

Abstract
Mycobacterium tuberculosis has a clonal population structure, and the Latin American-Mediterranean (LAM) family is one of the largest and most widespread within this species, showing evidence for remarkable pathobiology and a confusing phylogeny. Here, we applied robust phylogenetic markers to study the evolution of the LAM family and its major sublineages circulating in Russia and neighboring countries. A total of 250 M. tuberculosis isolates were confirmed to belong to the LAM family based on the analysis of the LAM-specific single-nucleotide polymorphisms (SNPs) in the Rv3062 and Rv0129c genes. At this stage, the family status was rectified for 121 isolates misleadingly assigned by CRISPR spoligotyping to non-LAM families (T1- or T5-RUS1). Consequently, the reestimated LAM prevalence rate increased 2-fold in Russia and Kazakhstan and 4-fold in Belarus. The majority (91.8 to 98.7%) of the LAM isolates from all three countries belonged to the LAM-RUS sublineage. In contrast, the Ibero-American LAM RD-Rio sublineage was identified in only 7 Russian isolates. Taken together, our findings and further analyses suggest a monophyletic origin of LAM-RUS: at a historically distant time, in Russia, in a small founding bacterial/human population. Its dissemination pattern and high prevalence rate in Northern Eurasia may indicate a long-term coexistence of the LAM-RUS sublineage and local human populations hypothetically leading to coadaptation and reduced pathogenicity of the relatively more ancient clones, such as spoligotype international type 254 (SIT254), compared to the more recent SIT252 and SIT266 clones. In contrast, rare LAM RD-Rio isolates were likely brought to Russia through occasional human contact. The spread of RD-Rio strains is not as global as commonly claimed and is determined largely by human migration flows (rather than by pathobiological properties of these strains). Consequently, a host population factor appears to play a major role in shaping the in situ dissemination pattern of the imported strains in an autochthonous population.
INTRODUCTION
Nothing in biology makes sense except in the light of evolution.
—Theodosius Dobzhansky (32)
Mycobacterium tuberculosis is doubtless an important pathogen from a human health viewpoint. Per se, it is also an interesting biological species. Under the umbrella name M. tuberculosis complex, it encompasses different ecotypes, its niches expanding well beyond that of the human host. It has long been a common yet true saying that M. tuberculosis is marked by a high level of genetic homogeneity (1). Over the past decade, an expansion of knowledge, greatly aided by whole-genome sequencing, has demonstrated a higher level of genetic variation than initially thought. On the other hand, notwithstanding the excellent added value of new-generation technologies, the previous use of the much simpler molecular typing methods more than 10 years ago delineated the phylogenetic structure of M. tuberculosis and identified most of its major families/genotypes (2). A Latin American-Mediterranean (LAM) family of M. tuberculosis was first suggested based on the phylogenetic analysis of a large spoligotyping data set, and its name reflects the strains' origins (2). The LAM family is part of the large and heterogeneous Euro-American lineage (one of six lineages of human M. tuberculosis [3]).
Within the LAM family, the best-known and most “popular” sublineage is RD-Rio, identified in Brazil several years ago based on deletion analysis and marked with two distant and large genomic deletions, RD174 (4) and RD-Rio (5). This LAM sublineage was a major focus of studies carried out in different settings across the Americas, Europe, and Africa (6, 7), and its global spread has been suggested (6); we discuss this issue of “global spread” below.
In Russia and Eastern Europe/Northern Eurasia as a whole, the LAM family has long remained in the shadow of the extensively studied Beijing genotype. Studies carried out in the central Russian region of Tula found a similarity of the IS6110-restriction fragment length polymorphism (RFLP) profiles of Russian LAM isolates, and it was suggested that these isolates be named the LAM-RUS family (8, 9). A specific IS6110 insertion in the plcA gene was identified as a characteristic feature of the LAM-RUS family (9). However, no further studies of this marker were carried out in either other Russian regions or neighboring countries. As a whole, no studies on the phylogeny of the LAM family and its sublineages were carried out in Russia and other countries of the former Soviet Union (FSU) to date.
A correct designation of lineages and sublineages of M. tuberculosis is important due to the known interlineage variation of pathobiological properties. CRISPR-based spoligotyping is still largely used to define LAM isolates. However, the inherent limitation of this method is the single target locus and the nonindependent and largely unknown mode of evolution of its information units (repeats/spacers). Consequently, an interpretation of large deleted blocks of spacers observed in spoligotyping profiles bears a huge amount of uncertainty, leading to an unresolvable ambiguity in the family assignment of such isolates. In this case, the use of phylogenetically robust markers, such as unique-event polymorphisms, presents a biologically meaningful and, hence, reliable approach.
Here, we applied robust evolutionary markers to estimate the true prevalence rate of the LAM family and its major sublineages, LAM-RUS and RD-Rio, in Russia and neighboring countries, to analyze its population structure, and, ultimately, to propose its large-scale evolutionary framework.
MATERIALS AND METHODS
Study samples.
The spoligotype database of the laboratory of molecular microbiology at St. Petersburg Pasteur Institute was used to select the isolates for this study. All isolates with the LAM prototype spoligotype international type 42 (SIT42) and isolates with spoligotyping profiles that could be derived from SIT42 were included. They represented Russia (n = 153, including 63 from St. Petersburg, 30/110 from Pskov [10], 8/28 from Tuva [alternatively spelled Tyva] [11], 32/102 from Karelia, 12/90 from Kaliningrad [12], 2/12 from Vologda, 2/12 from Murmansk, and 4/22 from Archangel), Belarus (88 of 194 isolates, all drug resistant [13]), Kazakhstan (35 of 158 isolates), Bulgaria (8 of 133 isolates [14]), and Vietnam (6 of 117 isolates [15]).
Except for isolates from St. Petersburg received in 2010 to 2013 from the Research Institute of Phthisiopulmonology and overdominated by drug-resistant and Beijing genotype isolates, all other collections were convenience or snapshot samples or were received for referral purposes.
