

Abstract
The nucleoid-associated protein EspR, a chromosome organizer, has pleiotropic effects on expression of genes associated with cell wall function and pathogenesis in Mycobacterium tuberculosis. In particular, EspR binds to several sites upstream of the espACD locus to promote its expression, thereby ensuring full function of the ESX-1 secretion system, a major virulence determinant. The N terminus of EspR contains the helix-turn-helix DNA-binding domain, whereas the C-terminal dimerization domain harbors residues involved in intersubunit interactions. While direct binding to DNA appears to be mediated by an EspR dimer-of-dimers, where two helix-turn-helix motifs remain free for long-range interactions, the mechanism of EspR higher-order organization and its impact on chromosome structure and gene expression are not understood. To investigate these processes, we identified seven amino acid residues using molecular dynamics and replaced them with Ala in order to probe interactions at either the dimer or the dimer-of-dimers interfaces. Arg70, Lys72, and Arg101 were important for protein stability and optimal DNA-binding activity. Moreover, the Arg70 mutant showed decreased dimerization in a mycobacterial two-hybrid system. To correlate these defects with higher-order organization and transcriptional activity, we used atomic force microscopy to observe different EspR mutant proteins in complex with the espACD promoter region. In addition, complementation of an M. tuberculosis espR knockout mutant was performed to measure their impact on EspA expression. Our results pinpoint key residues required for EspR function at the dimer (Arg70) and the dimer-of-dimers (Lys72) interface and demonstrate that EspR dimerization and higher-order oligomerization modulate espACD transcriptional activity and hence pathogenesis.
INTRODUCTION
EspR is an abundant DNA-binding protein of 14.7 kDa that is widely conserved among mycobacteria and other related actinomycetes. EspR was originally described to be a key activator of the virulence-associated ESX-1 secretion system by mediating direct upregulation of the espACD operon, which encodes three proteins (EspA, EspC, and EspD) required for proper ESX-1 activity (1–3). Genome-wide in vivo studies have revealed that in addition to the espACD locus, EspR binds hundreds of loci throughout the Mycobacterium tuberculosis chromosome, and multiple binding sites are frequently clustered (4). Associated with EspR binding sites are a variety of genes, sometimes topologically distant from each other but mainly serving a common biological function related to M. tuberculosis cell wall function and pathogenesis (4). Moreover, EspR is capable of influencing target gene expression in either a positive or negative manner. In particular, EspR appears able to regulate negatively its own expression under some conditions (1, 4). Thus, M. tuberculosis espR mutants are attenuated in a murine infection model (1), probably as a result of dysregulation of multiple cell wall and cellular components, including virulence determinants like the ESX-1 secretion system.
EspR is believed to act differently from a classical transcriptional regulator, since it often binds intragenic regions or sites far upstream of the transcriptional start site of related genes. Transcriptional regulation thus occurs over long distances of the chromosome by joining sites otherwise distant from each other into an active (or inactive) complex. Direct visualization of such complexes has been observed in atomic force micrographs, where EspR complexed with the full-length upstream region of espACD (1,357 bp) resulted in the formation of insulated DNA loops stabilized by EspR oligomers (5). Proteins such as EspR, playing a dual role in chromosome organization and gene expression, are termed nucleoid-associated proteins (NAPs) (6). NAPs are small and abundant DNA-binding proteins that control gene expression within the bacterial nucleoid via multiple possible mechanisms, including blockage of RNA polymerase's accessibility to promoter regions, displacement of other regulatory proteins, or assembly of transcriptionally active promoter conformations (6). Mechanisms of chromosome organization mediated by NAPs involve local perturbation of DNA architecture, such as DNA bending, wrapping, and bridging and protein oligomerization.
Most of the mechanistic studies of EspR have focused on its interaction with the critical ESX-1-related espACD locus (1, 4, 5, 7, 8), which is subject to complex control involving multiple regulators, including three NAP-like proteins (EspR, CRP, and Lsr2) (4, 9, 10), two members of two-component systems (PhoP and MprA) (11–13), and long-range interactions along the long (1,357 bp) espACD upstream region (7, 14). Although cis-regulatory elements of the espACD promoter have been extensively studied by Hunt et al. (7), the mechanism by which EspR upregulates espACD expression has not been elucidated. Critical to this process, however, is the ability of EspR to dimerize and form higher-order oligomers along several binding sites within the promoter region (4, 5, 8).
Structurally, EspR consists of two domains. The N-terminal DNA-binding domain (DBD) contains a typical helix-turn-helix (HTH) motif, whereas the C-terminal dimerization domain (CTDD) mediates the formation of the tight EspR dimer by providing a series of hydrophobic and polar interactions stabilizing the assembly of the two EspR subunits (5, 8). The C-terminally truncated form of EspR, EspRΔ10, indicates the importance of EspR dimerization for proper function. Indeed, removal of the last 10 C-terminal residues of EspR is sufficient for abrogation of its transcriptional activity (1). This phenotype is due to a decreased capacity to homodimerize as demonstrated by the crystal structure of EspRΔ10 and its biochemical characterization (5). DNase footprinting and gel shift experiments demonstrated that high-affinity binding occurred only when two regions of the espACD promoter, centered at positions −497 and −456 bp relative to the translational start site, were present on the same DNA fragment. Moreover, EspRΔ10 was unable to shift the same DNA fragment at the concentration tested, consistent with stable EspR dimerization being necessary for cooperative, high-affinity binding (5). Similar conclusions were drawn by Rosenberg et al., who showed cooperative, high-affinity binding of EspR proteins on a fragment of the espACD upstream region containing two EspR binding sites centered at −983 and −798 bp upstream of the espA start codon (8). In a previous study, we proposed a model for cooperative binding in which EspR binds DNA as a dimer-of-dimers, inserting one of two subunits of each dimer into two consecutive major grooves while at the same time forming a stable interface between the two EspR dimers (5). This conformation leaves one subunit from each EspR dimer free for further protein-protein or protein-DNA interactions, thereby promoting protein assembly into higher-order oligomers capable of bridging interactions between distant binding sites. As such, nucleoprotein complex formation would occur in a cooperative manner whereby binding of EspR dimer-of-dimers initiates at high-affinity sites and is extended by protein polymerization along DNA, possibly engaging nonspecific contacts with DNA.
In order to gain a more complete understanding of the molecular determinants governing EspR's mode of action, how it structures large DNA regions and influences transcription at specific promoters, we performed alanine mutagenesis of EspR residues identified in molecular dynamics (MD) simulations as key contributors to stability, flexibility, and oligomerization. The consequence for thermal stability and DNA-binding properties was characterized for each of these mutations. Then, selected mutants showing lower in vitro affinity for DNA than the wild-type (wt) protein were further characterized for oligomerization and structural effects on the 1.3-kb-long espACD promoter fragment in vitro, for homodimerization in live mycobacteria, and for transcriptional activity in M. tuberculosis cells.
