukuda S, Kurimoto M. 2000. Cloning and nucleotide sequence of a gene encoding a glycogen debranching enzyme in the trehalose operon from Arthrobacter sp. Q36. Biochim. Biophys. Acta 1476:377–381. 10.1016/S0167-4838(99)00253-8 [DOI] [PubMed] [Google Scholar]
- 15.Qu Q, Lee SJ, Boos W. 2004. TreT, a novel trehalose glycosyltransferring synthase of the hyperthermophilic archaeon Thermococcus litoralis. J. Biol. Chem. 279:47890–47897. 10.1074/jbc.M404955200 [DOI] [PubMed] [Google Scholar]
- 16.Van TT, Ryu SI, Lee KJ, Kim EJ, Lee SB. 2007. Cloning and characterization of glycogen-debranching enzyme from hyperthermophilic archaeon Sulfolobus shibatae. J. Microbiol. Biotechnol. 17:792–799 http://www.ncbi.nlm.nih.gov/pubmed/18051301 [PubMed] [Google Scholar]
- 17.Maruta K, Mitsuzumi H, Nakada T, Kubota M, Chaen H, Fukuda S, Sugimoto T, Kurimoto M. 1996. Cloning and sequencing of a cluster of genes encoding novel enzymes of trehalose biosynthesis from thermophilic archaebacterium Sulfolobus acidocaldarius. Biochim. Biophys. Acta 1291:177–181. 10.1016/S0304-4165(96)00082-7 [DOI] [PubMed] [Google Scholar]
- 18.Teste MA, Enjalbert B, Parrou JL, Prancois JM. 2000. The Saccharomyces cerevisiae YPR184w gene encodes the glycogen debranching enzyme. FEMS Microbiol. Lett. 193:105–110. 10.1111/j.1574-6968.2000.tb09410.x [DOI] [PubMed] [Google Scholar]
- 19.Torija MJ, Novo M, Lemassu A, Wilson W, Roach PJ, Francois J, Parrou JL. 2005. Glycogen synthesis in the absence of glycogenin in the yeast Saccharomyces cerevisiae. FEBS Lett. 579:3999–4004. 10.1016/j.febslet.2005.06.007 [DOI] [PubMed] [Google Scholar]
- 20.Park HS, Park JT, Kang HK, Cha H, Kim DS, Kim JW, Park KH. 2007. TreX from Sulfolobus solfataricus ATCC 35092 displays isoamylase and 4-α-glucanotransferase activities. Biosci. Biotechnol. Biochem. 71:1348–1352. 10.1271/bbb.70016 [DOI] [PubMed] [Google Scholar]
- 21.Woo EJ, Lee S, Cha H, Park JT, Yoon SM, Song HN, Park KH. 2008. Structural insight into the bifunctional mechanism of the glycogen-debranching enzyme TreX from the archaeon Sulfolobus solfataricus. J. Biol. Chem. 283:28641–28648. 10.1074/jbc.M802560200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim TJ, Kim MJ, Kim BC, Kim JC, Cheong TK, Kim JW, Park KH. 1999. Modes of action of acarbose hydrolysis and transglycosylation catalyzed by a thermostable maltogenic amylase, the gene for which was cloned from a Thermus strain. Appl. Environ. Microbiol. 65:1644–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JT, Park HS, Kang HK, Hong JS, Cha H, Woo JS, Kim JW, Kim MJ, Boos W, Lee S, Park KH. 2008. Oligomeric and funtional properties of a debranching enzyme (TreX) from the archaeon Sulfolobus solfataricus P2. Biocatal. Biotransf. 26:76–85. 10.1080/10242420701806652 [DOI] [Google Scholar]
- 24.Miller GL. 1959. Use of dinitrosalycylic acid reagent for determination of reducing sugars. Anal. Chem. 31:426–428. 10.1021/ac60147a030 [DOI] [Google Scholar]
- 25.Bradford M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 26.Kang HK, Cha H, Yang TJ, Park JT, Lee S, Kim YW, Auh JH, Okada Y, Kim JW, Cha J, Kim CH, Park KH. 2008. Enzymatic synthesis of dimaltosyl-β-cyclodextrin via a transglycosylation reaction using TreX, a Sulfolobus solfataricus P2 debranching enzyme. Biochem. Biophys. Res. Commun. 366:98–103. 10.1016/j.bbrc.2007.11.115 [DOI] [PubMed] [Google Scholar]
