

Abstract
EsxA (ESAT-6) and EsxB (CFP-10) are virulence factors exported by the ESX-1 system in mycobacterial pathogens. In Mycobacterium marinum, an established model for ESX-1 secretion in Mycobacterium tuberculosis, genes required for ESX-1 export reside at the extended region of difference 1 (RD1) locus. In this study, a novel locus required for ESX-1 export in M. marinum was identified outside the RD1 locus. An M. marinum strain bearing a transposon-insertion between the MMAR_1663 and MMAR_1664 genes exhibited smooth-colony morphology, was deficient for ESX-1 export, was nonhemolytic, and was attenuated for virulence. Genetic complementation revealed a restoration of colony morphology and a partial restoration of virulence in cell culture models. Yet hemolysis and the export of ESX-1 substrates into the bacteriological medium in vitro as measured by both immunoblotting and quantitative proteomics were not restored. We show that genetic complementation of the transposon insertion strain partially restored the translocation of EsxA and EsxB to the mycobacterial cell surface. Our findings indicate that the export of EsxA and EsxB to the cell surface, rather than secretion into the bacteriological medium, correlates with virulence in M. marinum. Together, these findings not only expand the known genetic loci required for ESX-1 secretion in M. marinum but also provide an explanation for the observed disparity between in vitro ESX-1 export and virulence.
INTRODUCTION
The ESAT-6 system-1 (ESX-1)/WXG-100 secretion system (Wss) is required for the virulence of both Gram-positive and mycobacterial pathogens (1–5). In mycobacterial pathogens the ESX-1 system likely promotes permeabilization of the phagosomal membrane allowing either the bacterial cell or bacterial products access to the cytosol of the macrophage (6–10). The ESX-1/Wss system is conserved and functional in nonpathogenic bacteria, where it promotes a range of activities, including conjugation (11–16).
Protein substrates are translocated across the mycobacterial cytoplasmic membrane by the ESX-1 export system (3, 5). Several genes are required for protein translocation, disruption of which results in loss of substrate secretion into the bacteriological medium (culture supernatant) in vitro (3–5, 17–26). It is unknown how the protein substrates are secreted across the mycolate outer membrane (MOM) out of the mycobacterial cell or if the known ESX genes promote MOM translocation. Likewise, it is not clear if the ESX-1 substrates are true exoproteins (released from the cell) or extrinsically associated with the MOM and shed into the bacteriological medium (27–29). Indeed, in addition to the culture supernatant, ESX-1 substrates have been localized to the mycobacterial cell wall and associated with the surface of the mycobacterial cell (28–31). It is unclear which population of ESX-1 substrates mediates virulence. The active secretion of ESX-1 substrates into the phagosome or cytosol of the macrophage has not been routinely observed.
In the human pathogen Mycobacterium tuberculosis, three genetic loci are required for substrate translocation. The largest locus is the extended region of difference 1 (RD1), which refers to a region of the M. tuberculosis genome that is absent from the Mycobacterium bovis BCG vaccine strains (3–5). The two additional loci include the espACD operon and espR (23, 24, 26).
Mycobacterium marinum is a pathogenic mycobacterial species that is established as a model for studying ESX-1 export in M. tuberculosis (32–35). In M. marinum the extended RD1 locus (MMAR_5439 through MMAR_5459, based on homology to the genes in M. tuberculosis) is the only genomic region reported to be required for ESX-1 export (18, 32, 34, 36–39). Transposon insertions within or targeted deletion of individual genes in the extended RD1 locus prevents the export of ESX-1 substrates, including EsxA (ESAT-6) and EsxB (CFP-10) (EsxA/B) in vitro and attenuates M. marinum in macrophage, amoeba, and zebrafish models of infection (32, 40, 41).
In general, genetic manipulations to the ESX-1 system which result in loss of substrate secretion into the culture supernatant in vitro lead to loss of virulence in infection models (initially described in references 3 and 5). Recent work from Chen et al. reported variants of EspA, an ESX-1 substrate in M. tuberculosis, which led to strains that were deficient for ESX-1 secretion as measured by Western blot analysis of culture filtrate proteins (42). Surprisingly, these M. tuberculosis strains were virulent in both macrophage and mouse infection models (42). While this result may be interpreted to mean that ESX-1 export and secretion are not required for virulence, there is long-standing evidence to the contrary. The mechanism underlying the apparent disparity between a loss of ESX-1 secretion and virulence remains undefined.
Here, we report the identification of a region of the M. marinum genome, outside the extended RD1 locus, required for ESX-1 export. We demonstrate that insertion of a transposon in the intergenic region between MMAR_1663 and MMAR_1664 in the M. marinum genome altered colony morphology, disrupted ESX-1-mediated hemolysis and substrate export into the bacteriological medium in vitro, and attenuated M. marinum in cell culture models of infection. The addition of a second copy of the MMAR_1663-MMAR_1668 genes led to complementation of colony morphology and a partial complementation of the virulence defect. However, ESX-1-dependent contact-dependent hemolysis and secretion of protein substrates into the bacteriological medium were not restored. Using a whole-colony ESX-1 secretion assay that measures the translocation of the EsxA and EsxB ESX-1 substrates to the cell surface and quantitative proteomics, we confirmed partial complementation of ESX-1 function. We propose that the translocation of EsxA and EsxB to the mycobacterial cell surface rather than export into the bacteriological medium correlates with virulence in M. marinum. Together, our findings expand the known genetic loci required for ESX-1 secretion and explain the observed disparity between in vitro ESX-1 export and virulence in M. marinum.
MATERIALS AND METHODS
Bacterial strains and cultures.
Mycobacterium marinum strains were grown at 30°C in Middlebrook 7H9 liquid broth (Sigma-Aldrich, St. Louis, MO) with 0.1% Tween 80 (Fisher Scientific, Pittsburgh, PA) or on Middlebrook 7H11 agar as described previously (19), supplemented with kanamycin (20 μg/ml; IBI, Poesta, IL) or hygromycin (50 μg/ml; EMD Millipore, Billerica, CA) as necessary. The mycobacterial strains used in this study are listed in Table 1. All strains were derived from the M. marinum M strain (ATCC BAA-535). The M. marinum strain 14 was constructed by introduction of the pMB272 suicide plasmid which carries the Mariner transposon (Tn) into the M strain (43). Mycobacterial transformation was performed using electroporation as previously described (43, 44). Following introduction of the plasmid, counter-selection was applied by adding sucrose to the agar, and growth was at 32°C to promote plasmid loss and transposon insertion into the genome as described previously (44). The transposon insertion site was identified by extracting and digesting the M. marinum genomic DNA and generating a plasmid library in pBluescript SK+ (Agilent Technologies, La Jolla, CA) (44). All restriction enzymes were purchased from New England BioLabs (NEB; Ipswich, MA) and used according to the manufacturer's recommendations. All plasmid preparations were made using an AccuPrep Plasmid MiniPrep DNA Extraction Kit (Bioneer, Alameda, CA). The plasmid including the genomic DNA bearing the transposon insertion was selected by introduction of the library into Escherichia coli DH5α and selecting for resistance to kanamycin. The insertion site was identified using the 821A (5′-CGCATCTTCCCGACAACGCAGACCGTTCC) and 822A (5′-TAATCGCGGCCTCGAGCAAGACGTTTCCCG) primers which anneal to the Tn and allow sequencing of the genomic regions flanking the transposon (43). All primers were ordered from Integrated DNA Technologies. In the M. marinum strain 14, the transposon inserted between a T/A dinucleotide (bases 2010906/2010907). The aph promoter on the Tn was in the reverse orientation to the predicted MMAR_1664 open reading frame (ORF). The ΔRD1 and the eccCb::Tn M. marinum strains were obtained from Eric J. Brown (18, 41).
TABLE 1.
Strains used in this study
| M. marinum strain | Genotype | Reference or source |
|---|---|---|
| M | Wild-type M. marinum | ATCC |
| 1663::Tn::1664 strain | M strain bearing a transposon insertion between the MMAR_1663 and MMAR_1664 genes | This study |
| 1663::Tn::1664/p′1663–1668 strain | Complemented; transposon insertion strain bearing an episomal plasmid carrying the C-terminal half of MMAR_1663 through MMAR_1668 | This study |
| ΔRD1 strain | M strain bearing a genomic deletion including ′eccCb-′espK | 41 |
| ΔesxBA strain | M strain bearing a deletion covering the esxB and esxA genes | 32 |
Construction of p′1663–1668 complementation plasmid.
