

Abstract
In this study we demonstrate a new form of immunoregulation: engagement on CD4+ T cells of the complement regulator CD46 promoted the effector potential of T helper type 1 cells (TH1 cells), but as interleukin 2 (IL-2) accumulated, it switched cells toward a regulatory phenotype, attenuating IL-2 production via the transcriptional regulator ICER/CREM and upregulating IL-10 after interaction of the CD46 tail with the serine-threonine kinase SPAK. Activated CD4+ T cells produced CD46 ligands, and blocking CD46 inhibited IL-10 production. Furthermore, CD4+ T cells in rheumatoid arthritis failed to switch, consequently producing excessive interferon-γ (IFN-γ). Finally, γδ T cells, which rarely produce IL-10, expressed an alternative CD46 isoform and were unable to switch. Nonetheless, coengagement of T cell antigen receptor (TCR) γδ and CD46 suppressed effector cytokine production, establishing that CD46 uses distinct mechanisms to regulate different T cell subsets during an immune response.
The suppression of immune responses to self antigen is vital to the limitation of autoimmunity. Similarly, the timely contraction of T cell responses to infection is critical for protection against immuno-pathologies arising from exuberant inflammation. The cytokine interleukin 10 (IL-10) is critical in immunosuppression1, inhibiting production of the proinflammatory cytokines tumor necrosis factor and IL-12 in macrophages and dendritic cells1,2 and suppressing the production of IL-2 and interferon-γ (IFN-γ) by effector T cells3. Thus, Il10−/− mice succumb to colitis because of their inability to regulate immune responses to gut flora4, and susceptibility to colitis has likewise been reported for human families carrying mutations in genes encoding IL-10 receptor chains5. Moreover, whereas IL-10 deficiency in mice accelerates clearance of infection with Toxoplasma gondii or Trypanosoma cruzi, such mice succumb to tissue damage caused by overproduction of proinflammatory cytokines2,6.
IL-10 can be produced by many cell types, including dendritic cells, macrophages, B cells and T cells, among which T helper type 2 (TH2) cells and adaptive regulatory T cells (Treg cells), such as type 1 Treg cells (Tr1 cells), have been suggested to be chief sources1,7,8. However, uncertainty as to the nature of critical IL-10-producing cells in vivo, particularly in humans, has been fueled by the realization that under certain conditions, large amounts of IL-10 can be secreted by some natural Treg cells9, IL-17-producing helper T cells10 and TH1 cells11–14. Indeed, IL-10-producing TH1 cells have sparked much interest because they seem to be key in regulating immune responses to certain infections14–17 and because their induction might be a mechanism by which tolerance is induced in the presence of persistent (self) antigen18. Thus, there is particular interest in understanding what regulates IL-10 production by TH1 cells.
CD46 is a ubiquitously expressed human type I transmembrane glycoprotein originally identified as a complement-regulatory protein. Coengagement of the T cell antigen receptor (TCR) and CD46 on human CD4+ T cells induces substantial IL-10 secretion, moderate IFN-γ production and expression of granzyme B and perforin19,20. Indeed, that phenotype, plus an IL-10-dependent, transcription factor Foxp3–independent ability to suppress bystander effector T cells, establishes the similarity of T cells activated with antibody to CD3 (anti-CD3) and anti-CD46 to Tr1 cells21. Moreover, both CD46-dependent induction of IL-10 and generation of Tr1 cells are highly dependent on exogenous IL-2 (refs.7,19,21), which suggests that CD46 might naturally be a key factor in the switch of TH1 cells to a Tr1-like phenotype; however, this has not been investigated directly.