MATERIALS AND METHODS
Molecular modeling.
MD simulations were performed with model systems of EspR and EspRΔ10 based on the EspR crystal structure (PDB 3QF3) and on a model system of EspR-DNA based on the dimer-of-dimers crystal structure (PDB 3QYX). The conformational ensembles generated by ∼70-ns-long MD simulations were investigated using standard trajectory analysis techniques, including principal component analysis, salt bridge and hydrogen bond persistence, and root mean square deviation (RMSD) (see Fig. S1 in the supplemental material). System setup and MD simulation protocols were computed as described previously (5). Energy decomposition of pairwise interactions at the protein-protein interface was performed using molecular mechanics/generalized Born surface area (MM/GBSA) (see Fig. S2) (15, 16). All calculations were performed using the parallelized version of MMPBSA.py implemented in the Amber 11 software program (17). An overall salt concentration of 0.15 M was used for general Born (GB) calculations; the program's default parameters were used in either case to calculate the nonpolar contribution. In all cases, 1 to 4 terms were included in the internal potential. Contributions of entropy to binding were not taken into account. Each MM/GBSA and energy decomposition calculation was performed with a set of 500 snapshots evenly taken from the last 30 ns of the equilibrated trajectory (one snapshot every 60 ps of MD simulation).
Bacterial strains and growth conditions.
M. tuberculosis H37Rv strains were grown in Middlebrook 7H9 broth (Difco) supplemented with 0.2% glycerol, 10% albumin-dextrose-catalase (ADC), and 0.05% Tween 80 or on solid 7H11 agar (Difco) supplemented with 0.5% glycerol and 10% oleic acid-albumin-dextrose-catalase (OADC). The M. tuberculosis H37RvΔespR strain was generated as described elsewhere (2). Mycobacterium smegmatis mc2155 strains were grown with the same media but with the difference that only 2% ADC or OADC enrichment was used. When necessary, kanamycin (25 μg/ml) and hygromycin B (50 μg/ml) were added to the Middlebrook medium. Escherichia coli TOP10 and BL21(DE3) cells were grown on Luria broth (LB) agar or broth supplemented with the appropriate antibiotic(s).
Site-directed mutagenesis.
Point mutants were generated using the QuikChange mutagenesis kit (Stratagene), following the manufacturer's protocol. Briefly, complementary oligonucleotides harboring the desired codon change to alanine were designed for use in PCRs using the plasmids pHis9gw-espR (for inducible protein overexpression in E. coli; see below), pMYespR (for inducible protein overexpression in M. tuberculosis; see below), or pUAB100-espR and pUAB200-espR (for protein overexpression in M. smegmatis in the two-hybrid system; see below) as template. Each mutation was confirmed by sequencing.
Protein expression and purification.
Wild-type and mutant pHis9gw-espR constructs used for expression of His9-tagged variants of EspR were transformed into E. coli BL21(DE3). Protein expression and purification were performed essentially as described previously (5). Briefly, cultures were grown in LB at 37°C till the exponential phase of growth (optical density at 600 nm [OD600], 0.5 to 0.8) prior to induction of protein overexpression with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) overnight at 16°C. Cell pellets were resuspended in lysis buffer (50 mM Tris [pH 8], 500 mM NaCl, 20 mM imidazole, 10% glycerol, 1% Tween 20, and 1 tablet of mini-protease inhibitor cocktail [Roche] per 20 ml of buffer), disrupted by sonication, and clarified by centrifugation. Purification of His9-tagged proteins was performed using Talon resin (Clontech Laboratories) affinity chromatography followed by size exclusion chromatography on a HiLoad 16/60 Superdex 75 column preequilibrated with 25 mM Tris (pH 8)–150 mM NaCl (Amersham Biosciences). Untagged proteins were separated from His9-tagged proteins by overnight thrombin-mediated cleavage of the His9 tag at 4°C followed by a second passage through the Talon affinity resin. The flowthrough was then concentrated to 1 to 2 mg ml−1, and protein purity was corroborated by SDS-PAGE. For SDS-PAGE analysis, the proteins were mixed with NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) and boiled for 10 min. Samples were resolved on NuPAGE 4 to 12% Bis-Tris gels (Invitrogen).
Thermal shift assay.
A real-time PCR machine (7900HT; Applied Biosystems) was used to heat the samples from 25°C to 95°C with a ramp rate of 1% and monitor the fluorescence of SYPRO orange (Invitrogen), used for tracking protein denaturation. Protein samples in 25 mM Tris buffer (pH 8)–150 mM NaCl in a reaction volume of 20 μl were incubated in fast 96-well plates (Applied Biosystems). Each reaction mixture contained 20 μM protein and 2.5× Sypro orange and was run at least in sextuplicate. Data analysis was performed as described by Niesen et al. (18). Melting temperatures (Tm) were calculated by fitting the sigmoidal melt curve to the Boltzmann equation using the GraphPad Prism software program (R2 values > 0.99). ΔTm was calculated using the Tm of wt EspR as a reference.
EMSA.
Electrophoretic mobility shift assays (EMSA) were performed to test the DNA-binding activity of EspR mutants essentially as described previously (5) using a 63-bp biotinylated double-stranded DNA (dsDNA) fragment (PespAI+II) covering one EspR binding region of the espACD promoter as a probe. Purified protein was incubated with 1 nM purified PCR product for 20 min at room temperature. Samples were electrophoresed onto a 6% (wt/vol) DNA retardation gel (Invitrogen), transferred onto a nylon membrane, and detected using the LightShift chemiluminescent EMSA kit (Pierce) according to the manufacturer's instructions.
AFM.
Atomic force microscopy (AFM) experiments were performed mainly as described before (5) but with some variations. The EspR-DNA binding reaction was performed by incubating 6 nM DNA with a 7.2 μM concentration of the EspR wt or mutant protein in a 10-μl reaction volume containing 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM dithiothreitol (DTT), 5% glycerol, and 1 mM EDTA for 10 min at room temperature. The mixture was then diluted five times in H2O containing MgCl2 (2 mM, final concentration) and deposited onto freshly cleaved mica. After 5 min of incubation, the mica surface was gently rinsed with 1 ml of Milli-Q water and subsequently gently blow-dried with filtered compressed air. AFM images were collected using a MultiMode scanning probe microscope (SPM) with a Nanoscope III controller (Veeco Instruments, Santa Barbara, CA, USA) operated in tapping mode in air. The AFM cantilevers used in air had a spring constant of 5 N m−1 (Veeco cantilevers; TAP150A) with resonance frequencies ranging between 120 and 160 kHz. All the recorded AFM images consist of 512 by 512 pixels with a scan frequency of ≤1 Hz. Images were simply flattened using the Nanoscope III software program (version 5.31; Veeco Instruments), magnified using Gwyddion software (version 2.22) (19), and no further image processing was carried out.