The p′1663–1668 plasmid was constructed by amplifying a 5.8-kb region from M. marinum M genomic DNA using the OGK29 (5′-GCTCTAGAGAAGTGGTCGATCTCGTATG) and OGK30 (5′-AAAACTGCAGGCGAAATGTTCGAGGATCTG) primers (XbaI and PstI restriction sites are underlined). The resulting PCR product included genomic DNA including the 3′ half of MMAR_1663 through the first ∼300 bp of MMAR_1669 as annotated in MycoBrowser (45). The PCR product was purified using an AccuPrep PCR Purification Kit (Bioneer, Alameda, CA). The resulting digested product was introduced into the pMV206Hyg shuttle vector (23, 46) using standard molecular biology approaches according to the manufacturer's instructions (NEB, Ipswitch MA). The plasmid was confirmed by sequencing analysis in the Notre Dame Genomics and Bioinformatics Core Facility (https://www3.nd.edu/∼genomics/). The resulting plasmid was introduced into the 14 M. marinum strain using electroporation. The resulting complementation strain was confirmed by amplifying the hygromycin resistance gene from the episomal plasmid following genomic DNA extraction using the OPC81 (5′-ACGAAAGGCTCAGTCGAAAG) and OPC82 (5′-TCCGCTGTGACACAAGAATC) primers.
M. marinum infection of amoebae and macrophage-like cells.
Acanthamoeba castellanii amoebae were cultured and maintained at room temperature in PYG-712 medium as previously described (40). M. marinum strains were cultured in vitro as described above. Twenty-four-well plates were seeded with 5 × 105 A. castellanii amoebae per well and infected with M. marinum at a multiplicity of infection (MOI) of 10. The infections were centrifuged briefly to ensure efficient contact with and uptake of bacteria. After 2 h, monolayers were treated with gentamicin (100 μg/ml; Research Products International [RPI], Mount Prospect, IL) for 1 h to eliminate extracellular bacteria. Infections and incubations were carried out at room temperature. The infected amoebae were incubated at room temperature for 24 h.
The mouse macrophage-like cell line RAW 264.7 was a gift from Jeff Schorey. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (HyClone, Logan, UT, USA), at 37°C under a humidified atmosphere of 95% air and 5% CO2. The cells were seeded in 24-well plates (Greiner Bio-One, Monroe, NC), at a density of 5 × 105 cells/well, 1 day prior to infection. The cells were infected with mycobacteria at an MOI of 10 for 2 h at 37°C in 5% CO2. The extracellular bacteria were removed as described above, and cells were further incubated at 37°C in 5% CO2 for 24 h.
Viability assays.
Cell viability analysis of A. castellanii was performed by treating infected monolayers with ethidium homodimer (4 μM) (Live/Dead Viability/Cytotoxicity Kit; Molecular Probes) for 30 min at room temperature, protected from light. The calcein-acetoxymethyl (calcein-AM) stain did not visibly stain the amoebae. Infected monolayers of RAW 264.7 cells were stained with ethidium homodimer 1 and calcein-AM (Live/Dead Viability/Cytotoxicity Kit; Life Technologies, Grand Island, NY) at 37°C in 5% CO2 for 1 h. For both amoebae and macrophages, imaging was performed using a Zeiss AxioObserver A1 inverted microscope with phase-contrast, rhodamine (red), and green fluorescent protein (GFP) filters. The relative cytotoxicity exhibited by infected cells was quantified by enumeration of dead cells in at least four fields chose randomly for each strain.
Hemolysis assay.
Hemolysis assays were described previously by Champion et al. (44). Briefly, M. marinum strains were grown in 25 ml of 7H9 broth (Middlebrook) and 0.1% Tween 80 with the appropriate antibiotics as required. Bacterial strains were normalized by number of cells as calculated by measuring the absorbance at the optical density at 600 nm (OD600). Bacterial cells and sheep red blood cells (sRBCs; BD biosciences, San Jose, CA) were washed with phosphate-buffered saline (PBS), mixed, collected by centrifugation, and incubated for 2 h at 30°C. Hemolysis was measured by changes in the absorbance at the OD405 using a SpectraMax M5 plate reader (Molecular devices, Sunnyvale, CA). Water and PBS were used as positive and negative controls, respectively.
ESX-1 secretion assay.
ESX-1 protein export assays to measure ESX-1-dependent translocation of EsxA and EsxB in the culture supernatant were performed as described previously (19, 44). Briefly, M. marinum cultures were grown to saturation in 7H9 defined broth and diluted to an OD600 of 0.8 in 50 ml of Sauton's defined broth with 0.05% Tween 80 and antibiotics as required. After 48 h of growth at 30°C, cells were harvested by centrifugation, and spent medium was filtered to generate culture filtrates. Whole-cell lysates were generated by lysing the mycobacterial cells with a Mini-Bead Beater-24 (BioSpec, Bartlesville, OK), and culture filtrates were concentrated and treated with phenylmethylsulfonyl fluoride (PMSF) as described previously (19, 44).
The protein fractions were used either for Western blot analysis or for targeted multiple-reaction monitoring (MRM) proteomics (see below). Protein concentrations were measured using a MicroBCA Protein Assay Kit (Thermo Scientific/Pierce, Rockford, IL); 10 μg of cell lysate and culture filtrate were separated using 4 to 20% Criterion TGX Precast Gels (Bio-Rad, Hercules, CA). Proteins were transferred to nitrocellulose and detected by Western blot analysis using LumiGLO Chemiluminescent Substrate (KPL, Gaithersburg, MD) for CFP-10 (NR-13801; BEI Resources, Manassas, VA) and ESAT-6 (AB26246; Abcam, Cambridge, MA) and FAP/MPT-32 (NR-13807; BEI Resources) antibodies as described by Champion et al. (44). A Li-Cor Odyssey Imaging System (LI-COR, Lincoln, Nebraska) was used to image the anti-RNA polymerase (RNAP) beta subunit antibody (AB12087; Abcam) in immunoblots. The following antibody dilutions were used in this study: 1:5,000 anti-RNAP beta subunit in blocking buffer for fluorescent Western blotting (Rockland, Gilbertsville, PA) and 1:5,000 anti-CFP-10, 1:5,000 anti-FAP/MPT-32, and 1:3,000 anti-ESAT-6 in 5% milk in PBS with 0.1% Tween 80 (PBST). Secondary antibodies were used at the following concentrations; 1:10,000 goat anti-mouse IgG DyLight 800 secondary antibody (35521; Pierce Biotechnology, Rockford, IL) in blocking buffer for RNAP immunoblots, 1:5,000 goat anti-mouse IgG horseradish peroxidase (HRP) conjugate secondary antibody (NB710-94938; Novus Biologicals, Littleton, CO) in 5% milk in PBST for ESAT-6 immunoblots, and 1:5,000 goat anti-rabbit IgG HRP conjugate secondary antibody (12-348; Millipore, Billerica, MA) in 5% milk in PBST for CFP-10 and FAP/MPT-32 immunoblots.
MS-MALDI.
Matrix-assisted laser desorption ionization (MALDI) secretion assays were performed essentially as described in our previous work (44). Briefly, ∼5-mm-sized colonies from bacteria grown for 7 days on Sauton's agar plates were removed with a toothpick to a 0.22-μm-pore-size centrifuge filter (Corning, NY). Fifty microliters of liquid chromatography-mass spectrometry (LC-MS)-grade water (Honeywell Burdick and Jackson, Wicklow, Ireland) was added and briefly vortexed on low (2/8 arbitrary units [AU]) and then centrifuged at 2,000 × g for 60 s. One microliter of sample from each M. marinum strain (M, ΔRD1 eccCb::Tn, 1663::Tn::1664, and complemented strains) was deposited onto an Au-MALDI target and dried, and 1 μl of saturated sinapic acid in 50% acetonitrile–0.1% trifluoroacetic acid (TFA) was applied as an overlay and allowed to dry. Myoglobin (10 μM) was applied as an external standard adjacent to the spots for analysis/calibration. MALDI spectra were acquired in linear mode on an AutoflexIII (Bruker, Billerica, MA) with a low-mass deflection set to 4,000 m/z and 300 nS of delay (47). A total of 1,000 spectra were summed at a laser frequency of 100 Hz. Spectra were processed using a 10-amu Gaussian smooth and a linear calibration to the [M + H]1+ and [M + 2H]2+ peaks of myoglobin as we described previously (44). Colony response was normalized using normalized peak intensities of secreted peaks at ∼7,500 m/z and 8,000 m/z.
Nano-UHPLC-MRM.