CD46 binds the opsonins C3b and C4b and functions as a cofactor in their proteolytic degradation by serine protease factor I (ref. 22). CD46 also functions as a receptor for several important human pathogens, such as Streptococcus pyogenes23,24. Four isoforms of CD46 arise by alternative splicing25. All isoforms contain four conserved complement control repeats, followed by variant forms of a highly O-glycosylated region (designated B and C), a transmembrane anchor, and one of two possible cytoplasmic domains, designated CYT-1 and CYT-2. Thus, the four commonly expressed CD46 isoforms are BC1 (denoting glycosylated regions B and C connected to CYT-1), BC2, C1 and C2. Both CYT-1 and CYT-2 contain kinase substrates and are tyrosine-phosphorylated after CD46 crosslinking of human CD4+ T cells26, which also activates the TCR adaptor proteins p120-CBL, Lat23 and Vav, the small GTPase Rac and the kinase Erk24, and additionally regulates expression of the receptor tyrosine kinase Flt3 and its ligand27. Thus CD46 activation has considerable intrinsic potential to regulate T cell function and has been reported as a T cell costimulator.
Notably, the interaction of T cells with antigen-presenting cells induces the secretion of complement proteins (such as C3, factor B and factor D) and the subsequent generation of complement-activation fragments28–30 by both cell populations. Such a scenario might provide local ligands for CD46, thereby affecting cell fate. Indeed, CD46 crosslinking by C3b and C4b during TCR activation can induce Tr1-like cells19. Likewise, the presence of S. pyogenes during TCR activation in vitro promotes IL-10-secreting Tr1-like cells31. Nonetheless, cultures of Tr1-like cells induced by CD46 engagement paradoxically also produce large amounts of IFN-γ and have other features of TH1 cells32, which again raises the question of whether CD46 engagement is critically involved in TH1-Tr1 switching19.
Although activation of conventional CD4+ T cells defines the adaptive response, it is now appreciated that unconventional T cells, of which γδ cells are a prototype, contribute substantially to the early phases of immune responses. Although such cells proliferate and show rapid and effusive effector function, little is known about how their potential to promote immunopathology is controlled. Indeed, there is no clear case for the existence of TCRγδ+Foxp3+ Treg cells under normal circumstances33 and scant evidence for the induction of IL-10+ Tr1-like γδ cells. Given the potential of CD46-mediated signals to induce IL-10 in adaptive T cells, it would be relevant to examine whether or not γδ cells are responsive to CD46-TCR costimulation as well.
Finally, we also considered the importance in vivo of CD46-mediated regulation of T cells by examining patients with inflammatory disease. By further analyzing the CD46-mediated signals that regulate cytokine production in human conventional and unconventional T cells, we establish here that the IL-2-dependent coactivation of human CD4+ T cells by TCR-CD46 was a powerful means by which to promote TH1 cells and then to switch them to IL-10 production. This pathway was absent from γδ T cells and was impaired in rheumatoid arthritis. At the same time, CD46 showed additional modes of immunosuppression, which permitted it to regulate an immune response across its temporal progression.
RESULTS
Regulation of IL-10 secretion by CD46 and IL-2
Although activation of purified human CD4+ T cells with stimulating monoclonal antibody (mAb) to CD3 and mAb to CD46 in the presence of IL-2 induces substantial IL-10 secretion and confers a suppressive phenotype19, we found that T cells activated with anti-CD3 and anti-CD46 also produced large amounts of IFN-γ19 (Fig. 1a). To better understand these seemingly paradoxical effects of CD46, we varied the strength of the activating signals during T cell activation (all experiments here used cells of human origin). Although changing the concentrations of the CD3- and CD46-specific mAbs did not substantially affect IFN-γ or IL-10 secretion measured 72 h after activation (data not shown), IL-10 production was regulated by exogenous IL-2 (Fig. 1b). Specifically, in a concentration of IL-2 of 0.5 U/ml or less, activation with anti-CD3 and anti-CD46 induced more IFN-γ secretion than that of T cells activated with either anti-CD3 alone or with anti-CD3 plus anti-CD28. Anti-CD3 and anti-CD46 also induced transient expression of IL-2, detectable at 24 h after activation in the absence of exogenous IL-2 (Fig. 1c). Increasing the concentration of IL-2 above 5 U/ml did not further increase IFN-γ production, but in IL-2 concentrations of 5–10 U/ml, T cells obtained from various donors and activated with anti-CD3 and anti-CD46 all showed considerable IL-10 secretion in addition to IFN-γ, with IL-10 production increasing with the amount of IL-2 (Fig. 1b). This was highly specific to IL-10, as no conditions promoted the production of IL-4, IL-5 or IL-17 (data not shown). CD46-induced IFN-γ peaked 24 h after activation and then steadily decreased, whereas IL-10 was barely detectable before 24 h and peaked at 72 h (Fig. 1c). Thus, CD46-mediated IFN-γ production preceded IL-10 secretion even in cultures containing high concentrations of IL-2 that are most conducive to IL-10 production.