To calibrate the system, EspR protein variants and the linear espACD promoter fragment (PespACD) were separately imaged by AFM, and the corresponding heights were measured using the NanoScope Analysis software program (Version 1.20; Bruker, 2010) (see Fig. S3 in the supplemental material). In protein-DNA binding experiments, proteins were considered DNA bound when the height measured at any point of the DNA molecule was at least 0.5 nm higher than the average height measured for the free DNA.
Each protein-DNA binding experiment was performed at least in duplicate. AFM images were obtained at several separate locations across the mica surface to ensure a high degree of reproducibility and were used for statistical analysis of protein-DNA complexes. Only protein-DNA complexes that were completely visible in the AFM image were considered for statistical analysis.
Inducible overexpression of EspR variants in M. tuberculosis H37RvΔespR cells.
Construction of the pMYespR plasmid for pristinamycin IA-inducible overexpression of espR in M. tuberculosis cells was described previously (4). The wt and mutant pMYespR plasmids, as well as the pMY769 empty plasmid, were transformed into the M. tuberculosis H37RvΔespR strain by electroporation. Clones were selected on 7H11 agar plates containing 100 μg/ml spectinomycin, and plasmid integration was verified by colony PCR. The resulting M. tuberculosis H37RvΔespR::pMYespR strains were grown to mid-log phase (OD600 of 0.4 to 0.6), diluted to an OD600 of 0.1, and split into two 25-ml volumes. Protein overexpression was induced with 2 μg/ml pristinamycin IA in one of the two 25-ml cultures. Then, both induced and noninduced cultures were incubated at 37°C with shaking for an additional 3 days prior to cell harvesting. For whole-cell Western blot analysis, bacteria were pelleted, washed with phosphate-buffered saline (PBS), and resuspended in PBS containing mini-protease inhibitor cocktail (1 tablet per 10 ml of PBS; Roche). After cell breaking by sonication, cell debris was pelleted and the supernatant filter sterilized. Total protein quantification was performed using the Bradford reagent (Sigma), and 10 μg of total protein lysates were separated on NuPAGE 4 to 12% Bis-Tris gels. Specific proteins were visualized by immunoblotting using rat polyclonal anti-EspR, rabbit polyclonal anti-EspA, and mouse monoclonal anti-GroEL2 (IT-70) as primary antibodies (2, 4).
Mycobacterial two-hybrid resazurin microtiter assay.
The episomal vector pUAB100, containing a Saccharomyces cerevisiae leucine-zipper GCN4-encoding gene fused in frame with fragments 1 and 2 of murine dihydrofolate reductase (mDHFR) via a C-terminal Gly-rich flexible linker (GCN4F[1,2]), and the integrating vector pUAB200, containing the GCN4 gene fused in-frame with fragment 3 of mDHFR via a C-terminal Gly-rich flexible linker (GCN4F[3]) (20) were used as template for cloning and as vector control for interaction experiments. A PCR amplicon containing the M. tuberculosis espR gene sequence flanked by BamHI and ClaI restriction sites was digested and used to replace the gcn4 gene by ligation into BamHI/ClaI-digested pUAB100, thus generating the episomal vector pUAB100-espR. Similarly, a PCR amplicon harboring espR flanked by MfeI and ClaI restriction sites was digested and used to replace the gcn4 gene by ligation into MfeI/ClaI-linearized pUAB200, thus generating the integrating vector pUAB200-espR. Ligation reactions were transformed into TOP10 ultracompetent cells and selected on LB agar supplemented with 50 μg/ml kanamycin (KAN) (pUAB100-espR) or 200 μg/ml hygromycin B (HYG) (pUAB200-espR). Gene integration was verified by sequencing. The pUAB100-espR and pUAB200-espR vectors were used as the template for site-directed mutagenesis, and again mutations were verified by sequencing. Plasmid pairs were transformed into M. smegmatis by electroporation, and transformants were selected on 7H11 agar containing 25 μg/ml KAN and 50 μg/ml HYG. The trimethoprim (TRIM) susceptibilities of all M. smegmatis clones were determined using the resazurin microtiter assay (REMA) (21). Briefly, M. smegmatis clones containing interacting plasmids were cultured in 7H9 containing 25 μg/ml KAN and 50 μg/ml HYG to mid-log phase, freshly diluted to an OD600 of 0.0005, and grown in a 96-well plate in the presence of serial 2-fold dilutions of TRIM (from 500 μg/ml to 0.5 μg/ml). In parallel, serial 2-fold dilutions of moxifloxacin (MFX) (from 1 μg/ml to 0.001 μg/ml) were assessed for each strain to ensure strict TRIM-dependent mechanisms of resistance. One well with bacteria only (no drug) was used as a control for each strain tested. Following a 24-h incubation period at 37°C, 10 μl of 0.025% resazurin solution was added to each well. The plate was further incubated at 37°C for 3 to 6 h, after which the fluorescence intensity was recorded using an Infinite 200 Pro microplate reader (Tecan) in bottom-reading mode with excitation at 560 nm and emission at 590 nm. Fluorescence signal was expressed as the percentage of the fluorescence intensity recorded from the “no drug” well. The MIC was defined as the highest TRIM concentration that results in background fluorescence signal.
RESULTS
Molecular modeling and selection of candidate residues for mutagenesis.
In a previous study, we solved the crystal structures of wt EspR (PDB 3QF3), the C-terminal deletion mutant EspRΔ10 (PDB 3QWG), and an EspR-DNA complex (PDB 3QYX) (5). Utilizing full-length (wt) and C-terminally truncated (Δ10) models based on the EspR dimer structure (PDB 3QF3) in molecular dynamics (MD) simulations, we first sought to assess the relationship between dimer flexibility and C-terminal dimerization domain (CTDD) integrity and correlate the orientations of the CTDD with respect to the DNA-binding domain (DBD) (5). A detailed energetic investigation was performed in the present study to identify hot spots at protein-protein interfaces and to quantify the contributions of specific residues to dimer stability and to the dimer-of-dimers arrangement. Analysis of the interactions between the two protomers enabled us to identify residues involved in persistent polar interactions along MD trajectories. These included salt bridges (Arg101-Glu88 [persistence index of 98%], Arg106-Asp123 [40%], and Arg106-Asp132 [30%]) and hydrogen bonds (Arg70-Phe68 [60%], Arg126-Arg106 [25%], and Arg126-His108 [15%]). A binding free energy decomposition based on a molecular mechanics/generalized Born surface area (MM/GBSA) analysis of the trajectories enabled us to further pinpoint the residues that most contribute to the stability of the EspR dimer interface, namely, Arg70, Arg101, Arg106, Asp123, Arg126, and Asp132 (see Fig. S2 in the supplemental material).