Targeted mass spectrometry via multiple-reaction monitoring (MRM) was performed essentially as described previously (19, 48, 49). Briefly, 50-μg fractions of culture filtrates or whole-cell lysates were denatured with 2,2,2-trifluoroethanol (2,2,2-TFE; Sigma, St. Louis, MO), reduced and alkylated (with dithiothreitol [DTT] and iodoacetamide [IAA], respectively), and then digested with trypsin (overnight at 1:50). Surface protein samples were digested identically; but there was approximately 10 to 20 μg of total protein, and approximately 250 ng was used per injection. After quenching, 5-μg aliquots were desalted using C18 ZipTips (Millipore, Boston, MA), and 3 μl each of a 13C15N stable-isotope version of EsxA and GroES peptides (0.71 μM and 0.65 μM) was added prior to drying. The internal tryptic sequence NH2-LAAAWGGSGSEAYR-OH (EsxA) was synthesized (New England Peptide, Gardner, MA) at >95% purity with [U-13C6, 15N4]arginine (residue in boldface). NH2-EKPQEGTVVAVGPGR-OH with [U-13C6, 15N4]arginine was synthesized for GroES. Both peptides were checked for purity, and MRM transitions were validated prior to use (see Fig. S3 in the supplemental material) (46, 50–52). Samples were resuspended into 20 μl of 0.5% formic acid, and 2 μl of this mixture (500 ng) was injected onto a 100-μm by 100-mm C18 BEH column (Waters, Milford, MA). This represents the injection of 213 fmol of EsxA and 195 fmol of GroES heavy-isotope peptides. Protein digests were separated using a 90-min gradient from 2 to 35% of solvents A and B (A, 0.1% formic acid; B, acetonitrile–0.1% formic acid) at 650 nl/min on an Eksigent 2D Ultra nano-ultrahigh-performance liquid chromatography (UHPLC) system (Eksigent, Dublin, CA). MRM transitions were determined from empirical LC-tandem MC (MS/MS) data and existing MRM data we have previously published (19, 44, 52).The transition list is included as Table S2 in the supplemental material. Mass spectrometry was performed on a QTrap 6500 running in triple quadrupole mode (AB Sciex, Foster City, CA). Dwell times, collision energy (CE), voltage, and other parameters are listed in Table S2. Each sample was injected in triplicate, with biological duplicates. Targeted proteomics peak areas were integrated with a single three-point Gaussian smooth using the MQ4 algorithm within Multiquant, version 2.2 (AB Sciex). Protein loading and cell lysis were normalized as described previously (19, 53), and absolute quantification was facilitated by the use of the stable heavy-isotope internal standards. Peak areas and associated statistics are available in Fig. S4 to S7 in the supplemental material. Approximate lower limits of quantification (LLOQs) were determined by scaling the observed signal-to-noise (S/N) ratio of ESAT-6 stable isotope peptide to an S/N limit of 3:1 to 5:1.
RESULTS
Identification of a smooth M. marinum strain.
We generated a transposon insertion library in M. marinum M consisting of ∼25,000 independent transposon (Tn) insertion strains (44). Using this library, we sought to identify additional genes required for ESX-1 secretion in M. marinum. In M. marinum loss of some ESX-1-associated genes has been reported to lead to smooth-colony morphology (18). By examining the colony morphology of Tn-bearing strains, we identified M. marinum strain 14, which exhibited a smooth-colony morphology following growth on 7H11 agar compared to the rough, wild-type phenotype of the M strain (Fig. 1A). Note that M. marinum strain 14 had a shiny appearance compared to wild-type M. marinum (Fig. 1A).
FIG 1.
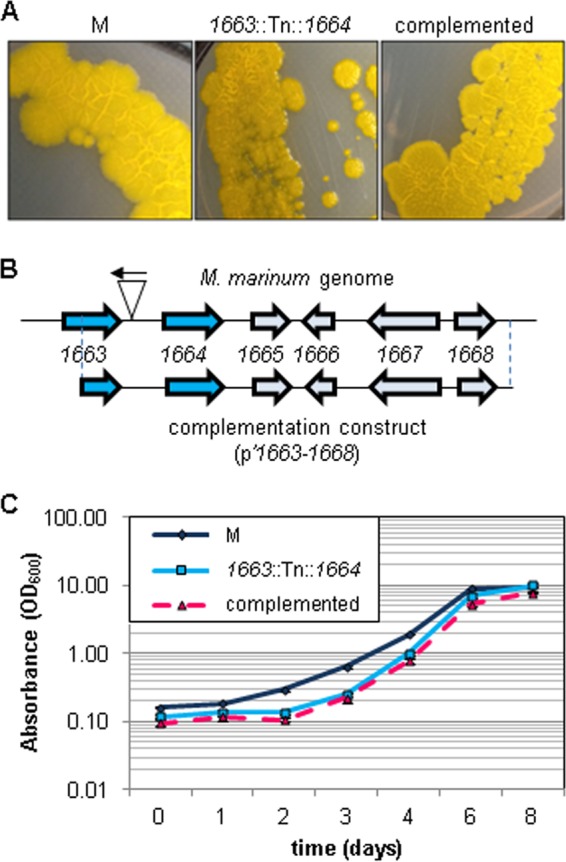
Identification and characterization of a smooth M. marinum strain. (A) Images of the indicated M. marinum strains grown on 7H11 agar plates showing the colony morphology phenotypes of the wild-type M strain, the Tn insertion strain, and the complemented strain. (B) The site of Tn insertion in the M. marinum strain 14 genome. The Tn inserted 120 bp upstream of the MMAR_1664 gene. The construct used for complementation is indicated below the locus and included the C-terminal half of the MMAR_1663 gene through the MMAR_1668 gene. (C) Growth as measured by absorbance at the OD600 of M. marinum cultures in 7H9 defined broth. Error bars represent standard deviations.
We identified the location of the Tn insertion in the M. marinum genome by isolating the Tn-containing DNA and defining the Mariner Tn/genome junction by DNA sequencing analysis (as described in Materials and Methods) (44). The Tn inserted between a T/A dinucleotide (bases 2010906/2010907) 120 bp upstream from the predicted start site for the MMAR_1664 gene as annotated by MycoBrowser (Fig. 1B) (45). We will refer to strain 14 as the 1663::Tn::1664 strain from this point forward to indicate the location of the Tn insertion in the M. marinum genome.
We attempted to determine which gene was transcriptionally affected by the Tn insertion using reverse transcriptase PCR. We were unable to detect transcription from MMAR_1663 or MMAR_1664 in wild-type M. marinum in vitro (data not shown).
To test if the colony morphology phenotype was due to the insertion of the Tn in the M. marinum genome, we introduced the region of the M. marinum M genome spanning from the middle of the MMAR_1663 gene through MMAR_1668 into a promoterless episomal plasmid (pMV206Hyg). We confirmed the presence of the complementation plasmid in the M. marinum transposon insertion strain using PCR. Introduction of the p′1663–1668 plasmid into the 1663::Tn::1664 M. marinum strain restored the rough-colony phenotype (Fig. 1A). From these data, we conclude that the transposon insertion was responsible for the smooth-colony phenotype observed in the 1663::Tn::1664 strain.
The orthologous locus in the M. tuberculosis genome includes several genes required for growth in vitro (54), including the ortholog of MMAR_1663, the gene directly upstream of the Tn insertion (Table 2). We therefore measured growth of the M, 1663::Tn::1664, and complemented strains in defined 7H9 broth to test if the 1663::Tn::1664 strain was defective for growth in vitro compared to the wild-type M strain. We observed a lag during logarithmic growth for the 1663::Tn::1664 strain compared to the M strain (Fig. 1C). Introduction of the complementation plasmid into the 1663::Tn::1664 strain failed to restore normal growth, likely because the complementation plasmid did not include all of the MMAR_1663 gene. Together, these data indicate that we have identified a novel locus in the M. marinum genome, disruption of which leads to a smooth-colony phenotype.
TABLE 2.
Comparison of the MMAR_1663-MMAR_1668 genomic regions in M. marinum and M. tuberculosis
| M. marinum gene | M. tuberculosis gene | % Amino acid identity | Proposed function (reference)a |
|---|---|---|---|
| MMAR_1663 | Rv3038c | 87 | Conserved hypothetical ortholog, MeT; essential for in vitro growth in M. tuberculosis (54) |
| MMAR_1664 | Rv3037c | 77 | Conserved hypothetical ortholog, SAM-MeT; essential for growth in macrophages in M. tuberculosis (63) |
| MMAR_1665 | Rv3036c (TB22.2) | 79 | Conserved hypothetical secreted/outer membrane protein; similar to MPT-64 |
| MMAR_1666 | Not conserved | NAb | Hypothetical membrane protein |
| MMAR_1667 | Rv3035 | 82 | Conserved protein; ortholog, pyrrolo-quinoline quinone/8-blade beta propeller |
| Essential for in vitro growth in M. tuberculosis (54) | |||
| MMAR_1668 | Rv3034c | 93 | Possible maltose O-transferase family; essential for in vitro growth in M. tuberculosis (54) |
Proposed functions collected from MycoBrowser, the TB Database, and NCBI BLAST. MeT, methyltransferase.