Genotyping.
Spoligotyping was performed according to standard protocols (16).
The LAM-specific single-nucleotide polymorphisms (SNPs) 309G>A in Rv0129c (6) and 1212C>G in Rv3062 (17) were studied by using PCR-RFLP assays with MboII and TaqI endonucleases, respectively. The particular LAM-specific alleles create additional sites for these specific restriction endonucleases. A 233-bp fragment of Rv3062 was amplified with primers 5′-CAAGCCGGTGCACACACTCGACT and 5′-GCCTAAGTTGGACGACGTAGCCG, followed by digestion with TaqI; the wild-type allele results in one major 214-bp digest, while the LAM isolate is defined by two major digests of 158 bp and 56 bp. A 189-bp fragment of Rv0129c was amplified with primers 5′-CAGTTCCAGGGCGGCGGACC and 5′-GTTGCTCTGCGAGGGCTGATACCA, followed by MboII digestion; the wild-type allele results in one major 189-bp fragment, while the LAM isolate is defined by two fragments of 108 bp and 81 bp.
An IS6110 insertion in the plcA gene (position 2630571 in the strain H37Rv genome) specific for the LAM-RUS subfamily was detected by using multiplex PCR, as described previously by Dubiley et al. (9).
An RD115 deletion (4) was detected by using multiplex PCR with forward primer GGATCCGTCCGGCAATATCG, forward inner primer GGCGAAGGTGGCGATGCTGG, and reverse primer CCGCCGCCTGTTCCCAGGAT. A 149-bp PCR product is amplified in the case of an RD115 deletion, and a 287-bp product is amplified in the case of an intact region.
An RD174 deletion (4) was detected by using multiplex PCR with forward primer GCTCGCGGTCCTCGTTGTCC, reverse inner primer TCACGCGCAATCTGCGACACCG, and reverse primer CGCAGCAAACCCACGCCGAAC. A 446-bp PCR product is amplified in the case of an RD115 deletion, and a 244-bp product is amplified in the case of an intact region.
An RD-Rio deletion was detected by using multiplex PCR as described previously by Gibson et al. (6).
Sequencing.
Direct sequencing of the PCR products was performed by using BigDye Terminator 3.1 chemistry (Applied Biosystems Inc.) with the respective primers and run on a 3730xl DNA analyzer (ABI).
Data analysis.
The spoligoprofiles were entered into Excel spreadsheets and compared with data in the SITVIT_WEB database (18) (http://www.pasteur-guadeloupe.fr:8081/SITVIT_ONLINE).
The SpolTools program (19, 20) (available at http://www.emi.unsw.edu.au/spolTools) was used to build the forest graph of the spoligotyping binary profiles.
A 2-by-2 χ2 test was used to detect any significant difference between the two groups. Yates-corrected χ2 and P values were calculated by using EpiCalc 2000 version 1.02 software (21).
RESULTS
Spoligotyping data available in our laboratory database (see “Study samples,” above) were screened for the major LAM prototype spoligoprofile (SIT42) and its possible derived profiles. The initial study collection included 290 “potential” LAM isolates from Russia (n = 153), Belarus (n = 88), Kazakhstan (n = 35), Bulgaria (n = 8), and Vietnam (n = 6). All spoligotyping profiles were compared to data in the SITVIT_WEB database (18), and a family was assigned based on the decision rules implemented in this database.
The isolates with different spoligoprofiles were further tested for the presence of LAM-specific SNPs in Rv3062 or Rv0129c. Based on this analysis, some isolates were identified as non-LAM isolates (n = 26) or as having a mix of LAM and non-LAM genotypes (n = 14) and were excluded from further study. The remaining 250 isolates from Russia (n = 135), Belarus (n = 81), Kazakhstan (n = 30), Bulgaria (n = 3), and Vietnam (n = 1), confirmed to be LAM isolates, were further tested for within-LAM-specific markers.
A comparison of the spoligotyping and LAM SNP analysis results (Table 1) permitted us to clarify a family status and reassign some isolates (in most instances, those of the T1- or T5-RUS1 family) to the LAM family. In all, spoligotyping correctly assigned 119 isolates to the LAM family and misleadingly assigned 121 isolates to non-LAM families (Table 1).
TABLE 1.