Figure 1 summarizes the structural mapping of these polar residues, which are highly conserved among EspR orthologs of closely related mycobacterial strains (Fig. 1A) and located at the interface between the two subunits of the EspR dimer (Fig. 1B). At the CTDD, Arg106 of helix α7 forms salt bridges with side chains of Asp123 and Asp132 on the opposite subunit. In addition, the carbonyl oxygen of Arg106 interacts with the side chain of Arg126 via H-bonding interactions. Altogether, this network of interactions defines the position of one CTDD with regard to the other. Also belonging to helix α7, the guanidinium group of Arg101 forms cation-π interactions with Trp91 and salt bridges with Glu88 on the α6 helix of the opposite subunit (Fig. 1B). Removal of these interactions was expected to decouple the orientation of the CTDD with respect to the DNA-binding domain and thus result in overall increased dimer flexibility. The sole direct N-terminal intersubunit interactions are formed by Arg70 of each subunit adopting a planar stacking conformation and H bonding to the carbonyl oxygen of Phe68 of the opposite subunit. The R70A mutation was anticipated to affect the positioning of the two DNA-binding domains relative to each other. We previously showed that EspR could bind DNA as a dimer-of-dimers, with one subunit from each dimer being capable of binding two consecutive DNA major grooves while forming a stabilizing dimer-dimer interface. Contributing mainly to this arrangement is Lys72, which forms intermolecular salt bridges with Asp78 and Glu80 on the opposite dimer subunit (5) (Fig. 1C).
FIG 1.

Structural mapping of the amino acids replaced by Ala in EspR. (A) Sequence alignment of EspR orthologs in different mycobacteria. Amino acid positions and secondary structure referring to the M. tuberculosis H37Rv EspR crystal structure (PDB 3QF3) are specified on the top. Amino acids selected for site-directed mutagenesis are highlighted in yellow, and the corresponding point mutation is indicated with an arrow at the bottom. The N-terminal helix-turn-helix DNA-binding domain and the C-terminal dimerization domain (CTDD) containing most of residues involved in intersubunit interactions are indicated. (B) Mapping of the residues replaced by Ala (yellow) onto the EspR dimer. Interactions involving these residues in the wt structure (PDB 3QF3) are shown in magnified insets. (C) Mapping of the Ala-replaced Lys72 (yellow) onto the EspR dimer-of-dimers bound to DNA as previously obtained by X-ray crystallography (PDB 3QYX) and molecular modeling (5). Interactions involving Lys72 at the dimer-of-dimers interface (blue-cyan) are shown in the magnified inset.
To test the significance of these predictions, a series of alanine mutations was constructed by site-directed mutagenesis of the designated polar residues and the EspR mutants produced in E. coli as N-terminal His9-tagged fusion proteins. For comparison, wt EspR and the EspRΔ10 C-terminal deletion mutant were produced under identical conditions and utilized throughout this study. All point mutant constructs were expressed at levels equivalent to that of the wt protein and eluted as single peaks, with associated volumes corresponding to protein dimers in size exclusion chromatography (SEC) purification. Consistent with findings of our previous study, SEC purification of the EspRΔ10 deletion mutant resulted in two peaks, corresponding to both dimeric and monomeric fractions (data not shown). Coomassie-stained SDS-PAGE gels of purified wt and mutant EspR (10 μM) revealed monomers along with SDS-resistant dimers (Fig. 2A), probably stabilized by the tight noncovalent hydrophobic interactions of the CTDD shared by all EspR point mutants analyzed. While the dimer-monomer ratios could not be measured accurately due to the limitations of the SDS-PAGE and protein quantitation methods used, these results generally point out the importance of hydrophobic residues within the CTDD for dimerization.
FIG 2.

Stability of the EspR wild type and mutants. (A) His9-EspR wt and mutants were expressed and purified as described in Materials and Methods. The arrows indicate their migration bands corresponding to the molecular masses of the monomeric and SDS-resistant dimeric forms in SDS-PAGE. MW, molecular size ladder. (B) Melting profiles of purified proteins as monitored by measuring Sypro orange fluorescence as a function of temperature with a real-time quantitative PCR (qPCR) instrument. Tm values were calculated by fitting the melting curve data to the Boltzmann model. ΔTm represents the change in the melting temperature of EspR mutants from that of the wt protein. Indicated values are averages ± SD for a minimum of six measurements of protein unfolding. Asterisks mark significant differences (P < 0.001) from results for wt EspR. A.U., arbitrary units.
Thermal stability of EspR mutants.
To evaluate more sensitively how these point mutations affected EspR stability, we examined purified His9-tagged wt and mutant EspR using the thermal shift assay (TSA). TSA involves the use of a fluorogenic molecule (here SYPRO orange) that binds to exposed hydrophobic sites of a protein undergoing temperature-induced denaturation and emits fluorescence as a result. By measuring changes in fluorescence intensity as a function of temperature, the folded and unfolded states of the protein can be distinguished. The stability of the protein is expressed by its melting temperature (Tm), which coincides with the transition midpoint of the fluorescence curve and corresponds to the equilibrium temperature at which half of the proteins remain folded. Figure 2B summarizes the melting profile of wt and mutant proteins tested under identical buffer and protein concentration conditions. Unfolding of wt and mutant EspR occurred over a broad temperature range and in some cases was not strictly monophasic, with a peak shoulder appearing at early stages of melting (Fig. 2B), possibly reflecting a transition between CTDD dissociation and individual melting of the N-terminal globular domains. Although the lack of a strict two-state transition curve precluded accurate Tm calculation, Tm values were estimated by treating the melting curves as sigmoidal curves and fitting them to the Boltzmann equation (18). The resulting Tm values were used to compare the thermal stabilities of the mutant proteins with that of wt EspR (ΔTm) (Fig. 2B).
The Tm for wt His9-EspR was 47.8°C and was above the Tm obtained with all EspR variants analyzed, with the exception of the His9-R106A variant, which displayed a slightly higher Tm of 49.8°C. The His9-K72A protein displayed a melting curve characterized by high initial fluorescence levels (Fig. 2B), suggesting that partial unfolding had already occurred at room temperature. Although this behavior may account for the relatively high standard deviation (1.7°C) for the corresponding Tm, the average Tm for His9-K72A (Tm = 47.0°C) was comparable to that for wt His9-EspR. Replacement of residue Asp123, Arg126, or Asp132, each of which interacts with Arg106 at the extremity of the CTDD, with Ala had a moderate impact on protein stability, as illustrated by the corresponding Tm values, ranging between 46.3 and 47.1°C. Interestingly, His9-EspRΔ10, from which Arg126 and Asp132 are absent, exhibited a much lower Tm of 40.6°C than the individual Tm of His9-R126A and His9-D132A mutants (Fig. 2B). These data confirmed, consistent with the MD simulations, that significant destabilization of the CTDD requires a perturbation of the hydrophobic core and/or removal of multiple polar interactions, while disruption of a single polar residue at the C-terminal extremity of EspR has only a moderate effect on dimer stability. Two EspR point mutants, His9-R70A and His9-R101A, exhibited a substantial destabilization (ΔTm < −5°C) compared to wt His9-EspR. These observations suggest that Arg70 and Arg101 are key residues impacting the flexibility of the EspR dimer, which in turn affects protein stability. Taken together, these results demonstrate a significant correlation between EspR conformation, flexibility, and thermal stability.