NA, not applicable.
Disruption of the MMAR_1663-MMAR_1668 locus leads to loss of ESX-1 export.
Because of the observed smooth-colony morphology phenotype, we tested if the 1663::Tn::1664 strain was deficient for ESX-1 export (18, 29). We measured ESX-1-dependent secretion of the EsxB and EsxA substrates into bacteriological medium in vitro using cell fractionation followed by Western blot analysis (Fig. 2A). The M strain produced and exported EsxA and EsxB into the culture filtrate (Fig. 2B). The deletion in the ΔRD1 M. marinum strain includes the esxBA genes, which encode EsxB and EsxA. Accordingly, the EsxB and EsxA proteins were not detected in the cell lysate or culture filtrate fractions. The 1663::Tn::1664 strain produced but did not export EsxB or EsxA, demonstrating a defect in ESX-1 export (Fig. 2B). The secretion of the Sec substrate, MPT-32, was not affected, indicating that the 1663::Tn::1664 strain was not generally defective for protein export. Despite the ability of the complementation plasmid to restore the colony morphology of the Tn insertion strain, it failed to restore ESX-1 secretion as measured by Western blot analysis (Fig. 2B).
FIG 2.
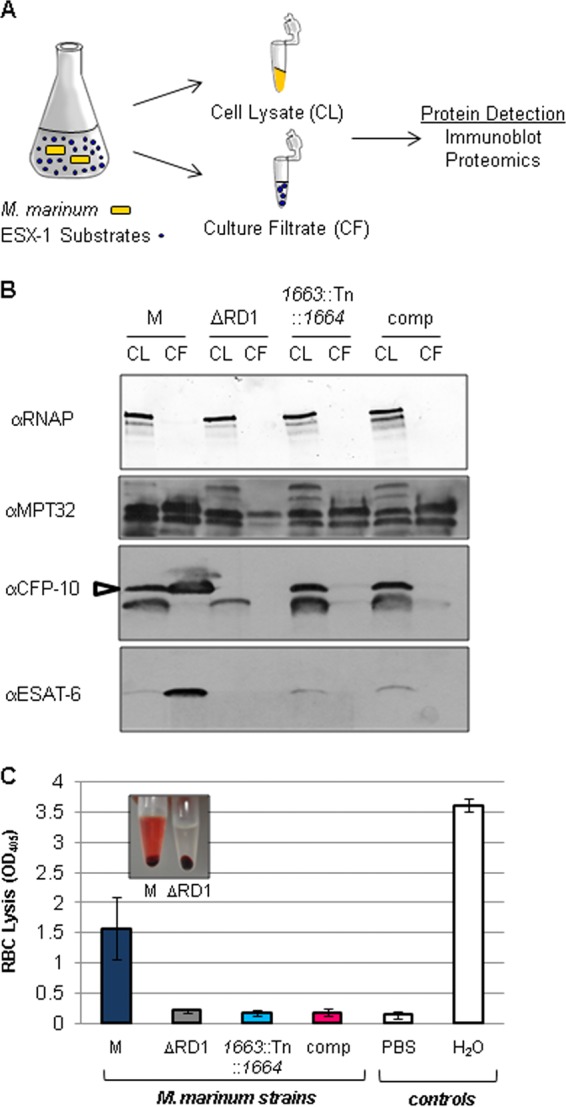
MMAR_1663-MMAR_1668 is required for ESX-1 export. (A) Schematic of the in vitro ESX-1 secretion assay. (B) M. marinum ESX-1 substrate production and secretion into the bacteriological medium as detected by Western blot analysis. The RNAP beta subunit served as a control for lysis and as a loading control for the cell lysate (CL); MPT-32 served as loading control for the culture filtrate (CF). The CFP-10 antibody detected the EsxB protein from M. marinum (indicated by the open arrow). A cross-reacting band is present below the EsxB band as determined by the lack of the top band in the ΔRD1 control strain. The ESAT-6 antibody detected the EsxA protein from M. marinum. The ΔRD1 strain served as a control for antibody specificity. α, anti; comp, complemented strain. (C) Hemolysis assay to measure ESX-1 function. PBS served as a negative control, and water served as a positive control for RBC lysis. Error bars represent the standard deviations. Representative experiments of three biological replicates are shown. The inset is a photograph of the hemolysis assay for the M and ΔRD1 strains.
Wild-type M. marinum lyses sheep red blood cells (sRBCs) in a contact-dependent, ESX-1-dependent manner (32, 55). Therefore, we also measured ESX-1 function by measuring hemolysis. The wild-type M strain lysed sRBCs, resulting in an increased OD405 (Fig. 2C, and inset). The ΔRD1 strain failed to lyse RBCs because of a nonfunctional ESX-1 system, resulting in an OD405 value comparable to that of the negative control for RBC lysis (PBS). Similarly, the M. marinum 1663::Tn::1664 strain failed to lyse RBCs, confirming a defect in the ESX-1 system. Consistent with our findings shown in Fig. 2B, the complementation plasmid did not restore hemolytic activity to the 1663::Tn::1664 strain. Taken together, these data indicate that the M. marinum 1663::Tn::1664 strain was deficient for ESX-1 export in vitro. However, although the complementation plasmid restored the colony morphology phenotype of the 1663::Tn::1664 strain, it did not restore ESX-1 export using accepted in vitro ESX-1 secretion assays.
The MMAR_1663-MMAR_1668 locus is required for M. marinum virulence.
The M. bovis BCG vaccine strain bears a genomic deletion that includes the RD1 locus, which carries several orthologs of genes required for ESX-1 export in M. tuberculosis (3–5, 56, 57). In M. bovis BCG, restoration of EsxA and EsxB export led to a visible change in M. bovis colony morphology (29). Because we were able to complement the M. marinum colony morphology phenotype but not the ESX-1 secretion defect, we sought to further characterize the 1663::Tn::1664 and complemented strains. With few exceptions, loss of ESX-1-dependent secretion as measured by the in vitro assays described in Fig. 2 correlates with loss of virulence in ex vivo and in vivo infection models (3, 5, 32, 42, 58). Based on our finding that the 1663::Tn::1664 strain was deficient for ESX-1-dependent secretion, we tested if this strain was attenuated for virulence in cell culture infection models.
We previously demonstrated that infection of Acanthamoeba castellanii with M. marinum led to amoeba lysis in an ESX-1-dependent manner (40). We infected amoebae with M. marinum strains at an MOI of 10 and measured cytolysis after 24 h using ethidium homodimer 1 (EthD-1). EthD-1 is a viability stain that cannot cross intact cellular membranes. Upon disruption of the cell membrane, EthD-1 binds DNA and emits red fluorescence. By 24 h postinfection wild-type M. marinum M lysed the amoebae, resulting in red fluorescence and destruction of the amoeba monolayer (see Fig. S1 in the supplemental material). Because the RD1 deletion strain (ΔRD1) is deficient for ESX-1 export (41), it did not lyse the amoeba monolayer and failed to lead to fluorescence of EthD-1 (see Fig. S1). Like the ΔRD1 strain, the 1663::Tn::1664 strain failed to lyse the amoeba monolayer and did not result in EthD-1 fluorescence. Importantly, the complemented strain lysed the amoeba monolayer and resulted in increased EthD-1 fluorescence (see Fig. S1).
Uptake, replication, and the cytosolic access of M. marinum leads to ESX-1-dependent cytolysis in macrophages (6, 8, 10, 32). We infected RAW 264.7 cells with M. marinum at an MOI of 10 and measured cytolysis at 24 h following infection. We measured cell viability using calcein-AM, a membrane-permeable dye that is cleaved by esterases in the cytoplasm of living cells. Following cleavage, the resulting product emits green fluorescence and is retained within the cell. EthD-1 was used to stain lysed RAW cells. As we found in the amoeba infection model, infection of RAW cells with wild-type M. marinum led to decreased viability (less green signal) and increased lysis (more red signal) compared to the uninfected control (Fig. 3A). The ΔesxBA strain is deficient for ESX-1 secretion and failed to lyse the macrophage monolayer to appreciable levels. Infection with the 1663::Tn::1664 strain resulted in phenotypes similar to infection with the ΔesxBA strain. As we observed for the amoeba infection, infection of RAW cells with the M. marinum complemented strain led to decreased viability and lysis of the RAW cell monolayer. By quantifying the EthD-1-stained cells (Fig. 3B), we conclude that the complementation plasmid partially restored the ability of the 1663::Tn::1664 strain to lyse RAW cells. Together, these data indicate that the MMAR_1663-MMAR_1668 region is essential for M. marinum virulence. Moreover, these data reveal that the complementation strain, which was phenotypically ESX-1 deficient by two independent in vitro measures of ESX-1 function, partially restored mycobacterial virulence.