Genotypes of the studied M. tuberculosis LAM isolates from different Russian regions and other countries
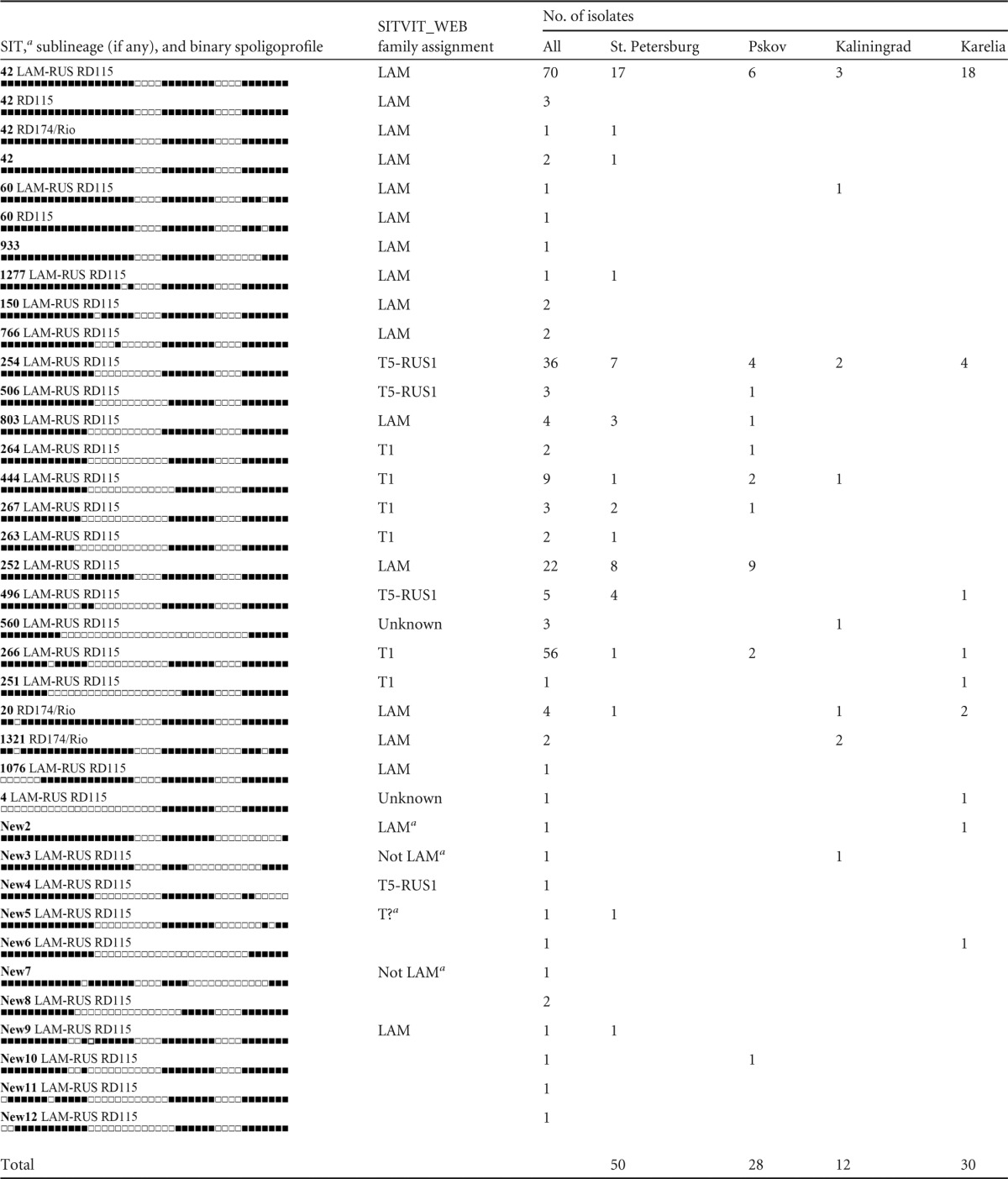

SIT, spoligotype international type according to the SITVIT_WEB database. Most “new” profiles (except for new4 and new9) not found in the SITVIT_WEB family were assigned to a family based on decision rules (18).
The 250 isolates robustly assigned to the LAM family by the above-described SNP analysis were tested for the presence of the specific deletions RD174, RD115, and RD-Rio and the LAM-RUS-specific plcA::IS6110 insertion. Sequencing of the RD115 deletion region was performed for a selection of isolates with different spoligotypes, including those with large deleted blocks of spacers (SIT42, SIT506, SIT254, and SIT252). A comparison of the obtained sequences to data in GenBank and the original RD115 sequence position (4) showed that this was a true RD115 deletion. It was identical in the studied isolates and in LAM isolates with complete genomes in GenBank, namely, strains CTRI-2 from Russia (LAM-RUS subfamily) and KZN4207, KZN605, and KZN1435 from South Africa (LAM-KZN subfamily), as well as isolates with available draft genomes, including 3 more KZN isolates and 5 isolates from Panama (22).
A distribution analysis of the above-described markers across spoligotypes showed (i) the copresence of RD174 and RD-Rio, (ii) the mutually exclusive occurrence of RD174/RD-Rio and RD115, and (iii) the presence of RD115 in all LAM-RUS isolates, while on the other hand, a number of isolates with RD115 did not belong to the LAM-RUS family (Table 1).
SIT42 included isolates with different kinds of deletions (nonsimultaneously), while other SITs included only isolates with one particular deletion, either RD115 or RD174. SIT60 included two isolates with an RD115 deletion, only one of which belonged to the LAM-RUS family. The polyphyletic origin of the SIT60 profile from SIT42/RD115 and SIT42/RD115/LAM-RUS isolates, i.e., an independent deletion of spacer 40 in the unrelated isolates, can be explained by convergent evolution of the direct repeat (DR) locus.
Relationships of the spoligotyping profiles of isolates from the largest collections in this study (Russia, Kazakhstan, and Belarus) were estimated as a forest graph (Fig. 1). The graph was built separately only for LAM-RUS isolates that made up the majority of the studied collection. This graph was manually complemented with a few other SITs representing branches other than the LAM-RUS sublineage within the LAM family. For this reason, the major ancient prototype SIT42 is represented by several nodes, based on RD analysis. In spite of limitations of the gap model, inherent to the SpolTools algorithm when applied to profiles with large deleted blocks, the forest graph seems to present a clear and unidirectional evolution of the LAM-RUS subfamily in the confines of the former Soviet Union.
FIG 1.

Relationships of the spoligotypes identified in this study in LAM isolates, within the RD framework, evaluated as a forest graph. Binary spoligoprofiles are shown schematically for some major types in the bottom right inset. Numbers in italics in country-specific segments of the pie charts show percentages of the strains of this particular type in the total collection of a given country. The “spoligoforest” burst output was generated with the SpolTools program. Solid edges are unique relationships between spoligotypes. Dotted lines have a probability of <0.5, while dashed lines have a probability of >0.5 (see Table 1 for types and profiles used).
DISCUSSION
This study reevaluated the rate of the pathobiologically important Latin American-Mediterranean family of M. tuberculosis and its major sublineages in areas across the former Soviet Union and estimated its framework phylogeny placed into the wider spatiotemporal context.
The tested SNPs in Rv3062 and Rv0129c were previously validated to be specific for the LAM family with diverse collections of reference and clinical isolates (6, 17, 24). A search in GenBank also showed the presence of these LAM-specific alleles only in isolates with an unambiguous LAM spoligotype signature. This is why these SNPs present unique-event, unidirectional polymorphisms, and their use offers a robust evolutionary approach for the detection of the LAM family and, in particular, for resolving ambiguous spoligotypes with large deleted blocks of spacers.