DNA-binding affinity and higher-order protein-DNA complexes.
To determine the DNA-binding activities of the selected mutant proteins, purified His9-tagged variants of EspR were examined by electrophoretic mobility shift assays (EMSA) using as a probe a DNA fragment of the espACD promoter region (PespAI+II fragment) that was previously demonstrated to bind EspR specifically (5). His9-EspRΔ10, His9-R70A, His9-K72A, and His9-R101A mutants did not form protein-DNA complexes at the concentration tested, while all other EspR variants showed a binding activity similar to that of the wt protein (Fig. 3A).
FIG 3.

DNA-binding activities of EspR mutants. Electrophoretic mobility shift assay (EMSA) using a 1 nM concentration of a specific 63-bp dsDNA fragment of the espA promoter (5) and indicated amounts of purified wt or mutated His9-tagged EspR (A) or untagged versions of the EspR wt or variants (B) displaying no shift in panel A. “B” means bound protein (protein-DNA complex). The EspRΔ10, R70A, R101A, and K72A mutants are characterized by a smaller-mobility shift and a lower affinity for DNA than the wt protein.
To rule out the possibility that the His9 tag affected the ability of EspR variants to bind DNA, we proteolytically cleaved the His9 tag and used untagged protein in further analysis. Titration experiments demonstrated that the EspRΔ10, R70A, K72A, and R101A proteins had lower affinity for the espACD promoter fragment than wt EspR but were still able to shift the DNA fragment at high protein concentrations (Fig. 3B). Nevertheless, species shifted by these EspR variants displayed a fast and smeared migration, indicating that the stoichiometry and/or the stability of protein-DNA complexes differed from those induced by the wt protein (Fig. 3B). These results indicate that Arg70, Lys72, Arg101, and the last 10 C-terminal residues of EspR are important for formation of high-order protein-DNA complexes and optimal DNA-binding activity in vitro.
Visualization of EspR mutants in complex with DNA by atomic force microscopy.
In our previous study, we applied AFM to directly observe individual structures of EspR-DNA complexes (5). To obtain deeper insight into the molecular mechanisms of EspR action, we again used AFM to systematically compare protein-DNA complexes induced by wt EspR and the EspRΔ10, R70A, K72A, and R101A variants when mixed with the full 1.36-kb espACD upstream region (PespACD) at identical protein-DNA concentration ratios (Fig. 4). An unrelated nonspecific DNA fragment of similar size (1.26 kb) was also included as a negative control (5).
FIG 4.

AFM imaging of DNA-EspR (wild-type and mutant) complexes. (A) wt EspR (7.2 μM) incubated with PespACD (6 nM) shows large amounts of DNA-protein complexes with frequent DNA loop formation. In contrast, the R70A, K72A, and EspRΔ10 mutants exhibit significantly lower affinity for the PespACD DNA than wt EspR and show almost no DNA loop formation under identical conditions; the R101A mutant exhibits lower DNA affinity than wt EspR but still forms some DNA loops that are similar to those formed by wt EspR. The first two rows show zoomed images (305 by 305 nm) of selected DNA molecules from the corresponding (same column) representative large-field AFM images (3 by 3 μm) on the third row. (B) Quantification of PespACD binding and DNA loop formation mediated by EspR wt and mutants as measured by AFM. The number of DNA molecules analyzed for each protein-DNA binding reaction is indicated. The standard error bars indicate the variation in the number of “bound” versus “unbound” DNA molecules observed in the different AFM images for a given protein-DNA binding reaction. (C) Comparison of the percentages of looped DNA structures (± standard errors) induced by the different proteins tested.
In agreement with EMSA analysis, quantification of free and DNA-bound proteins (see Materials and Methods) showed that the EspRΔ10, R70A, K72A, and R101A mutants had lower affinity for the PespACD fragment than wt EspR. Of the protein-bound DNA molecules, “relaxed” and “looped” structures were distinguished (Fig. 4B and C). Typical complexes as observed in these experiments are shown in Fig. 4. Consistent with our previous AFM imaging experiments, looped DNA structures were frequently (27%) observed in protein-DNA complexes containing wt EspR. In contrast, the PespACD-R70A and PespACD-K72A complexes were essentially incapable of forming DNA loops, with most of the DNA-bound protein gathering at isolated regions across the PespACD fragment while the overall DNA architecture appeared relaxed (Fig. 4). Similarly, the C-terminal deletion mutant EspRΔ10 also caused globular DNA aggregation and little structuring effect on PespACD, while the R101A mutant, although showing lower DNA-binding affinity than wt EspR, retained the ability to form some nucleoprotein loops (22%). Higher-order protein organization leading to DNA loop stabilization was confirmed to be sequence specific, since wt EspR and its variants displayed low affinity for the control DNA fragment (DNA-bound proteins at <40%), which appeared mainly relaxed when mixed with each of the EspR variants tested for (see Fig. S4 in the supplemental material).
Activation of EspA expression.
The M. tuberculosis H37RvΔespR strain fails to produce EspA and consequently shows functional defects of ESX-1 secretion (1). To correlate the DNA-binding and DNA-structuring deficiencies of the EspR variants analyzed above with their transcriptional activities in M. tuberculosis, we measured EspA expression in the M. tuberculosis H37RvΔespR strain complemented with a plasmid overexpressing either espR+ or espR mutants under the control of a pristinamycin IA-inducible promoter (4). Immunoblot analysis showed that introduction of espR+ but not espRΔ10 in M. tuberculosis H37RvΔespR restored EspA expression (Fig. 5). This defect of EspA expression was accompanied by low detection of EspRΔ10, probably due to decreased protein stability and/or reduced affinity for the polyclonal anti-EspR antibody. Similarly, no EspA was detected in the cell lysate of M. tuberculosis H37RvΔespR strains complemented with the R70A or K72A protein variant. The observed decrease in protein concentration for both mutants confirmed that these two point mutations affected EspR folding and/or stability. However, the levels of both EspR variants were still comparable to endogenous EspR levels detected in H37Rv (4), suggesting that the R70A and K72A mutations interfere with espACD expression beyond the apparent deviation in protein concentration. In contrast, although the R101A mutant showed DNA-binding deficiency in vitro, its overexpression in M. tuberculosis H37RvΔespR restored EspA to levels equivalent to those induced by wt EspR. A possible explanation for this phenotype is that overexpression of this particular EspR mutant is sufficient to compensate for the reduced affinity to DNA, as suggested in a study of Ler, a member of the H-NS family of NAPs in enteropathogenic Escherichia coli (22).
FIG 5.