FIG 3.
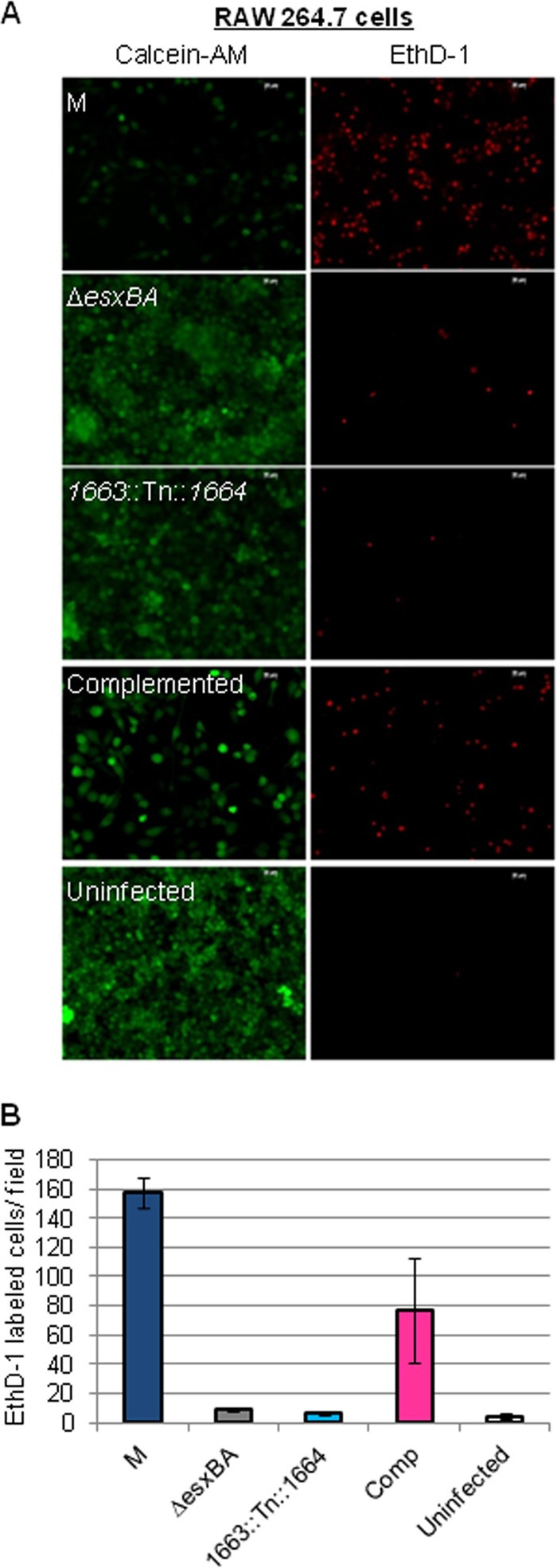
The MMAR_1663-MMAR_1668 region of the M. marinum genome is required for virulence. M. marinum infection of the RAW 264.7 cell line was performed at an MOI of 10. Images were acquired at 24 h postinfection with a 20× objective on a Zeiss Axio Observer microscope. Scale bar, 20 mm. EthD-1 staining showed membrane permeabilization of the macrophages, resulting in red fluorescence. Calcein-AM labeled live macrophages following infection. (B) Quantitation of the infection data shown in panel A. Red EthD-1-stained cells per field were counted; four independent fields from each infection were counted, and the counts were averaged. Error bars indicate the standard deviations between fields.
Cell-associated EsxAB are physiologically relevant for virulence.
An M. tuberculosis strain that failed to export ESAT-6 (EsxA) and CFP-10 (EsxB) into the culture supernatant in vitro but remained virulent in both cell-based and mouse models of infection was recently reported (42). One potential explanation for this finding was that the M. tuberculosis strain exported ESX-1 substrates to a level below the limits of detection of Western blot analysis (42). EsxA and EsxB have been detected not only in the bacteriological medium during in vitro growth but also in the cell wall and associated with the mycobacterial cell surface (28–31). Therefore, an alternate hypothesis to explain the observed disparity between ESX-1 export in vitro and virulence is that cell-associated EsxA and EsxB, not EsxA and EsxB in the culture supernatant, mediate virulence.
To test this hypothesis we measured ESX-1 substrates on the cell surface using a whole-colony MALDI-MS secretion assay we developed previously (44). For the whole-colony ESX-1 assay (Fig. 4A), the M. marinum strains were grown on Sauton's agar to induce ESX-1 export. We performed an aqueous protein extraction, which is highly enriched for proteins that are extrinsically associated with the mycobacterial cell. We measured the levels of EsxA and EsxB on the mycobacterial cell surface using MALDI-time of flight (TOF) MS (44). We detected high levels of both EsxA and EsxB on the surface of the wild-type M. marinum strain (Fig. 4B). We did not detect EsxA or EsxB on the surface of the ΔRD1 strain which bears a deletion including the esxBA genes. The eccCb::Tn M. marinum strain has a Tn insertion in the eccCb gene, which encodes an ATPase required for ESX-1-dependent secretion (3, 5, 18, 32). This strain produces but does not secrete EsxB and EsxA and served as a control for lysis in the MALDI assay (44). Accordingly, EsxB and EsxA were not detected on the surface of the eccCb::Tn strain. Consistent with the loss of ESX-1 export, EsxB and EsxA were not detected on the surface of the 1663::Tn::1664 strain. However, strong peaks for both EsxB and EsxA were detected in the proteins isolated from the surface of the complemented strain (Fig. 4B).
FIG 4.

Complementation partially restores the translocation of EsxA and EsxB to the mycobacterial cell surface. (A) Schematic of the whole-colony MALDI MS ESX-1 secretion assay. (B) MALDI peaks for EsxA and EsxB generated from surface proteins from M. marinum strains. The ΔRD1 strain lacks the genes encoding EsxA and EsxB and served as a control for specificity. The eccCb::Tn strain produces but does not export EsxA or EsxB and served as a control for lysis. (C) Differential quantitative mass spectrometry-based analysis of surface protein (SP) complementation from the wild-type, ΔRD1, mutant, and complemented strains. Relative quantification was determined by analysis of tryptic digests of washed colony extracts (surface proteins) compared using LC-MS/MS (MRM-based) measurement. Lysis and normalization were performed using levels of GroES and GroEL. The ΔRD1 strain served as a negative ESX-1 and lysis control. Standard error is shown from triplicate analysis. (D) Absolute levels of EsxA and GroES from colony surface proteins from the wild-type (WT), ΔRD1, 1663::Tn::1664, and complemented strains. Quantification was determined by LC-MS/MS (MRM-based) analysis of tryptic digests with the addition of 13C15N stable heavy-isotope versions of peptides from each protein. EsxA values shown are normalized to the GroES values shown. Standard error is shown from triplicate analyses. Large colonies yielded about 10 to 20 μg of total soluble protein.
To address the question of the magnitude of complementation observed at the cell surface, we used targeted mass spectrometry with heavy-isotope standards to probe the surface-associated EsxA and EsxB proteome quantitatively. Surface proteins were collected from each strain as in the MALDI assay. We readily detected EsxA and EsxB in surface protein fractions from the wild-type M strain, consistent with the MALDI detection (Fig. 4C). We did not detect EsxA on the surface of the ΔRD1 strain as expected. We detected low levels of EsxB on the cell surface of the ΔRD1 strain, corresponding to 5% of the EsxB present on the surface of the wild-type strain. Low levels of EsxA and EsxB were present on the cell surface of the M. marinum 1663::Tn::1664 strain, corresponding to 31% and 19%, respectively, of the EsxA and EsxB present on the surface of the wild-type strain. Indeed, small peaks corresponding to both EsxA and EsxB were present in the MALDI spectra for this strain. Consistent with the MALDI data shown in Fig. 4B, the complemented strain showed a partial restoration of both EsxA and EsxB, corresponding to 53% and 70% of the wild-type levels, respectively. Based on absolute quantification of EsxA in the surface protein fractions, we confirmed modest but significant complementation of EsxA on the surface of the complemented strain (Fig. 4D). When normalized to typical Western blot loading controls, low-level complementation of EsxA was measured (17.9 ng/colony in the complemented versus 10.4 ng/colony in the 1663::Tn::1664 strain). Together, these data indicate that complementation of the 1663::Tn::1664 strain led to a partial restoration of ESX-1-mediated transport of EsxB and EsxA to the surface of the mycobacterial cell.