Here, a family status was rectified for some large spoligotypes (SIT254 and SIT266) that would be assigned to the T family based on spoligotyping decision rules alone. As a whole, the LAM prevalence rate was reestimated in all studied areas within the former Soviet Union. Across Russia, it increased from 20.6% (suggested by SITVIT_WEB [Table 1]) to 29.4% in Karelia, from 14.5% to 25.5% in Pskov, from 7.8% to 13.3% in Kaliningrad, and from 10.7% to 28.6% in Tuva. Beyond Russia, it increased from 9.4% to 19.5% in Kazakhstan and from 10.8% to 41.8% in Belarus.
We compared the spoligoprofiles detected in Russian LAM isolates against data in the SITVIT_WEB database and other relevant publications (6–9, 25–28) (Tables 2 and 3). For simplicity, we assumed a monophyletic nature of all derived SITs, except for SIT42; in other words, for example, while SIT20 was found in this and other studies to belong to RD-Rio, we assumed all SIT20 isolates in SITVIT_WEB to represent RD-Rio. Regarding the prevalence of the LAM-RUS family, its direct estimation was not possible by using data reported in the literature, and we used LAM-RUS-specific spoligotypes found in this study as a proxy. In particular, this comparison showed that some LAM-RUS SITs also circulate in other countries (with a high rate of historical or recent immigration), but they make up a negligible rate compared to their local LAM populations (Tables 2 and 3) (25–28).
TABLE 2.
Distribution of the LAM sublineages and SITs in this study and in selected world regionsd
| SIT, sublineage, and/or genomic deletion | No. of isolates/total no. of LAM isolates tested |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RUa this study | RU, Tulab | BEa | KZa | BGa | BRc | CRc | USAc | ZAc | TNc | NLc | |
| 42 | 1 | 1 | |||||||||
| 933 | 1 | ||||||||||
| Orphans/new | 1 | 1 | 1 | ||||||||
| 42, RD-Rio, RD174 | 1 | 45/52 | 11/11 | 7/16 | 1/1 | 0/46 | 5/19 | ||||
| 60, RD-Rio, RD174 | 3/6 | 0/19 | 1/1 | ||||||||
| 20, RD-Rio, RD174 | 4 | 20/20 | 3/3 | 5/6 | 15/15 | 3/3 | |||||
| 1321, RD-Rio, RD174 | 2 | ||||||||||
| 42, RD115 | 1 | 1 | |||||||||
| 60, RD115 | 1 | ||||||||||
| 42, LAM-RUS, RD115 | 46 | 6 | 12 | 12 | |||||||
| 60, LAM-RUS, RD115 | 1 | ||||||||||
| 1277, LAM-RUS, RD115 | 1 | ||||||||||
| 150, LAM-RUS, RD115 | 2 | 1 | |||||||||
| 766, LAM-RUS, RD115 | 2 | ||||||||||
| 254, LAM-RUS, RD115 | 20 | 3 | 6 | 10 | 4 | ||||||
| 506, LAM-RUS, RD115 | 2 | 1 | |||||||||
| 803, LAM-RUS, RD115 | 4 | ||||||||||
| 264, LAM-RUS, RD115 | 2 | ||||||||||
| 444, LAM-RUS, RD115 | 5 | 1 | 3 | ||||||||
| 267, LAM-RUS, RD115 | 3 | ||||||||||
| 263, LAM-RUS, RD115 | 2 | ||||||||||
| 252, LAM-RUS, RD115 | 17 | 27 | 5 | ||||||||
| 496, LAM-RUS, RD115 | 5 | ||||||||||
| 560, LAM-RUS, RD115 | 3 | ||||||||||
| 266, LAM-RUS, RD115 | 4 | 1 | 52 | ||||||||
| 251, LAM-RUS, RD115 | 1 | 1 | |||||||||
| 1076, LAM-RUS, RD115 | 1 | ||||||||||
| 4, LAM-RUS, RD115 | 1 | ||||||||||
| Orphans/new, LAM-RUS-RUS | 6 | 2 | 2 | ||||||||
| Total RD-Rio (all SITs considered) | 7/135 | 0/39 | 0/81 | 0/30 | 0/3 | 131/254 | 34/42 | 33/51 | 37/117 | 0/46 | 22/72 |
| Total LAM-RUS (all SITs considered) | 124/135 | 39/39 | 80/81 | 29/30 | 1/3 | ||||||
Data from this study.
Data reported by Gibson et al. (6). Gibson et al. did not show data on RD115, which is why we show the numbers of RD-Rio isolates/all tested LAM isolates in a single cell. Mixed RD-Rio and wild-type isolates were not considered.
Data reported by Ignatova et al. (8). Ignatova et al. did not test for RD115, but we assumed that all LAM-RUS isolates have this deletion; for this reason, LAM-RUS RD115 data from Ignatova et al. are shown.
SIT, spoligotype international type according to the SITVIT_WEB database (12). Abbreviations: RU, Russia; BE, Belarus; KZ, Kazakhstan; BG, Bulgaria; BR, Brazil, CR, French Guiana and Caribbean; ZA, South Africa, TN, Tunisia; NL, Netherlands.
TABLE 3.