Figure 1.

IL-2 regulates TH1 versus Tr1 effector function in CD4+ T cells activated with anti-CD3 and anti-CD46. (a) IFN-γ and IL-10 in supernatants of purified T cells not activated (NA) or activated for 72 h with various combinations of mAb to CD3 (α-CD3), mAb to CD28 (α-CD28) and mAb to CD46 (α-CD46; horizontal axis) plus IL-2 (25 U/ml). (b) IFN-γ and IL-10 in supernatants of purified T cells left unactivated (-) or activated for 72 h with various combinations of mAbs as in a plus various concentrations of IL-2 (wedge). (c) IL-2, IL-10 and IFN-γ in supernatants of purified T cells not activated or activated for 0–120 h (horizontal axis) with anti-CD3 plus anti-CD46 plus no exogenous IL-2 (IL-2 analysis) or IL-2 at 50 U/ml (IFN-γ or IL-10 analysis). (d) Cytokine secretion at 36 h by purified CD4+ T cells not activated or activated with various combinations of mAbs to CD3, CD28 and CD46 (above plots) plus IL-2 (dose, left margin) or neutralizing mAb to IL-2. Numbers in plots indicate percent IFN-γ+IL-10− cells (top left), IFN-γ+IL-10+ cells (top right) or IFN-γ−IL-10+ cells (bottom right). (e) Proliferation of freshly purified CD4+ T cells mixed with supernatants of T cells that were activated for 36 h with anti-CD3 and anti-CD46 in the presence of IL-2 (50 U/ml), separated by flow cytometry based on their secretion profile (IFN-γ+IL-10−, IFN-γ+IL-10+ or IFN-γ−IL-10+) and cultured independently for further 18 h (low IL-2); the mixtures were activated with anti-CD3 plus anti-CD28 with neutralizing mAb to IL-10 (α-IL-10) or isotype-matched control antibody (isotype) and assessed at day 6 (middle and right). Far left (three bars), proliferation of freshly purified T cells activated with anti-CD3 and anti-CD28 in the presence of fresh media or supernatants of T cells activated for 72 h with anti-CD3 and anti-CD28. A490, absorbance at 490 nm. *P < 0.05 and **P < 0.01 (Student’s t-test). Data are representative of eight (a) or three (c,e) experiments (mean ± s.d.), eight experiments with three independent donors (b) or six experiments (d).