Complementation does not restore EsxA and EsxB secretion to the culture supernatant.
To definitively show that the complemented strain failed to restore secretion of EsxA and EsxB into the culture supernatant, we prepared protein fractions as described in the ESX-1 secretion assay in Fig. 2A. We then used the targeted detection of EsxA and EsxB proteins with an MRM-based proteomics approach to quantify the production and secretion of EsxA and EsxB into the culture supernatant (19). To determine the absolute quantity of EsxA and GroES and to define the lower limits of detection and sensitivity of this approach, we included stable heavy-isotope versions of tryptic peptides from EsxA and GroES as absolute internal standards to utilize stable-isotope dilution quantitation.
As shown in Fig. 5A, we readily detected the production and secretion of EsxA and EsxB into the culture supernatant by the wild-type M strain. The production and secretion of EsxA corresponded to 5,828 ± 332 ng/mg of whole-cell lysate and 1.056 × 104 ± 634 ng/mg of culture supernatant (Fig. 5A and B). Deletion of the RD1 region resulted in loss of detection of EsxA in both the whole-cell lysate and the culture supernatant. In the ΔRD1 strain, we observed a 50-fold decrease in the levels of EsxB in the cell lysate and a 500-fold decrease in the levels of EsxB in the culture supernatant. The residual signals were likely due to a second identical EsxB protein in M. marinum encoded outside the RD1 locus (MMAR_0187), which prevented us from accurately quantifying EsxB using stable heavy-isotope peptides, and was also observed in the data shown in Fig. 4B and C. For both EsxA and EsxB, the 1663::Tn::1664 strain and the complemented strains showed a 30 to 40% decrease in the protein levels in the cell lysate. This decrease corresponded to 3,143 ± 204 ng/mg and 3,001 ± 93 ng/mg of cell lysate for the 1663::Tn::1664 and complemented strains, respectively. Secretion levels of EsxA and EsxB were comparable in the culture supernatant to levels less than 1% of the wild-type strain. Absolute quantitation of EsxA in the culture supernatant was 28 ± 4.3 ng/mg and 40 ± 3.7 ng/mg of culture supernatant for the transposon insertion and complemented strains, respectively, which does not appear significant compared to the wild-type strain.
FIG 5.

Differential and absolute quantification of ESX-1 substrates in M. marinum. Relative levels of EsxA, EsxB, and MMAR_2929 in whole-cell lysates and culture supernatants from the ΔRD1, 1663::Tn::1664, and complemented M. marinum strains were compared to levels of the wild-type M strain. Error bars represent the average percent coefficient of variation of the cell lysates (CL) and culture filtrates (CF). (B) Absolute levels of EsxA and GroES in whole-cell lysates and culture supernatants generated from the wild-type, ΔRD1, 1663::Tn::1664, and complemented strains. Quantification was determined by analyzing tryptic digests by LC-MS/MS triple-quadrupole MRM-based proteomics with 13C15N stable heavy-isotope versions of peptides from each protein. Our linear dynamic range was 4 orders of magnitude, and the LLOQs were approximately 2.5 ng/mg for EsxA and 250 pg/mg for GroES. Error shown is the percent coefficient of variation of the measured value. *, MRM value; **, bicinchoninic acid protein assay of bulk material.
These experiments were controlled by measuring relative changes in MMAR_2929, a secreted protein bearing an Sec signal sequence (59) (Fig. 5A). We observed a relative increase in MMAR_2929 in culture supernatants generated from both the 1663::Tn::1664 and the complemented strains compared to the wild-type strain. Levels of GroES were used as an internal control and were extremely consistent between strains, with an average quantity of 7,160 ng ± 475 ng of GroES/mg of total cell lysate (Fig. 5B). Levels of GroES were used as a lysis marker in culture supernatants and were consistent for all strains other than the ΔRD1 strain, which exhibited less lysis and was corrected for in subsequent quantification. EsxA and EsxB have large signal-to-noise ratios by targeted proteomics in the wild-type M strain (>15,000:1 at 3σ), and we observed more than 3 orders of linear dynamic range, compared to an exact isotopic standard. Based on these data, we estimate the lower limits of detection for this assay of EsxA to be better than 3 ng/mg of culture filtrate. We would thus reliably be able to detect and quantify EsxA production and export at less than 0.05% of the levels we detected from the wild-type strain. Together, these findings demonstrate that the complementation of the Tn insertion in the MMAR_1663-MMAR_1668 locus restored the localization of EsxA and EsxB to the mycobacterial cell surface but not the export of EsxA and EsxB into the culture supernatant.
DISCUSSION
The presence of EsxA, EsxB, and other ESX-1 substrates in the bacteriological medium has been routinely used as a measure of ESX-1 export in several mycobacterial species. This assay has been central in defining the components and the substrates of the ESX-1 system as well as the molecular mechanisms underlying substrate selection and export (initial examples include references 3 and 5). Indeed, in the vast majority of reports, the secretion of the EsxA and EsxB substrates into bacteriological medium matches the virulence phenotype of the mycobacterial strain. The presence of EsxA and EsxB in the culture supernatant correlates with virulence while the absence of these proteins correlates with attenuation. There are four reports in which in vitro ESX-1 secretion as detected by Western blot analysis on culture filtrates did not match the virulence phenotypes in a variety of infection models (42, 58, 60, 61). In three of these reports, M. tuberculosis strains that still secreted EsxA and EsxB into the bacteriological medium were attenuated for virulence (58, 60, 61). In 2013, Chen et al. constructed EspA variants in M. tuberculosis which resulted in a loss of ESX-1 export into the culture supernatant in vitro by Western blot analysis, yet the strains were as virulent as wild-type M. tuberculosis in both mouse and macrophage infection models (42). Here, in our attempts to complement an M. marinum transposon insertion strain with a defect in ESX-1 export, we uncovered a disparity similar to that reported in the study of Chen et al.: loss of EsxA and EsxB export into the culture supernatant did not correspond to attenuation. This is the first example of a disparity between in vitro export and virulence in M. marinum. Our studies indicate that the disparity observed is a direct consequence of the assay used to measure ESX-1 export. We reconciled this apparent disparity by measuring ESX-1-mediated translocation of EsxA and EsxB to the M. marinum cell surface (44). We have definitively shown that in the complemented strain, EsxA and EsxB were exported to the cell surface of M. marinum but not released into the bacteriological medium.
In M. tuberculosis, both EsxA (ESAT-6) and EsxB (CFP-10) have been localized to the cell wall and to the cell surface (28–31). Moreover, the exogenous addition of EsxA to ESX-1-deficient, irradiated M. tuberculosis H37Rv led to reassociation of EsxA to the cell surface and lytic activity against alveolar cells (28). Here, we provide the first example of a mycobacterial strain that translocates the EsxA and EsxB proteins to the cell surface but not into the bacteriological medium in vitro. We show that this M. marinum strain is virulent and retains the ability to infect and cause cytolysis of both macrophages and amoebae, ESX-1-mediated functions that have been ascribed to EsxA (4, 28, 39). Interestingly, we found that EsxB was more readily detected on the mycobacterial cell surface than EsxA. This result is in line with the study of Kinhikar et al. which identified CFP-10 (EsxB) on the cell surface of M. tuberculosis but reported a paucity of ESAT-6 (EsxA) (28).
It is not clear if the ESX-1 substrates are true exoproteins (released from the cell) or extrinsically associated with the MOM and shed, either actively or passively, into the bacteriological medium in vitro. We propose, in line with the findings by Kinhikar et al., that it is likely that translocation of EsxA and EsxB to the mycobacterial cell surface, rather than the active secretion or passive shedding into the bacteriological medium, correlates with virulence in M. marinum (28, 29). While it is not yet resolved if ESX-1 substrates remain cell associated in vivo, association of EsxA and EsxB with the mycobacterial cell surface may explain why the active export of ESX-1 substrates into the phagosome or cytosol of the macrophage has not been routinely observed.
In considering why the complemented strain exported ESX-1 substrates to the cell surface but did not release them into the culture supernatant, we can propose at least two ideas. First, the release of EsxA and EsxB into the culture medium could be active, and the locus we have identified could be involved in this process. However, if this model were correct, we would have expected to see a partial restoration of EsxA and EsxB in the culture supernatant. Alternatively, the levels of EsxA/B on the cell surface in the complemented strain are lower than those in the wild-type strain. It could simply be that ESX-1 substrates are passively shed in the culture supernatant. Less protein on the surface could translate to a lack of shedding into the medium.