Distribution of the SITs found in this study in the SITVIT_WEB database
| SITa | No. of isolatesc |
||
|---|---|---|---|
| Total | FSU and surrounding areasb | Rest of the world | |
| 933 | 8 | 5 (4 RUS, 1 FIN) | 1 PER, 2 USA |
| 60 | 181 | 1 (1 RUS) | 2 ARG, 3 AUT, 2 BEL, 28 BRA, 4 CUB, 2 DEU, 2 ALG, 1 ESP, 8 FRA, 1 UK, 7 GMB, 5 GNB, 2 GUF, 8 ITA, 1 JPN, 3 MOR, 1 MDG, 3 MEX, 1 MOZ, 2 NLA, 2 POR, 2 PAR, 8 SAU, 1 SEN, 9 TUN, 8 USA, 11 VEN, 40 ZAF, 1 ZAM |
| 20 | 594 | 10 (3 RUS, 4 EST, 1 FIN, 1 GEO, 1 TUR) | 2 ARG, 2 AUS, 1 AUT, 15 BEL, 83 BRA, 5 GUA, 11 GUF, 26 HAI, 7 MEX, 1 PER, 2 PHI, 49 POR, 1 PAR, 3 SWE, 1 TUN, 26 VEN, 24 ZAF, 2 ZAM, 7 CAM, 5 CUB, 10 DEU, 10 ESP, 17 FRA, 1 UK, 10 GNB, 3 GUB, 5 ITA, 1 MOZ, 11 MLW, 1 NMB, 13 NLA, 3 TNZ, 154 USA, 1 VNM |
| 1321 | 11 | 1 ARG, 2 BRA, 1 GUF, 2 IND, 3 POR, 1 USA | |
| 1277 | 16 | 2 ARG, 2 BRA, 8 DOM, 1 ESP, 3 USA | |
| 150 | 74 | 3 (1 RUS, 1 FIN, 1 TUR) | 6 ARG, 22 BEL, 7 BRA, 1 ALG, 1 EGY, 9 POR, 5 VEN, 3 MOR, 8 FRA, 2 GUF, 6 ITA, 1 MEX |
| 766 | 23 | 12 (5 EST, 1 FIN, 1 TUR) | 1 BNG, 1 FRA, 1 NLA, 1 POR, 7 SAU |
| 254 | 121 | 75 (50 RUS, 9 GEO, 2 AZE, 1 KAZ, 6 LAT, 1 FIN, 1 POL, 5 TUR) | 9 ZAF, 1 TAN, 12 USA, 1 SAU, 1 FRA, 1 ESP, 1 DEN, 2 DEU, 2 BRA, 1 BEL, 10 AUT, 1 NLA, 3 ITA |
| 506 | 4 | 2 (2 RUS) | 1 USA, 1 MEX |
| 803 | 26 | 18 (7 RUS, 2 EST, 1 LAT, 5 GEO, 2 ARM, 1 POL) | 1 BEL, 1 BRA, 1 CHN, 2 VNM, 1 SAU, 1 SWE, 1 USA |
| 264 | 45 | 44 (22 RUS, 3 LAT, 8 GEO, 10 POL, 1 TUR) | 1 DEU |
| 444 | 33 | 3 (1 RUS, 1 GEO, 1 KAZ) | 26 USA, 1 DEU, 3 AUT |
| 267 | 15 | 7 (2 RUS, 1 UKR, 4 GEO) | 2 DEU, 1 BEL, 3 USA, 2 LBY |
| 263 | 5 | 5 (4 RUS, 1 FIN) | |
| 252 | 42 | 39 (36 RUS, 3 LAT) | 3 SAU |
| 496 | 6 | 6 (5 RUS, 1 GEO) | |
| 560 | 6 | 4 (3 RUS, 1 GEO) | 1 USA, 1 BRA |
| 266 | 14 | 13 (12 RUS, 1 LAT) | 1 ALG |
| 251 | 13 | 7 (6 RUS, 1 FIN) | 2 USA, 3 MOR, 1 NLA |
| 1076 | 7 | 3 (3 RUS) | 2 BRA, 1 IDN, 1 MOZ |
SIT, spoligotype international type. SIT42 (which may include RD-Rio, RD115, RD115/LAM-RUS, or other sublineages) and SIT4 (which may represent different families due to convergent evolution) were excluded.
Countries of the former Soviet Union, Finland, Poland, Romania, Bulgaria, Turkey, and Iran.
Abbreviations: ALG, Algeria; ARG, Argentina; ARM, Armenia; AUS, Australia; AUT, Austria; AZE, Azerbaijan; BEL, Belgium; BNG, Bangladesh; BRA, Brazil; CAM, Cameroon; CHN, China; CUB, Cuba; DEN, Denmark; DEU, Germany; DOM, Dominican Republic; EGY, Egypt; ESP, Spain; EST, Estonia; FIN, Finland; FRA, France; GEO, Republic of Georgia; GMB, Gambia; GNB, Guinea Bissau; GUA, Guadeloupe; GUF, French Guiana; HAI, Haiti; IDN, Indonesia; IND, India; ITA, Italy; JPN, Japan; KAZ, Kazakhstan; LAT, Latvia; LBY, Libya; MDG, Madagascar; MEX, Mexico; MLW, Malawi; MOR, Morocco; MOZ, Mozambique; NLA, Netherlands; NMB, Namibia; PAR, Paraguay; PER, Peru; PHI, Philippines; POL, Poland; POR, Portugal; RUS, Russia; SAU, Saudi Arabia; SEN, Senegal; SWE, Sweden; TNZ, Tanzania; TUN, Tunisia; TUR, Turkey; UKR, Ukraine; VEN, Venezuela; VNM, Vietnam; ZAF, South Africa; ZAM, Zambia.
The LAM-RUS family in Northern Eurasia, a historically established, overwhelming host.
LAM-RUS isolates in this study represented distant locations across Russia and the FSU (Fig. 2). The LAM-primordial SIT42 was highly dominated by LAM-RUS isolates: 52 of 55 in Russia, 12 of 13 in Belarus, and 12 of 13 in Kazakhstan. In this light, the following should be noted. First, the LAM-RUS branch of the LAM family is found almost exclusively in Russia and not elsewhere. Second, the LAM-RUS branch makes up almost all of the LAM strains in Russia, and the ancient LAM type SIT42 is dominated by LAM-RUS isolates in Russian collections. Third, LAM-RUS heterogeneity is observed already at the spoligotype level (known to be a low-level discriminatory marker). Taken together, this implies a monophyletic origin of the LAM-RUS branch (specific plcA::IS6110 insertion in the SIT42 subpopulation): in Russia, at a historically distant time, in a single small founding bacterial population. This event was followed, through human bottleneck circumstance, by long-term in situ evolution and divergence. Its dissemination and prevalence within the large area of the former Soviet Union may be indicative of the specific long-term coexistence of the LAM-RUS branch and local human populations hypothetically leading to coadaptation and reduced pathogenicity of relatively more ancient clones, such as SIT254 versus the more recent SIT252 and SIT266 (see below).