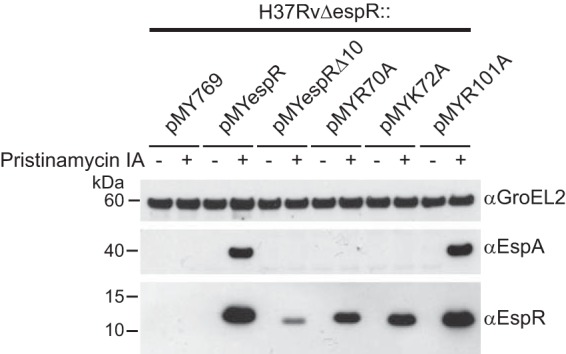
Activation of EspA transcription mediated by the EspR wild type and mutants. Protein expression analysis of the M. tuberculosis H37RvΔespR strain complemented with an empty vector (pMY769) or a vector carrying an espR variant (pMYespR, pMYespRΔ10, pMYR70A, pMYK72A, and pMYR101A) under the control of a pristinamycin IA-inducible promoter (4, 27). Equal amounts (10 μg) of total protein from cell lysates were analyzed. GroEL2 was used as a loading control.
Toward a high-throughput assay to assess homodimerization in live mycobacterial cells.
We utilized a two-hybrid system (also called a mycobacterial protein fragment complementation assay [M-PFC]) developed by Singh et al. (20) to evaluate the homodimerization capacity of the EspR variants that showed low binding affinity for DNA and to establish a high-throughput assay to screen for compounds that inhibit homodimerization. This system has been used previously to demonstrate M. tuberculosis protein-protein interactions (20) and has the advantage of functioning in a mycobacterial intracellular environment using the fast-growing M. smegmatis as a host. This assay is based on a murine dihydrofolate reductase (mDHFR), a 21-kDa monomeric enzyme that catalyzes the reduction of dihydrofolate to tetrahydrofolate for the biosynthesis of nucleotides and certain amino acids. Enzyme activity is reconstituted only when two proteins fused to complementary fragments of mDHFR, F[1,2] and F[3], are interacting. Protein-protein interaction can thus be detected by bacterial growth on medium containing trimethoprim (TRIM), an antibiotic that selectively targets bacterial DHFR.
To demonstrate that EspR homodimerization can be detected with this system, we coexpressed espR cloned as C-terminal fusions to fragments F[1,2] mDHFR and F[3] mDHFR in M. smegmatis (here designated EspRF[1,2] and EspRF[3]). In addition, the previously described (20) yeast leucine-zipper GCN4-mediated mDHFR reconstitution was assessed as a positive control. Strains coexpressing EspRF[1,2]/EspRF[3] and GCN4F[1,2]/GCN4F[3] combinations grew on 7H11 HYG/KAN/TRIM plates (data not shown), indicating that association of the two fused EspR subunits reconstituted functional mDHFR activity to levels comparable to those obtained with the GCN4F[1,2]/GCN4F[3] pair. In contrast, M. smegmatis coexpressing either EspRF[1,2]/GCN4F[3] or GCN4F[1,2]/EspRF[3] combinations was unable to grow on HYG/KAN/TRIM-containing medium, thus confirming the specificity of the assay.
In order to develop a sensitive and quantitative assay of EspR homodimerization, we adapted the EspR two-hybrid system to a 96-well plate resazurin microtiter assay (REMA). Briefly, M. smegmatis carrying the plasmid pairs described above was cultured in 96-well plates in the presence of serial TRIM dilutions. The strength of protein-protein association was evaluated by measuring bacterial viability using REMA (Fig. 6A). To ensure that bacterial survival was TRIM dependent, strain susceptibility to moxifloxacin (MFX), a DNA gyrase inhibitor, was also evaluated by REMA (Fig. 6).
FIG 6.

Determination of homodimerization using a mycobacterial two-hybrid assay. Resazurin microtiter assay (REMA) with M. smegmatis expressing pairs of proteins fused to fragments 1 and 2 (F[1,2]) or fragment 3 (F[3]) of mDHFR. (A) Equal amounts of the three M. smegmatis strains were seeded in two rows containing serial 2-fold dilutions of trimethoprim (TRIM) (from 500 μg/ml to 0.5 μg/ml) or moxifloxacin (MFX) (from 1 μg/ml to 0.001 μg/ml). The last column on the right contains untreated cells. (B) Percentage of resazurin turnover measured in the plate shown in panel A as a function of drug concentration. Filled lozenges, GCN4F[1,2]/GCN4F[3] combination; empty circles, EspRF[1,2]/GCN4F[3] combination; filled circles, EspRF[1,2]/EspRF[3] combination. Low TRIM susceptibility correlates with target protein-protein interaction (dimerization) as a result of proper mDHFR reconstitution. Note that all M. smegmatis strains have similar susceptibilities to MFX, confirming that the system is TRIM dependent.
From Fig. 6, it can be clearly seen that EspRF[1,2]/EspRF[3] and GCN4F[1,2]/GCN4F[3] interacting partners conferred TRIM resistance. In contrast, the EspRF[1,2]/GCN4F[3] combination showed an approximately 100-fold-lower MIC for TRIM, confirming that mDHFR reconstitution required specific interaction of target proteins. We then introduced the two Ala substitutions at the dimer (R70A and R101A) or dimer-of-dimers (K72A) interface that caused decreased DNA-binding affinity into the EspRF[1,2] and EspRF[3] expression plasmids by site-directed mutagenesis and assayed their ability to homodimerize by determining the TRIM MIC by REMA (Table 1). In parallel, EspRΔ10 dimerization was also assayed using the same system for comparison. As expected, deletion of the last 10 C-terminal residues disrupted EspR dimerization in vivo, as evidenced by the TRIM MIC (= 8 to 16 μg/ml) for the strain carrying the EspRΔ10F[1,2]/EspRΔ10F[3] combination, analogous to the strain carrying the EspRF[1,2]/GCN4F[3] negative-control combination. In contrast, the dimerization capacity of the R70A protein (TRIM MIC = 63 μg/ml) was intermediate between those of wt and the negative control, while the R101A and K72A mutants allowed for levels of dimerization similar to that of wt EspR (TRIM MIC = 250 to 500 μg/ml). Thus, it appears that the Arg70 residue but not Arg101 and Lys72 contributes significantly to the stability of the EspR dimer in vivo. Moreover, these results confirm that the lack of transcriptional activity of the K72A mutant is not due to a major defect in dimerization.
TABLE 1.
Dimerization capacity of pairs of EspR mutants as measured by mycobacterial two-hybrid REMA
| Protein partners | Trimethoprim MIC (μg/ml) |
|---|---|
| EspRF[1,2]/EspRF[3] | 250–500 |
| EspRF[1,2]/GCN4F[3] | 8 |
| EspRΔ10F[1,2]/EspRΔ10F[3] | 8–16 |
| R70AF[1,2]/R70AF[3] | 63 |
| R101AF[1,2]/R101AF[3] | 250–500 |
| K72AF[1,2]/K72AF[3] | 250–500 |
DISCUSSION
EspR is a key global regulator influencing M. tuberculosis pathogenesis, in part due to its role as an activator of the major virulence determinant, the ESX-1 secretion system, and thereby represents an attractive drug target (23). Previous studies have shown a direct relationship between transcriptional activity of EspR and conserved protein residues of the N-terminal DNA-binding domain, as well as C-terminal residues involved in the dimerization domain (1, 5, 8). While the link between the DNA-binding domain and transcriptional activity appears obvious, the direct relationship between EspR-EspR intersubunit interactions and transcriptional activity is elusive, and an understanding of the subjacent molecular mechanisms could provide strategies for allosteric perturbation.