Successive induction of IFN-γ+ and IL-10+ cells
We next investigated the single-cell dynamics of the secretion of IFN-γ and IL-10 induced by anti-CD3 and anti-CD46. We activated purified CD4+ T cells with mAb to CD3 alone or in combination with mAb to CD28 plus mAb to CD46 in the presence of a neutralizing mAb to IL-2, or either a low dose (5 U/ml) or high dose (50 U/ml) of IL-2. We measured active cytokine secretion 36 h after activation (Fig. 1d). Blockade of IL-2 inhibited cytokine production under every condition analyzed. However, in the presence of IL-2, activation with anti-CD3 and anti-CD46 or with anti-CD3, anti-CD28 and anti-CD46 induced three discrete T cell populations with distinct secretion profiles: IFN-γ+IL-10− cells, IFN-γ+IL-10+ cells and IFN-γ−IL-10+ cells (Fig. 1d). Cultures with IL-2 at a concentration of 5 U/ml contained greater frequencies of IFN-γ-secreting cells (17.4% ± 5.2% IFN-γ+IL-10− and IFN-γ+IL-10+ cells in cultures activated with anti-CD3 and anti-CD46) than IL-10-producing T cells (12.0% ± 1.8% IFN-γ+IL-10+ and IFN-γ−IL-10+ cells), whereas IL-2 at a concentration of 50 U/ml induced substantially more IL-10+ cells (24.7% ± 5.8% IFN-γ+IL-10+ and IFN-γ−IL-10+ cells). In contrast, although cultures stimulated with anti-CD3 and anti-CD28 also contained all three T cell populations secreting IFN-γ and IL-10, there was no IL-2- dependent change in the ratio of IFN-γ-producing T cells to IL-10+ T cells, and the total number of IL-10-producing cells remained low even in high concentrations of IL-2 (Fig. 1d). Although these results (Fig. 1d) reflect assays at 36 h, cellular cytokine staining at 12 h, 72 h and 90 h after stimulation also mirrored the results of our kinetic study reported above (Fig. 1c): at 24 h, coactivation of cells with anti-CD3 and anti-CD46 promoted mostly IFN-γ+IL-10− cells, even in high concentrations of IL-2, with the peak of IL-10-producing cells arising subsequent to this. By 90 h, the overall number of cytokine-secreting cells decreased substantially (data not shown).
We used purified CD4+ T cell populations that at minimum included naive and memory CD4+ T cells, as well as natural Treg cells, for the experiments described above. To exclude the possibility that the observed CD46-elicited populations reflected selective reactivation and/or population expansion of one or more of these subsets, we applied anti-CD3–anti-CD46 activation protocols to highly purified naive CD4+ T cells (CD4+CD45RA+CD45RO−CD127−CCR7−CD25−). As before, we detected IFN-γ+IL-10−, IFN-γ+IL-10+ and IFN-γ−IL-10+ cells in the presence of high concentrations of IL-2 for 36 h (Supplementary Fig. 1). However, activation of naive T cell cultures induced a lower frequency of IL-10+ cells than IFN-γ+ cells (1.6% ± 0.7% versus 5.6% ± 1.9%; Supplementary Fig. 1). Conversely, the use of anti-CD3 and anti-CD46 to restimulate naive cells previously activated and expanded with mAb to CD3 and mAb to CD46 substantially increased the proportion of both IFN-γ+IL-10+ and IFN-γ−IL-10+ T cells (Supplementary Fig. 1, right). Moreover, the frequency of IFN-γ+IL-10+ cells and IFN-γ−IL-10+ cells was significantly increased by each additional restimulation with anti-CD3 and anti-CD46 (P < 0.01). Again, we detected no IL-4 or IL-5 in any of these cultures (data not shown). Secondary stimulation with anti-CD3 and anti-CD46 also increased IL-10 production, albeit to a lesser extent, from T cells initially activated with anti-CD3 alone (Supplementary Fig. 1, left). In short, the IL-2-dependent emergence over time of IL-10-producing cells does not obviously reflect the selective response of a preexisting subset.