Genetic complementation in mycobacteria is often difficult. We show here that complementation is not straightforward and that a negative result should not be taken to mean that complementation has failed. Clearly, the success of the complementation depends highly on the assay used for detection. Although we achieved partial complementation, this strain served as a valuable tool in dissecting how commonly used ESX-1 export assays correlate with each other and with virulence. Our study also raises the question of how widely used in vitro assays reflect ESX-1 export that occurs within a host cell. Based on our findings, colony morphology phenotypes and whole-colony secretion assays correlate with M. marinum virulence. The correlation between hemolytic activity and secretion of ESX-1 substrates into the bacteriological medium with virulence is less clear.
Finally, we report a novel locus outside the extended RD1 region in the M. marinum genome that is required for ESX-1-mediated export. Importantly, our study proves that there are additional genes outside the extended RD1 locus which affect ESX-1 secretion in M. marinum. There have been several genetic screens designed to identify genes required for ESX-1 export and ESX-1 substrates. There has never been a direct, saturating screen to identify all of the known ESX-1 components or substrates in any organism. Indeed, screens to find secreted proteins have specifically missed ESX-1 substrates (62). A screen for ESX-1 genes in M. marinum using hemolysis assays identified genes only in the extended RD1 locus (32). This screen was not saturating and, based on our findings here (Fig. 2B), may not reflect the type of ESX-1 export that mediates virulence. Hemolysis has historically correlated with both virulence and the presence of ESX-1 substrates in the culture supernatant (see, for example, reference 32). Here, we report the first disparity between these assays. Importantly, the partially complemented strain was nonhemolytic but caused intermediate levels of cytolysis in macrophage-like cells. These findings highlight the point that the mechanisms underlying hemolysis and cytolysis may be distinct.
Reintroduction of the 3′ half of MMAR_1663 and the entirety of MMAR_1664-MMAR_1668 to the 1663::Tn::1664 Tn insertion strain led to a restoration of colony morphology, a partial restoration of the translocation of EsxA and EsxB to the M. marinum surface, and a partial restoration of ESX-1-mediated virulence in cell culture models of infection. As shown in Table 2, several genes surrounding the transposon insertion are essential for growth in vitro in M. tuberculosis, including the ortholog of the gene immediately upstream of the transposon insertion, MMAR_1663 (54). The transposon insertion strain exhibited a lag in growth compared to the wild-type M strain, indicating that the transposon likely had some effect on MMAR_1663 (Fig. 1C). The complementation plasmid contained the 3′ half of the MMAR_1663 gene and failed to restore the growth phenotype (Fig. 1B and C). Yet the complementation plasmid partially restored virulence and export (Fig. 3 and Fig. 4), implicating genes other than MMAR_1663 in ESX-1 export. Importantly, these findings demonstrate that the lag in growth was not entirely responsible for the attenuation observed in both amoebae and macrophages. While we have not determined which individual gene or genes are specifically required for ESX-1 secretion and virulence in this study, based on the position of the transposon insertion and the orientation of the annotated genes on the genome (45), it is probable that either MMAR_1664, MMAR_1665, or both genes are required.
In M. tuberculosis, the ortholog of MMAR_1664 is Rv3037c (77% identical at the amino acid level) (Table 2). The closest orthologs of MMAR_1664 and Rv3037c resemble S-adenosylmethionine (SAM)-dependent methyltransferases (MeTs). Rv3037c was identified as required for M. tuberculosis infection of macrophages using the transposon site hybridization (TraSH) approach (63). It is not known how Rv3037c contributes to M. tuberculosis virulence, but this gene has not previously been linked to ESX-1 export. We would hypothesize, based on our findings, that the Rv3037c gene and/or other genes at the locus promote ESX-1 export in M. tuberculosis.
MMAR_1665 is also conserved in M. tuberculosis. MMAR_1665 is 79% identical to Rv3036c (TB22) at the amino acid level. TB22 is a protein likely secreted by the Sec secretion system based on the presence of a predicted N-terminal signal sequence (59). Because we were unsuccessful in determining if MMAR_1664 and MMAR_1665 are operonic, the contribution of MMAR_1665 to ESX-1 is unclear.
Finally, considering that the Tn insertion mapped to an intergenic region, there remains a formal possibility that the transposon insertion disrupted a small regulatory RNA located between MMAR_1663 and MMAR_1664. Determining the precise mechanism of how the transposon disrupts ESX-1 export will be the focus of future investigations.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this study was supported by the National Institute of Allergies and Infectious Diseases of the National Institutes of Health under award numbers R21AI092484 and R01AI106872 to P.A.D.C. G.M.K. is supported by the GLOBES fellowship program funded by Integrative Graduate Education, Research, and Traineeship Training Grant 504495 from the National Science Foundation. G.C.H. was supported by a Summer Undergraduate Research Fellowship from Notre Dame's College of Science. F.M.M. is supported by a Postdoctoral Fellowship from the Eck Institute of Global Health at the University of Notre Dame.
We thank the Mass Spectrometry and Proteomics Facility and the Genomics and Bioinformatics Facility at the University of Notre Dame for their assistance. We thank members of the Champion Lab and faculty from the Department of Biological Sciences for critical feedback.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 7 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01502-14.
REFERENCES
- 1.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. U. S. A. 102:1169–1174. 10.1073/pnas.0405620102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J. Bacteriol. 190:7004–7011. 10.1128/JB.00458-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 51:359–370. 10.1046/j.1365-2958.2003.03844.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U. S. A. 100:12420–12425. 10.1073/pnas.1635213100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A. 100:13001–13006. 10.1073/pnas.2235593100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 14:1287–1298. 10.1111/j.1462-5822.2012.01799.x [DOI] [PubMed] [Google Scholar]
- 7.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480. 10.1016/j.chom.2012.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 8:e1002507. 10.1371/journal.ppat.1002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanley SA, Johndrow JE, Manzanillo P, Cox JS. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178:3143–3152 [DOI] [PubMed] [Google Scholar]
- 10.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129:1287–1298. 10.1016/j.cell.2007.05.059 [DOI] [PubMed] [Google Scholar]
- 11.Akpe San Roman S, Facey PD, Fernandez-Martinez L, Rodriguez C, Vallin C, Del Sol R, Dyson P. 2010. A heterodimer of EsxA and EsxB is involved in sporulation and is secreted by a type VII secretion system in Streptomyces coelicolor. Microbiology 156:1719–1729. 10.1099/mic.0.037069-0 [DOI] [PubMed] [Google Scholar]
- 12.Coros A, Callahan B, Battaglioli E, Derbyshire KM. 2008. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol. Microbiol. 69:794–808. 10.1111/j.1365-2958.2008.06299.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flint JL, Kowalski JC, Karnati PK, Derbyshire KM. 2004. The RD1 virulence locus of Mycobacterium tuberculosis regulates DNA transfer in Mycobacterium smegmatis. Proc. Natl. Acad. Sci. U. S. A. 101:12598–12603. 10.1073/pnas.0404892101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fyans JK, Bignell D, Loria R, Toth I, Palmer T. 2013. The ESX/type VII secretion system modulates development, but not virulence, of the plant pathogen Streptomyces scabies. Mol. Plant Pathol. 14:119–130. 10.1111/j.1364-3703.2012.00835.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray TA, Krywy JA, Harold J, Palumbo MJ, Derbyshire KM. 2013. Distributive conjugal transfer in mycobacteria generates progeny with meiotic-like genome-wide mosaicism, allowing mapping of a mating identity locus. PLoS Biol. 11:e1001602. 10.1371/journal.pbio.1001602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baptista C, Barreto HC, Sao-Jose C. 2013. High levels of DegU-P activate an Esat-6-like secretion system in Bacillus subtilis. PLoS One 8:e67840. 10.1371/journal.pone.0067840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect. Immun. 74:88–98. 10.1128/IAI.74.1.88-98.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsson F, Joshi SA, Rangell L, Brown EJ. 2009. Polar localization of virulence-related Esx-1 secretion in mycobacteria. PLoS Pathog. 5:e1000285. 10.1371/journal.ppat.1000285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Champion PA, Champion MM, Manzanillo P, Cox JS. 2009. ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol. Microbiol. 73:950–962. 10.1111/j.1365-2958.2009.06821.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JM, Boy-Rottger S, Dhar N, Sweeney N, Buxton RS, Pojer F, Rosenkrands I, Cole ST. 2012. EspD is critical for the virulence-mediating ESX-1 secretion system in Mycobacterium tuberculosis. J. Bacteriol. 194:884–893. 10.1128/JB.06417-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Converse SE, Cox JS. 2005. A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J. Bacteriol. 187:1238–1245. 10.1128/JB.187.4.1238-1245.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daleke MH, van der Woude AD, Parret AH, Ummels R, de Groot AM, Watson D, Piersma SR, Jimenez CR, Luirink J, Bitter W, Houben EN. 2012. Specific chaperones for the type VII protein secretion pathway. J. Biol. Chem. 287:31939–31947. 10.1074/jbc.M112.397596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 102:10676–10681. 10.1073/pnas.0504922102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacGurn JA, Raghavan S, Stanley SA, Cox JS. 2005. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol. Microbiol. 57:1653–1663. 10.1111/j.1365-2958.2005.04800.x [DOI] [PubMed] [Google Scholar]