FIG 2.

Geographic distribution of the LAM family and its sublineages and major spoligotypes. Numbers in italics show percentages of LAM isolates in the local population of M. tuberculosis (also roughly reflected by the circle size). The prevalence of LAM isolates in St. Petersburg was estimated to be 25% based on an analysis of the IS6110-RFLP database of our laboratory in the light of previously reported LAM-RUS IS6110-RFLP profiles (23). Data on Tula were reported previously (8, 23). Map source, Free World Maps (http://www.freeworldmaps.net/pdf/world/ApianGlobular1.pdf).
Geographic mapping of the prevalence of LAM sublineages and major types within the LAM-RUS branch showed variation for (i) LAM as a whole and (ii) its major spoligotypes and sublineages across studied settings (Fig. 2). SIT42 and SIT254 are found in all dispersed areas, which may suggest their earlier emergence (apparent in the case of ancient SIT42) than the less ubiquitous SIT252 and SIT266. Similarly, in SITVIT_WEB, major derived LAM-RUS types represent mainly the FSU and its neighbors (Table 2). The more geographically diverse SIT254 may have emerged earlier (and hence had more time to spread) than SIT252 and SIT266, which are limited to the European part of the former Soviet Union. Whether this implies the very recent emergence of SIT252 and SIT266 remains to be elucidated with larger sample sizes from more locations across Russia and the former Soviet Union by using high-resolution typing, including next-generation sequencing.
The LAM RD-Rio sublineage in Northern Eurasia, a recent(?), rare stranger.
While the LAM-RUS branch was studied only by one Russian group, the RD-Rio sublineage has attracted much more attention since its discovery (5–7, 29), but evidence of its increased virulence and transmissibility appears controversial (30, 31). In this study, only a few isolates belonged to RD-Rio (7 such isolates) (Fig. 1). They represented only northwestern Russia but not other parts of Russia, Belarus, or Kazakhstan (this study) or central Russia (8). More specifically, these rare RD-Rio isolates were from St. Petersburg, Karelia, and Kaliningrad (Fig. 2). It is tempting to speculate that, unlike innermost Pskov, Tula, and Siberian Tuva, these areas are more open to longer and/or more active international contact and hence have had more chances to import “alien” isolates.
The prevalence data taken together (Fig. 2), the westernmost city of Kaliningrad (formerly Königsberg) appears to be least affected by LAM as a whole but, in contrast, features the highest prevalence rate of RD-Rio strains. Interestingly, RD-Rio strains were statistically more prevalent in Kaliningrad than in St. Petersburg (P = 0.04) (odds ratio [OR], 8.0; 95% confidence interval [CI], 1.2 to 54.9). This is especially remarkable in view of the lowest prevalence of LAM isolates as a whole in the former setting. Kaliningrad forms an exclave on the Baltic Sea inside the European Union separated from mainland Russia by Lithuania and Poland (Fig. 2). As we noted in our previous article focused on Kaliningrad (12), “population of the area has changed completely since 1946 when northern part of the former East Prussia was transferred to USSR … virtually closed from foreign contact from 1950s until 1991, Kaliningrad today is a busy Russian-European crossroad.” SIT20 is characteristic of strains in the Americas, both South America (Brazil being a focus) and North America, due to immigration. The particular immigration flows may also explain the high or visible RD-Rio prevalence in some European countries marked by close contact with South America (Table 2) (6, 7). While SIT20 is a large type, SIT1321 is found in only 11 globally dispersed isolates in SITVIT_WEB (Americas, India, and Portugal), and neither is from the FSU or its neighbors. The latter notion points to an external source of the RD-Rio isolates in Russia.
Concluding remarks.
In summary, the LAM-RUS family is both an endemic and historical M. tuberculosis family in Russia, a likely area of its distant origin. There is a clear indication of high prevalence of LAM-RUS isolates in the reestimated doubled LAM pool in the FSU and their limited circulation in the world, as sporadic isolates of different spoligotypes.
In contrast, the LAM RD-Rio sublineage is found at a high prevalence rate (up to 50%) in LAM isolates in the Ibero-American and Caribbean regions, where it likely originated and initially disseminated. It is also identified at a high or at least noticeable rate in the regions marked by high immigration from these regions (such as the United States and the Netherlands) and is sharply reduced to sporadic isolates in the areas with a historical prevalence of LAM-RUS isolates. Unlike historical and endemic LAM-RUS isolates, the sporadic RD-Rio isolates were likely brought to Russia/Northern Eurasia through occasional human contact/travel. The spread of the LAM RD-Rio lineage is not so global as commonly claimed but is determined largely by human migration flows (rather than by pathobiological properties of these strains). Consequently, we suggest that the genetic background of the host population plays a crucial role in in situ dissemination of the imported strains in an autochthonous, naive population.
The LAM-RUS subfamily is a major LAM subfamily across Russia and the FSU but apparently not in neighboring countries. The link between the LAM-RUS isolates and some SITs demonstrated in this study offers a good practical proxy to take a new look at and to reevaluate previously reported spoligotype data. At the same time, due to the convergent evolution of the DR locus and possible homoplasy of some spoligotyping profiles, one should specifically test the plcA::IS6110 insertion. This is desirable in cases of some particular derived types, such as SIT254 and SIT266, and necessary in cases of the ancient type SIT42, which may include isolates of different RD-defined sublineages.
It has not escaped our notice that an identical RD115 deletion is shared by geographically distant isolates from Eurasia, the Americas, and Africa, namely, the LAM-RUS and LAM-KZN subfamilies and some Panama LAM strains. This implies a very distant origin of the LAM-RD115 “grand sublineage” and raises a question about the human populations in which it could occur; an answer to this question remains a challenge for the future.