We then assessed the capacity of the various cell populations induced by anti-CD3 and anti-CD46 to regulate T cell activation. We activated CD4+ T cells through ligation of CD3 and CD46 and subsequently sorted them into IFN-γ+IL-10−, IFN-γ+IL-10+ and IFN-γ−IL-10+ subsets, which we then cultured for 18 h in IL-2 at a concentration of 1 U/ml. The supernatants of these three cell types continued to contain large amounts of IFN-γ, approximately equal amounts of IFN-γ and IL-10, and large amounts of IL-10 with negligible IFN-γ, respectively (data not shown). When we cultured freshly purified CD4+ T cells in the presence of those supernatants together with immobilized crosslinking anti-CD3 and anti-CD28, their proliferation (measured at day 6) was substantially inhibited by the supernatants of the IFN-γ+IL-10+ cells induced by anti-CD3 and anti-CD46 and of the IFN-γ−IL-10+ T cells (Fig. 1e). The inhibition was not elicited by supernatants of IFN-γ+IL-10− T cells or by supernatants of T cells activated for 72 h by anti-CD3 and anti-CD28 (Fig. 1e). IL-10 upregulated by anti-CD3 and anti-CD46 is the main soluble mediator of this effect, as regulation was almost completely blocked by a neutralizing mAb to IL-10.
CD46-induced IL10 originates in a TH1 subset
To assess whether CD46-induced IFN-γ+IL-10+ and IFN-γ−IL-10+ T cells originated from the IFN-γ+IL-10− TH1 cells that first arose or whether each cell population arose with different kinetics from separate cell populations (Fig. 2a), we undertook two sets of experiments. First we activated purified CD4+ T cells with anti-CD3 and anti-CD46 in a high concentration of IL-2 in the presence of neutralizing mAb to IFN-γ, a condition known to inhibit TH1 development8. As expected, after 48 h we observed a significant decrease in the number of IFN-γ+IL-10− T cells (from 14% ± 4% to 4% ± 2.5%; Fig. 2b). However, this treatment also substantially decreased the percentage of anti-CD46-induced IFN-γ+IL-10+ cells (from 16% ± 2% to 2.5% ± 1.5%) and of IFN-γ−IL-10+ cells (from 18% ± 3% to 1.5% ± 0.5%). Thus, limiting TH1 differentiation limited the IL-10 switch. IL-10 neutralization did not decrease the cell number of any of the T cell populations induced but instead slightly increased their representation (IFN-γ+IL-10− cells, from 14% ± 4% to 17.5 ± 4%; IFN-γ+IL-10+ cells, from 16% ± 2% to 19.5% ± 6.5%; IFN-γ−IL-10+ cells, from 18% ± 3% to 18.5% ± 2.5%; Fig. 2b). These data suggest at minimum that the continuous production of IL-10 initially induced via anti-CD3, anti-CD46 and IL-2-mediated signals does not depend on positive feedback by IL-10 itself.
Figure 2.
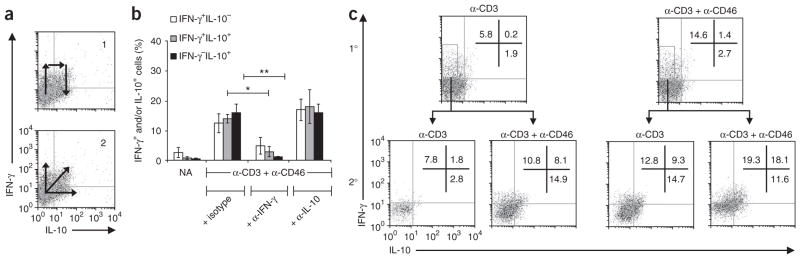
CD46–IL-2 signals induce a switch from a TH1 phenotype to a suppressive Tr1 phenotype in CD4+ T cells. (a) Flow cytometry of the induction of IL-10-secreting T cells by anti-CD46 from an initial TH1 effector cell (1) or from a distinct cell subset (2). (b) TH1 lineage induction in purified CD4+ T cells left not activated or activated with anti-CD3 and anti-CD46 plus IL-2 (50 U/ml IL-2) and isotype-matched control antibody or neutralizing mAb to IFN-γ or IL-10, presented as the percentage of cytokine-positive (Cyt+) cells: IFN-γ+IL-10−, IFN-γ+IL-10+ or IFN-γ−IL-10+ (key). *P < 0.05 and **P < 0.01 (Student’s t-test). Data are representative of three experiments (mean ± s.d.). (c) Secretion of IFN-γ and IL-10 by IFN-γ+IL-10− cells isolated by flow cytometry from purified T cells activated for 36 h (primary stimulation (1°)) with anti-CD3 alone or with anti-CD3 and anti-CD46 in the presence of IL-2 (5 U/ml); the IFN-γ+IL-10− populations were expanded for 4 d with IL-2 (5 U/ml), then restimulated (above plots; secondary stimulation (2°)) and assayed 18 h later. Numbers in plots indicate percent IFN-γ+IL-10− cells (top left), IFN-γ+IL-10+ cells (top right) or IFN-γ−IL-10+ cells (bottom right). Data are representative of four experiments.