In this study, we performed MD simulations to identify hot spot residues at the dimer and dimer-of-dimers interfaces and then evaluated the impact of these residues on the biological and biophysical properties of EspR by generating and characterizing seven Ala-substituted point mutants. We consistently observed that small changes promoted by replacement of polar residues with Ala at the dimer interface had little effect on dimerization in vitro. In spite of the cooperative role of Arg106, Asp123, Arg126, and Asp132 in forming a network of charged and polar interactions on the external surface of the C-terminal four-helical bundle (helices α7 and α8 of each protomer at the CTDD), none of these residues resulted in deficient protein dimerization when they were replaced with Ala, possibly due to the compensatory effect of the remaining polar and hydrophobic interactions that stabilize the CTDD.
In contrast, replacement of Arg70, Lys72, and Arg101 with Ala resulted in proteins with altered DNA-binding ability. However, like the C-terminally truncated protein EspRΔ10, the R70A, K72A, and R101A EspR variants were still able to bind the espACD promoter fragment in vitro, albeit at lower affinity, and to form complexes with DNA of stoichiometries different from that for the wt protein. This suggested that defects in the oligomerization process may cause this decreased affinity and that modulation of DNA binding could be mediated through higher-order organization.
Dimerization seems necessary but not sufficient for optimal DNA-binding activity. Thus, like the C-terminal deletion in EspRΔ10, mutation of Arg70 to Ala perturbed EspR's dimerization capacity in a mycobacterial two-hybrid system, which in turn affected protein stability, DNA-binding and DNA-structuring activity in vitro, as well as transcriptional activity in M. tuberculosis. In contrast, a similar outcome for protein stability and DNA-binding affinity did not correlate with dimerization defects or lack of transcriptional activity of the R101A protein, and the effects on DNA architecture related to this protein were close to those induced by wt EspR. Arg101 forms π-stacking interactions and salt bridges with Trp91 and Glu88, respectively, on the opposite protomer. This residue lies in the middle of helix α7, which contains six additional residues (four hydrophobic and two polar) involved in interactions holding opposing helices α7 and α8 of each EspR protomer together (5). Arg101, however, points alone toward the long helix α6, connecting the N-terminal and C-terminal domains of each subunit, suggesting an effect on dimer conformation preponderant over dimer stability. Upon elimination of Arg101, the hinge between the CTDD and the DBD is further loosened and the overall dimer mobility increased, which possibly explains the reduced thermal stability and the impaired DNA binding capacity of the R101A mutant. In contrast, Arg70 residues, which interact with each other in a planar stacking conformation and form H bonds to the carbonyl oxygen of Phe68 of the opposite subunit, appear to be more directly involved in dimerization because they represent a unique direct contact point between adjacent DBD domains. Moreover, residues involved in stabilizing the dimer-of-dimers interface lie at boundaries of helix α4, and the localization of Arg70 at the C-terminal end of helix α4 may also indicate a structural interplay; a protomer with R70A substitution orientated with the recognition helix α3 bound to the major groove of DNA may undergo a reorientation of the DBD that might not be compatible to interact with another protomer bound to the adjacent major groove.
We found that mutants with the K72A substitution, which was expected to affect the formation of the EspR dimer-of-dimers on DNA, exhibited DNA-binding and DNA-structuring defects in vitro and failed to functionally restore EspA expression in M. tuberculosis. Protein instability and deficient dimerization do not seem to cause these effects, since the K72A protein was readily detected in cell lysates of M. tuberculosis H37RvΔespR complemented with the K72A protein and the protein efficiently dimerized in M. smegmatis (as monitored with the two-hybrid system). This suggests that in vivo defects of the K72A mutant are due to the destabilization of protein-protein interactions mediated by the Lys72 residue between EspR dimers. However, we cannot exclude the possibility that this mutation also affects interactions with other proteins.
EspR has been categorized as a NAP (4). Proteins of this family exhibit chromosome structural capacities usually involving high-order protein oligomerization and a large number of subsequent cooperative protein-DNA contacts. Alteration of higher-order structures at promoters to fine-tune expression outcomes have been suggested for NAPs such as H-NS (24). Here we report a direct correlation between defects in structuring the espACD promoter in vitro and a lack of transcriptional activation of EspA expression in vivo. Importantly, an M. tuberculosis H37RvΔespR strain overexpressing the R70A or K72A EspR variant expresses no EspA, and visualization of complexes of the respective purified proteins with the espACD promoter fragment shows defects in DNA loop organization. The same conclusion was also true for EspRΔ10. From these results, a model of EspR-mediated promoter organization involves EspR first dimerizing and then nucleating at one or more high-affinity sites of the espACD promoter before further cooperatively polymerizing to link more distant DNA sequences to mediate transcriptionally active DNA loop formation. Blockage of any of these steps would result in EspR dysfunction. At AFM resolution, it is unclear how EspR mediates the interaction between the adjacent DNA duplexes, and the specific oligomeric state cannot be deduced. In one possible model, all four DNA-binding domains of the EspR dimer-of-dimers engage with DNA simultaneously. Extension of this conformation to a series of adjacent EspR dimer-of-dimers would create a protein multimer onto which two DNA helices could be intertwined and then connected by a loop.
In light of the present results and of the observation that EspR levels vary during the bacterial cell cycle (4), it is tempting to speculate that modulation of the EspR concentration constitutes a regulatory mechanism to alter EspR higher-order organization and therefore EspR activity. Importantly, overexpression of the R101A EspR variant in the M. tuberculosis H37RvΔespR strain induced expression of EspA to levels similar to those observed upon overexpression of wt EspR, suggesting that the DNA binding defects of the R101 protein could be compensated by high protein concentration. Moreover, AFM data showed that a high concentration of R101A protein could structure the PespACD fragment similarly to what is observed for wt EspR. Another possible scenario is that additional events, such as ligand or coregulatory protein binding or posttranslational modification affecting the higher-order organization of EspR, also play a role in modulating EspR activity. An example of such a regulatory mechanism is the formation of the Hha/H-NS complex, which results in alteration of H-NS's oligomerization, required for targeted gene silencing in Salmonella (25).