ACKNOWLEDGMENTS
We thank Boris Vishnevsky, Tatiana Otten, Larisa Chernousova, Natalia Vasilenko, Ho Minh Li, Bakhytkul Zhakipbaeva, Nadya Markova, Violeta Valcheva, and Yuri Markelov for providing some of the isolates or DNA samples.
Footnotes
Published ahead of print 28 February 2014
REFERENCES
- 1.Musser JM, Amin A, Ramaswamy S. 2000. Negligible genetic diversity of mycobacterium tuberculosis host immune system protein targets: evidence of limited selective pressure. Genetics 155:7–16 http://www.genetics.org/content/155/1/7.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sola C, Filliol I, Legrand E, Mokrousov I, Rastogi N. 2001. Mycobacterium tuberculosis phylogeny reconstruction based on combined numerical analysis with IS1081, IS6110, VNTR, and DR-based spoligotyping suggests the existence of two new phylogeographical clades. J. Mol. Evol. 53:680–689. 10.1007/s002390010255 [DOI] [PubMed] [Google Scholar]
- 3.Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, Nicol M, Niemann S, Kremer K, Gutierrez MC, Hilty M, Hopewell PC, Small PM. 2006. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 103:2869–2873. 10.1073/pnas.0511240103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, Goguet de la Salmoniere YO, Aman K, Kato-Maeda M, Small PM. 2004. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc. Natl. Acad. Sci. U. S. A. 101:4865–4870. 10.1073/pnas.0305634101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazzarini LC, Huard RC, Boechat NL, Gomes HM, Oelemann MC, Kurepina N, Shashkina E, Mello FC, Gibson AL, Virginio MJ, Marsico AG, Butler WR, Kreiswirth BN, Suffys PN, Lapa e Silva JR, Ho JL. 2007. Discovery of a novel Mycobacterium tuberculosis lineage that is a major cause of tuberculosis in Rio de Janeiro, Brazil. J. Clin. Microbiol. 45:3891–3902. 10.1128/JCM.01394-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibson AL, Huard RC, Gey van Pittius NC, Lazzarini LC, Driscoll J, Kurepina N, Zozio T, Sola C, Spindola SM, Kritski AL, Fitzgerald D, Kremer K, Mardassi H, Chitale P, Brinkworth J, Garcia de Viedma D, Gicquel B, Pape JW, van Soolingen D, Kreiswirth BN, Warren RM, van Helden PD, Rastogi N, Suffys PN, Lapa e Silva J, Ho JL. 2008. Application of sensitive and specific molecular methods to uncover global dissemination of the major RDRio sublineage of the Latin American-Mediterranean Mycobacterium tuberculosis spoligotype family. J. Clin. Microbiol. 46:1259–1267. 10.1128/JCM.02231-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.David S, Duarte EL, Leite CQ, Ribeiro JN, Maio JN, Paixão E, Portugal C, Sancho L, Germano de Sousa J. 2012. Implication of the RD(Rio) Mycobacterium tuberculosis sublineage in multidrug resistant tuberculosis in Portugal. Infect. Genet. Evol. 12:1362–1367. 10.1016/j.meegid.2012.04.021 [DOI] [PubMed] [Google Scholar]
- 8.Ignatova A, Dubiley S, Stepanshina V, Shemyakin I. 2006. Predominance of multi-drug-resistant LAM and Beijing family strains among Mycobacterium tuberculosis isolates recovered from prison inmates in Tula Region, Russia. J. Med. Microbiol. 55:1413–1418. 10.1099/jmm.0.46575-0 [DOI] [PubMed] [Google Scholar]
- 9.Dubiley S, Kirillov E, Ignatova A, Stepanshina V, Shemyakin I. 2007. Molecular characteristics of the Mycobacterium tuberculosis LAM-RUS family prevalent in central Russia. J. Clin. Microbiol. 45:4036–4038. 10.1128/JCM.01217-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokrousov I, Vyazovaya A, Otten T, Zhuravlev V, Pavlova E, Tarashkevich L, Krishevich V, Vishnevsky B, Narvskaya O. 2012. Mycobacterium tuberculosis population in northwestern Russia: an update from Russian-EU/Latvian border region. PLoS One 7:e41318. 10.1371/journal.pone.0041318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matrakshin AG, Mes'ko EM, Beliakova NK, Andreevskaia SN, Smirnova TG, Larionova EE, Kuz'min AV, Mokrousov IV, Pospelov LE, Chernousova LN. 2004. Genotypic characteristics of Mycobacterium tuberculosis strains from the Republic of Tyva. Probl. Tuberk. Bolezn. Legk. 2004:37–40 (In Russian) [PubMed] [Google Scholar]
- 12.Mokrousov I, Otten T, Zozio T, Turkin E, Nazemtseva V, Sheremet A, Vishnevsky B, Narvskaya O, Rastogi N. 2009. At Baltic crossroads: a molecular snapshot of Mycobacterium tuberculosis population diversity in Kaliningrad, Russia. FEMS Immunol. Med. Microbiol. 55:13–22. 10.1111/j.1574-695X.2008.00470.x [DOI] [PubMed] [Google Scholar]
- 13.Vasilenko N, Vyazovaya A, Mokrousov I, Limeschenko E, Semenov V, Narvskaya O. 2006. Spacer oligonucleotide typing of drug-resistant Mycobacterium tuberculosis circulating on the territory of Belarus. Immunopat. Allergol. Infektol. 2006:70–74 (In Russian) [Google Scholar]
- 14.Valcheva V, Mokrousov I, Narvskaya O, Rastogi N, Markova N. 2008. Molecular snapshot of drug-resistant and drug-susceptible Mycobacterium tuberculosis strains circulating in Bulgaria. Infect. Genet. Evol. 8:657–663. 10.1016/j.meegid.2008.06.006 [DOI] [PubMed] [Google Scholar]