Second, we activated purified CD4+ T cells with anti-CD3 alone or with anti-CD3 and anti-CD46 in a low concentration of IL-2 and isolated the resulting IFN-γ+IL-10− cells. When, after 4 d of population expansion, we restimulated the cells with anti-CD3 and anti-CD46, they produced a high frequency of IFN-γ+IL-10+ and IFN-γ−IL-10+ cells (Fig. 2c). Together these two data sets suggest that anti-CD46-induced IL-10-secreting T cells obligatorily derive from an initial TH1 phase. In agreement with that, anti-CD3–anti-CD46–mediated reactivation of TH1 cells primed through activation of naive T cells with anti-CD3 and anti-CD28 in the presence of IL-12 and neutralizing mAb to IL-4 (strong TH1-skewing conditions) also promoted IFN-γ+IL-10+ and IFN-γ−IL-10+ cells (data not shown).
Anti-CD3- and anti-CD46-induced IL-10 cells retain TH1 markers
T cell lineages are characterized by specific transcription factor expression and activation: TH1 differentiation is accompanied by phosphorylation of STAT1 and STAT4 and expression of T-bet34,35; TH2 cells require STAT6 activation and GATA-3 expression34,35; and natural Treg cells are characterized by Foxp3 expression9. When we activated purified CD4+ T cells for 36 h with anti-CD3 and anti-CD46 in a high concentration of IL-2, all resultant cells had a predominantly TH1 profile (Table 1). In mice, IL-10 secretion by TH1 cells requires activation of the kinase Erk18,36. Likewise, CD46-dependent, TCR-independent induction of Erk1 and Erk2 in human CD4+ T cells has been reported24. All three T cell populations induced with anti-CD46 contained large amounts of phosphorylated Erk1 and Erk2 (Table 1). Signaling through the IL-2 receptor complex induces Janus kinase–mediated phosphorylation of STAT5. Consistent with the fact that anti-CD46-induced IL-10 production is IL-2 dependent, we detected phosphorylated STAT5 in IFN-γ+IL-10− and IFN-γ+IL-10+ cells, although it was lower in IFN-γ−IL-10+ T cells, perhaps consistent with their limited proliferative capacity19 (data not shown).
Table 1.
Expression profiles of key T cell lineage transcription factors in CD46-induced effector (TH1) and suppressor (Treg) cell populations
| Transcription factor | IFN-γ+IL-10−(TH1) | IFN-γ+IL-10+ (Treg) | IFN-γ−IL-10+ (Treg) | TH1a | TH2a |
|---|---|---|---|---|---|
| T-bet | +++ | +++ | +++ | ++++ | − |
| GATA-3b | −/+ | + | + | −/+ | ++++ |
| Foxp3 | − | − | − | − | − |
| p-STAT4 | ++ | +++ | ++ | ++++ | ++ |
| p-STAT5 | +++ | +++ | + | ++++ | ++ |
| p-STAT6 | + | + | −/+ | + | ++++ |
| p-Erk | ++ | +++ | +++ | ++++ | ++ |
| ICER/CREM | +++ | ++ | + | ++ | ++ |
IL-10- and IFN-γ-secretion assays plus intracellular flow cytometry staining for T cell lineage markers in purified CD4+ T cells activated for 36 h with anti-CD3 and anti-CD46 in a high concentration of IL-2; transcription factor expression is presented relative to the expression in TH1 or TH2 cells as follows: ++++, highest; +++, strong; ++, moderate; +, low; −/+, weak but detectable; −, undetectable; p-, phosphorylated.