- 25.Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, Cox JS. 2010. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 7:210–220. 10.1016/j.chom.2010.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. 2008. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454:717–721. 10.1038/nature07219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chagnot C, Zorgani MA, Astruc T, Desvaux M. 2013. Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front. Microbiol. 4:303. 10.3389/fmicb.2013.00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, Hsu T, Jacobs WR, Jr, Laal S. 2010. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol. Microbiol. 75:92–106. 10.1111/j.1365-2958.2009.06959.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46:709–717. 10.1046/j.1365-2958.2002.03237.x [DOI] [PubMed] [Google Scholar]
- 30.Berthet FX, Rasmussen PB, Rosenkrands I, Andersen P, Gicquel B. 1998. A Mycobacterium tuberculosis operon encoding ESAT-6 and a novel low-molecular-mass culture filtrate protein (CFP-10). Microbiology 144:3195–3203. 10.1099/00221287-144-11-3195 [DOI] [PubMed] [Google Scholar]
- 31.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. 2005. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J. Immunol. 174:3570–3579 [DOI] [PubMed] [Google Scholar]
- 32.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. 2004. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol. Microbiol. 53:1677–1693. 10.1111/j.1365-2958.2004.04261.x [DOI] [PubMed] [Google Scholar]
- 33.Shiloh MU, DiGiuseppe Champion PA. 2010. To catch a killer. What can mycobacterial models teach us about Mycobacterium tuberculosis pathogenesis? Curr. Opin. Microbiol. 13:86–92. 10.1016/j.mib.2009.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PL, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, Cole ST. 2008. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18:729–741. 10.1101/gr.075069.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tobin DM, Ramakrishnan L. 2008. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol. 10:1027–1039. 10.1111/j.1462-5822.2008.01133.x [DOI] [PubMed] [Google Scholar]
- 36.Stoop EJ, Schipper T, Huber SK, Nezhinsky AE, Verbeek FJ, Gurcha SS, Besra GS, Vandenbroucke-Grauls CM, Bitter W, van der Sar AM. 2011. Zebrafish embryo screen for mycobacterial genes involved in the initiation of granuloma formation reveals a newly identified ESX-1 component. Dis. Model Mech. 4:526–536. 10.1242/dmm.006676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi SA, Ball DA, Sun MG, Carlsson F, Watkins BY, Aggarwal N, McCracken JM, Huynh KK, Brown EJ. 2012. EccA1, a component of the Mycobacterium marinum ESX-1 protein virulence factor secretion pathway, regulates mycolic acid lipid synthesis. Chem. Biol. 19:372–380. 10.1016/j.chembiol.2012.01.008 [DOI] [PubMed] [Google Scholar]
- 38.McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, Cox JS, Brown EJ. 2007. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 3:e105. 10.1371/journal.ppat.0030105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY. 2008. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect. Immun. 76:5478–5487. 10.1128/IAI.00614-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy GM, Morisaki JH, Champion PA. 2012. Conserved mechanisms of Mycobacterium marinum pathogenesis within the environmental amoeba, Acanthamoeba castellanii. Appl. Environ. Microbiol. 78:2049–2052. 10.1128/AEM.06965-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. 2004. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2:e367. 10.1371/journal.pbio.0020367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen JM, Zhang M, Rybniker J, Basterra L, Dhar N, Tischler AD, Pojer F, Cole ST. 2013. Phenotypic profiling of Mycobacterium tuberculosis EspA point mutants reveals that blockage of ESAT-6 and CFP-10 secretion in vitro does not always correlate with attenuation of virulence. J. Bacteriol. 195:5421–5430. 10.1128/JB.00967-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao LY, Groger R, Cox JS, Beverley SM, Lawson EH, Brown EJ. 2003. Transposon mutagenesis of Mycobacterium marinum identifies a locus linking pigmentation and intracellular survival. Infect. Immun. 71:922–929. 10.1128/IAI.71.2.922-929.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Champion MM, Williams EA, Kennedy GM, Champion PA. 2012. Direct detection of bacterial protein secretion using whole colony proteomics. Mol. Cell. Proteomics 11:596–604. 10.1074/mcp.M112.017533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kapopoulou A, Lew JM, Cole ST. 2011. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb.) 91:8–13. 10.1016/j.tube.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 46.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. 2003. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U. S. A. 100:6940–6945. 10.1073/pnas.0832254100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edmondson RD, Russell DH. 1996. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass measurement accuracy by using delayed extraction. J. Am. Soc. Mass Spectrom. 7:995–1001. 10.1016/1044-0305(96)00027-X [DOI] [PubMed] [Google Scholar]
- 48.Gooyit M, Peng Z, Wolter WR, Pi H, Ding D, Hesek D, Lee M, Boggess B, Champion MM, Suckow MA, Mobashery S, Chang M. 2014. A chemical biological strategy to facilitate diabetic wound healing. ACS Chem. Biol. 9:105–110. 10.1021/cb4005468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rund SS, Bonar NA, Champion MM, Ghazi JP, Houk CM, Leming MT, Syed Z, Duffield GE. 2013. Daily rhythms in antennal protein and olfactory sensitivity in the malaria mosquito Anopheles gambiae. Sci. Rep. 3:2494. 10.1038/srep02494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schubert OT, Mouritsen J, Ludwig C, Rost HL, Rosenberger G, Arthur PK, Claassen M, Campbell DS, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann SH, Moritz RL, Aebersold R. 2013. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13:602–612. 10.1016/j.chom.2013.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun L, Li Y, Champion MM, Zhu G, Wojcik R, Dovichi NJ. 2013. Capillary zone electrophoresis-multiple reaction monitoring from 100 pg of RAW 264.7 cell lysate digest. Analyst 138:3181–3188. 10.1039/c3an00287j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Wojcik R, Dovichi NJ, Champion MM. 2012. Quantitative multiple reaction monitoring of peptide abundance introduced via a capillary zone electrophoresis-electrospray interface. Anal. Chem. 84:6116–6121. 10.1021/ac300926h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Champion PA, Stanley SA, Champion MM, Brown EJ, Cox JS. 2006. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313:1632–1636. 10.1126/science.1131167 [DOI] [PubMed] [Google Scholar]
- 54.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7:e1002251. 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King CH, Mundayoor S, Crawford JT, Shinnick TM. 1993. Expression of contact-dependent cytolytic activity by Mycobacterium tuberculosis and isolation of the genomic locus that encodes the activity. Infect. Immun. 61:2708–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284:1520–1523. 10.1126/science.284.5419.1520 [DOI] [PubMed] [Google Scholar]
- 57.Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. 1999. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol. Microbiol. 32:643–655. 10.1046/j.1365-2958.1999.01383.x [DOI] [PubMed] [Google Scholar]
- 58.Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, Ramsdell TL, Lopez MF, Behar SM, Sarracino DA, Fortune SM. 2010. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog. 6:e1000957. 10.1371/journal.ppat.1000957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 60.Bottai D, Majlessi L, Simeone R, Frigui W, Laurent C, Lenormand P, Chen J, Rosenkrands I, Huerre M, Leclerc C, Cole ST, Brosch R. 2011. ESAT-6 secretion-independent impact of ESX-1 genes espF and espG1 on virulence of Mycobacterium tuberculosis. J. Infect. Dis. 203:1155–1164. 10.1093/infdis/jiq089 [DOI] [PubMed] [Google Scholar]
- 61.Brodin P, de Jonge MI, Majlessi L, Leclerc C, Nilges M, Cole ST, Brosch R. 2005. Functional analysis of early secreted antigenic target-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex-formation, virulence and immunogenicity. J. Biol. Chem. 280:33953–33959. 10.1074/jbc.M503515200 [DOI] [PubMed] [Google Scholar]
- 62.McCann JR, McDonough JA, Sullivan JT, Feltcher ME, Braunstein M. 2011. Genome-wide identification of Mycobacterium tuberculosis exported proteins with roles in intracellular growth. J. Bacteriol. 193:854–861. 10.1128/JB.01271-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 102:8327–8332. 10.1073/pnas.0503272102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.