- 27.Yang SJ, Lee HS, Kim JW, Lee MH, Auh JH, Lee BH, Park KH. 2006. Enzymatic preparation of maltohexaose, maltoheptaose, and maltooctaose by the preferential cyclomaltooligosaccharide (cyclodextrin) ring-opening reaction of Pyrococcus furiosus thermostable amylase. Carbohydr. Res. 341:420–424. 10.1016/j.carres.2005.11.031 [DOI] [PubMed] [Google Scholar]
- 28.Park JT, Shim JH, Tran PL, Hong IH, Yong HU, Oktavina EF, Nguyen HD, Kim JW, Lee TS, Park SH, Boos W, Park KH. 2011. Role of maltose enzymes in glycogen synthesis by Escherichia coli. J. Bacteriol. 193:2517–2526. 10.1128/JB.01238-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hers HG, Verhue W, van Hoof F. 1967. The determination of amylo-1,6-glucosidase. Eur. J. Biochem. 2:257–264. 10.1111/j.1432-1033.1967.tb00133.x [DOI] [PubMed] [Google Scholar]
- 30.Robyt JF, Mukerjea R. 1994. Separation and quantitative determination of nanogram quantities of maltodextrins and isomaltodextrins by thin-layer chromatography. Carbohydr. Res. 251:187–202. 10.1016/0008-6215(94)84285-X [DOI] [Google Scholar]
- 31.Kawai R, Igarashi K, Kitaoka M, Ishii T, Samejima M. 2004. Kinetics of substrate transglycosylation by glycoside hydrolase family 3 glucan (1→3)-β-glucosidase from the white-rot fungus Phanerochaete chrysosporium. Carbohydr. Res. 339:2851–2857. 10.1016/j.carres.2004.09.019 [DOI] [PubMed] [Google Scholar]
- 32.Gusakov AV, Sinitsyn AP, Goldsteins GH, Klyosov AA. 1984. Kinetics and mathematical model of hydrolysis and transglycosylation catalysed by cellobiase. Enzyme Microb. Technol. 6:275–283. 10.1016/0141-0229(84)90130-3 [DOI] [Google Scholar]
- 33.Tabata S, Hizukuri S. 1992. Properties of yeast debranching enzyme and its specificity toward branched cyclodextrins. Eur. J. Biochem. 206:345–348. 10.1111/j.1432-1033.1992.tb16933.x [DOI] [PubMed] [Google Scholar]
- 34.Meléndez-Hevia E, Waddell TG, Shelton ED. 1993. Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem. J. 295:477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.König H, Skorko R, Zillig W, Reiter WD. 1982. Glycogen in thermoacidophilic archaebacteria of the genera Sulfolobus, Thermoproteus, Desulfurococcus and Thermococcus. Arch. Microbiol. 132:297–303. 10.1007/BF00413378 [DOI] [Google Scholar]
- 36.Shim JH, Park JT, Hong JS, Kim KW, Kim MJ, Auh JH, Kim YW, Park CS, Boos W, Kim JW, Park KH. 2009. Role of maltogenic amylase and pullulanase in maltodextrin and glycogen metabolism of Bacillus subtilis 168. J. Bacteriol. 191:4835–4844. 10.1128/JB.00176-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meléndez R, Meléndez-Hevia E, Cascante M. 1997. How did glycogen structure evolve to satisfy the requirement for rapid mobilization of glucose? A problem of physical constraints in structure building. J. Mol. Evol. 45:446–455 [DOI] [PubMed] [Google Scholar]
- 38.Barford D, Hu SH, Johnson LN. 1991. Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J. Mol. Biol. 5:233–260 [DOI] [PubMed] [Google Scholar]
- 39.Oikonomakos NG, Skamnaki VT, Tsitsanou KE, Gavalas NG, Johnson LN. 2000. A new allosteric site in glycogen phosphorylase a as a target for drug interactions. Structure 8:575–584. 10.1016/S0969-2126(00)00144-1 [DOI] [PubMed] [Google Scholar]
- 40.Montero M, Eydallin G, Viale AM, Almagro G, Muñoz FJ, Rahimpour M, Sesma MT, Baroja-Fernández E, Pozueta-Romero J. 2009. Escherichia coli glycogen metabolism is controlled by the PhoP-PhoQ regulatory system at submillimolar environmental Mg2+ concentrations, and is highly interconnected with a wide variety of cellular processes. Biochem. J. 424:129–141. 10.1042/BJ20090980 [DOI] [PubMed] [Google Scholar]