- 15.Mokrousov I, Jiao WW, Valcheva V, Vyazovaya A, Otten T, Ly HM, Lan NN, Limeschenko E, Markova N, Vyshnevskiy B, Shen AD, Narvskaya O. 2006. Rapid detection of the Mycobacterium tuberculosis Beijing genotype and its ancient and modern sublineages by IS6110-based inverse PCR. J. Clin. Microbiol. 44:2851–2856. 10.1128/JCM.00705-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden JDA. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35:907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abadia E, Zhang J, dos Vultos T, Ritacco V, Kremer K, Aktas E, Matsumoto T, Refregier G, van Soolingen D, Gicquel B, Sola C. 2010. Resolving lineage assignation on Mycobacterium tuberculosis clinical isolates classified by spoligotyping with a new high-throughput 3R SNPs based method. Infect. Genet. Evol. 10:1066–1074. 10.1016/j.meegid.2010.07.006 [DOI] [PubMed] [Google Scholar]
- 18.Demay C, Liens B, Burguiere T, Hill V, Couvin D, Millet J, Mokrousov I, Sola C, Zozio T, Rastogi N. 2012. SITVITWEB—a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect. Genet. Evol. 12:755–766. 10.1016/j.meegid.2012.02.004 [DOI] [PubMed] [Google Scholar]
- 19.Reyes JF, Francis AR, Tanaka MM. 2008. Models of deletion for visualizing bacterial variation: an application to tuberculosis spoligotypes. BMC Bioinformatics 9:496. 10.1186/1471-2105-9-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang C, Reyes JF, Luciani F, Francis AR, Tanaka MM. 2008. spolTools: online utilities for analyzing spoligotypes of the Mycobacterium tuberculosis complex. Bioinformatics 24:2414–2415. 10.1093/bioinformatics/btn434 [DOI] [PubMed] [Google Scholar]
- 21.Gilman J, Myatt M. 1998. EpiCalc 2000, version 102 Brixton Books, London, United Kingdom [Google Scholar]
- 22.Lanzas F, Karakousis PC, Sacchettini JC, Ioerger TR. 2013. Multidrug-resistant tuberculosis in Panama is driven by clonal expansion of a multidrug-resistant Mycobacterium tuberculosis strain related to the KZN extensively drug-resistant M. tuberculosis strain from South Africa. J. Clin. Microbiol. 51:3277–3285. 10.1128/JCM.01122-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubiley S, Ignatova A, Mukhina T, Nizova A, Blagodatskikh S, Stepanshina V, Shemyakin I. 2010. Molecular epidemiology of tuberculosis in the Tula area, central Russia, before the introduction of the directly observed therapy strategy. Clin. Microbiol. Infect. 16:1421–1426. 10.1111/j.1469-0691.2009.03102.x [DOI] [PubMed] [Google Scholar]
- 24.Homolka S, Projahn M, Feuerriegel S, Ubben T, Diel R, Nübel U, Niemann S. 2012. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS One 7:e39855. 10.1371/journal.pone.0039855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smit PW, Haanperä M, Rantala P, Couvin D, Lyytikäinen O, Rastogi N, Ruutu P, Soini H. 2013. Molecular epidemiology of tuberculosis in Finland, 2008-2011. PLoS One 8:e85027. 10.1371/journal.pone.0085027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krawczyk M, Brzostek A, Gorna A, Knapska K, Ziolkiewicz M, Wojtasik A, Dziadek J. 2011. Epidemiological analysis of Mycobacterium tuberculosis strains isolated in Lodz, Poland. Int. J. Tuberc. Lung Dis. 15:1252–1258. 10.5588/ijtld.10.0718 [DOI] [PubMed] [Google Scholar]
- 27.Jagielski T, Augustynowicz-Kopec E, Zozio T, Rastogi N, Zwolska Z. 2010. Spoligotype-based comparative population structure analysis of multidrug-resistant and isoniazid-monoresistant Mycobacterium tuberculosis complex clinical isolates in Poland. J. Clin. Microbiol. 48:3899–3909. 10.1128/JCM.00572-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Augustynowicz-Kopeć E, Jagielski T, Kozińska M, Kremer K, van Soolingen D, Bielecki J, Zwolska Z. 2012. Transmission of tuberculosis within family-households. J. Infect. 64:596–608. 10.1016/j.jinf.2011.12.022 [DOI] [PubMed] [Google Scholar]
- 29.Dalla Costa ER, Lazzarini LC, Perizzolo PF, Díaz CA, Spies FS, Costa LL, Ribeiro AW, Barroco C, Schuh SJ, da Silva Pereira MA, Dias CF, Gomes HM, Unis G, Zaha A, Almeida da Silva PE, Suffys PN, Rossetti ML. 2013. Mycobacterium tuberculosis of the RDRio genotype is the predominant cause of tuberculosis and associated with multidrug resistance in Porto Alegre City, South Brazil. J. Clin. Microbiol. 51:1071–1077. 10.1128/JCM.01511-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazzarini LC, Spindola SM, Bang H, Gibson AL, Weisenberg S, da Silva Carvalho W, Augusto CJ, Huard RC, Kritski AL, Ho JL. 2008. RDRio Mycobacterium tuberculosis infection is associated with a higher frequency of cavitary pulmonary disease. J. Clin. Microbiol. 46:2175–2183. 10.1128/JCM.00065-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbosa CB, Lazzarini LC, Elias AR, Leung JA, Ribeiro SB, da Silva MG, Duarte RS, Suffys P, Gomes HM, Kritski AL, Lapa e Silva JR, Ho JL, Boéchat N. 2012. Tuberculosis caused by RDRio Mycobacterium tuberculosis is not associated with differential clinical features. Int. J. Tuberc. Lung Dis. 16:1377–1382. 10.5588/ijtld.11.0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobzhansky T. 1973. Nothing in biology makes sense except in the light of evolution. Am. Biol. Teacher 35:125–129 http://www.jstor.org/stable/4444260 [Google Scholar]