The bacterial two-hybrid system presented here provides us with an assay that can be used to screen compound or peptide libraries against EspR homodimerization directly in a mycobacterial environment. The wide TRIM susceptibility window between the strains encoding dimerization-proficient and dimerization-impaired EspR proteins together with the specific correlations between EspR homodimerization, TRIM concentration, and bacterial survival allow for a sensitive and quantitative assay that can be performed in a high-throughput manner (20, 26). Screening and characterizing potential antivirulence compounds targeting EspR will be the focus of further research.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stefanie Boy-Röttger and Philippe Busso for technical assistance and Adrie Steyn for providing the M-PFC system.
The research leading to these results has received funding from the European Community's Seventh Framework Programme ([FP7/2007-2013]) under grant agreement no. 201762 and the Swiss National Science Foundation under grant no. 31003A-125061.
Footnotes
Published ahead of print 14 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00039-14.
REFERENCES
- 1.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. 2008. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454:717–721. 10.1038/nature07219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JM, Boy-Rottger S, Dhar N, Sweeney N, Buxton RS, Pojer F, Rosenkrands I, Cole ST. 2012. EspD is critical for the virulence-mediating ESX-1 secretion system in Mycobacterium tuberculosis. J. Bacteriol. 194:884–893. 10.1128/JB.06417-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 102:10676–10681. 10.1073/pnas.0504922102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasco B, Chen JM, Hartkoorn R, Sala C, Uplekar S, Rougemont J, Pojer F, Cole ST. 2012. Virulence regulator EspR of Mycobacterium tuberculosis is a nucleoid-associated protein. PLoS Pathog. 8:e1002621. 10.1371/journal.ppat.1002621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasco B, Stenta M, Alonso-Sarduy L, Dietler G, Peraro MD, Cole ST, Pojer F. 2011. Atypical DNA recognition mechanism used by the EspR virulence regulator of Mycobacterium tuberculosis. Mol. Microbiol. 82:251–264. 10.1111/j.1365-2958.2011.07813.x [DOI] [PubMed] [Google Scholar]
- 6.Dillon SC, Dorman CJ. 2010. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8:185–195. 10.1038/nrmicro2261 [DOI] [PubMed] [Google Scholar]
- 7.Hunt DM, Sweeney NP, Mori L, Whalan RH, Comas I, Norman L, Cortes T, Arnvig KB, Davis EO, Stapleton MR, Green J, Buxton RS. 2012. Long-range transcriptional control of an operon necessary for virulence-critical ESX-1 secretion in Mycobacterium tuberculosis. J. Bacteriol. 194:2307–2320. 10.1128/JB.00142-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg OS, Dovey C, Tempesta M, Robbins RA, Finer-Moore JS, Stroud RM, Cox JS. 2011. EspR, a key regulator of Mycobacterium tuberculosis virulence, adopts a unique dimeric structure among helix-turn-helix proteins. Proc. Natl. Acad. Sci. U. S. A. 108:13450–13455. 10.1073/pnas.1110242108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon BR, Li Y, Wang L, Sintsova A, van Bakel H, Tian S, Navarre WW, Xia B, Liu J. 2010. Lsr2 is a nucleoid-associated protein that targets AT-rich sequences and virulence genes in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 107:5154–5159. 10.1073/pnas.0913551107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickman L, Scott C, Hunt DM, Hutchinson T, Menendez MC, Whalan R, Hinds J, Colston MJ, Green J, Buxton RS. 2005. A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol. Microbiol. 56:1274–1286. 10.1111/j.1365-2958.2005.04609.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalo-Asensio J, Mostowy S, Harders-Westerveen J, Huygen K, Hernandez-Pando R, Thole J, Behr M, Gicquel B, Martin C. 2008. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One 3:e3496. 10.1371/journal.pone.0003496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 60:312–330. 10.1111/j.1365-2958.2006.05102.x [DOI] [PubMed] [Google Scholar]
- 13.Pang X, Samten B, Cao G, Wang X, Tvinnereim AR, Chen XL, Howard ST. 2013. MprAB regulates the espA operon in Mycobacterium tuberculosis and modulates ESX-1 function and host cytokine response. J. Bacteriol. 195:66–75. 10.1128/JB.01067-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxton RS, Green J, Hunt DM, Kahramanoglou C, Stapleton MR, Sweeney NP. 2012. Long range transcriptional control of virulence critical genes in Mycobacterium tuberculosis by nucleoid-associated proteins? Virulence 3:408–410. 10.4161/viru.20918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. 10.1021/ar000033j [DOI] [PubMed] [Google Scholar]
- 16.Noskov SY, Lim C. 2001. Free energy decomposition of protein-protein interactions. Biophys. J. 81:737–750. 10.1016/S0006-3495(01)75738-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. 2005. The Amber biomolecular simulation programs. J. Comput. Chem. 26:1668–1688. 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niesen FH, Berglund H, Vedadi M. 2007. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2:2212–2221. 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 19.Nečas D, Klapetek P. 2012. Gwyddion: an open-source software for SPM data analysis. Cent. Eur. J. Phys. 10:181–188. 10.2478/s11534-011-0096-2 [DOI] [Google Scholar]
- 20.Singh A, Mai D, Kumar A, Steyn AJ. 2006. Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc. Natl. Acad. Sci. U. S. A. 103:11346–11351. 10.1073/pnas.0602817103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palomino JC, Martin A, Camacho M, Guerra H, Swings J, Portaels F. 2002. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 46:2720–2722. 10.1128/AAC.46.8.2720-2722.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yerushalmi G, Nadler C, Berdichevski T, Rosenshine I. 2008. Mutational analysis of the locus of enterocyte effacement-encoded regulator (Ler) of enteropathogenic Escherichia coli. J. Bacteriol. 190:7808–7818. 10.1128/JB.00663-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JM, Pojer F, Blasco B, Cole ST. 2010. Towards anti-virulence drugs targeting ESX-1 mediated pathogenesis of Mycobacterium tuberculosis. Drug Discov. Today Dis. Mech. 7:e25–e31. 10.1016/j.ddmec.2010.09.002 [DOI] [Google Scholar]
- 24.Ali SS, Beckett E, Bae SJ, Navarre WW. 2011. The 5.5 protein of phage T7 inhibits H-NS through interactions with the central oligomerization domain. J. Bacteriol. 193:4881–4892. 10.1128/JB.05198-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali SS, Whitney JC, Stevenson J, Robinson H, Howell PL, Navarre WW. 2013. Structural insights into the regulation of foreign genes in Salmonella by the Hha/H-NS complex. J. Biol. Chem. 288:13356–13369. 10.1074/jbc.M113.455378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mai D, Jones J, Rodgers JW, Hartman JL, IV, Kutsch O, Steyn AJ. 2011. A screen to identify small molecule inhibitors of protein-protein interactions in mycobacteria. Assay Drug Dev. Technol. 9:299–310. 10.1089/adt.2010.0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forti F, Crosta A, Ghisotti D. 2009. Pristinamycin-inducible gene regulation in mycobacteria. J. Biotechnol. 140:270–277. 10.1016/j.jbiotec.2009.02.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.