Purity of TH1 and TH2 cells assessed as percent IFN-γ+IL-4- cells (TH1) or IFN-γ−IL-4+ cells (TH2) after restimulation with anti-CD3 and anti-CD28 (>85% in all assays).
GATA-3 mRNA assessed by quantitative real-time RT-PCR analysis because of the lack of a suitable antibody.
Regulation of IL-2 secretion by CD3-CD46 signals
Drawing further parallels with TH1 cells, we examined IL-2 expression, as it is a TH1 hallmark reportedly lost by TH1 cells that have the ability to also secrete IL-10 (refs. 18,36). We had detected IL-2 expression in CD4+ T cell cultures activated with anti-CD3 and anti-CD46 (Fig. 1c), and when we examined them in greater detail, we found that >85% of IFN-γ+ cells actively secreted IL-2 at 36 h after activation with anti-CD3 and anti-CD46 (Fig. 3a), whereas this was true for <25% of IL-10-secreting cells19 (Fig. 3a). Of those, 50–60% coexpressed IFN-γ (data not shown). Thus, cells activated with anti-CD3 and anti-CD46 shared with TH1 cells production of IL-2 that ceased with induction of the IL-10-secreting state.
Figure 3.

CD46-mediated signals contribute to the regulation of IL-2 expression by CD4+ T cells. (a) Secretion of IFN-γ, IL-10 and IL-2 by T cells either not activated or stimulated for 36 h with anti-CD3 and anti-CD46. Numbers in plots in top row indicate percent IFN-γ+IL-2− cells (top left), IFN-γ+IL-2+ cells (top right) or IFN-γ−IL-2+ cells (bottom right); numbers in bottom row indicate percent IL-10+IL-2− cells (top left), IL-10+IL-2+ cells (top right; also IFN-γ+ (data not shown)) or IL-10−IL-2+ cells (bottom right). (b) Intracellular flow cytometry analysis of ICER/CREM expression after 36 h of no activation or activation with various combinations of mAbs to CD3, CD28 or CD46 (key) or isotype-matched control antibody (Isotype control). (c) Immunoblot analysis of the nuclear translocation of ICER/CREM, assessed in cytoplasmic (C) and nuclear (N) protein fractions of purified T cells either not activated or activated for 24 or 48 h with anti-CD3 and anti-CD46 (3+46). Right margin, molecular size in kilodaltons (kDa). (d) Chromatin-immunoprecipitation analysis of the binding of ICER/CREM to the IL2 promoter in IFN-γ+IL-10− cells (IFN-γ+) and IFN-γ−IL-10+ cells (IL-10+) induced with anti-CD3 and anti-CD46, presented as PCR amplification of an IL2 promoter–specific sequence from genomic DNA (control; left) or from DNA precipitated with anti-ICER/CREM (PP DNA; right). Right margin, size in base pairs (bp). Data are representative of three (a,b,d) or two (c) experiments.
Gene-array comparison of primary CD4+ T cells activated with anti-CD3 and anti-CD46 versus those activated with anti-CD3 and anti-C28 showed substantially more mRNA for the transcriptional regulators ICER and CREM (which both control IL-2 transcription and cannot be distinguished in FACS analysis with the antibodies used; thus, called ‘ICER/CREM’ here) in cells activated with anti-CD46 (data not shown). Translocation of ICER/CREM to the nucleus attenuates transcription of the gene encoding IL-2 (ref. 37). Flow cytometry confirmed that ligation of CD3 and CD46 induced ICER/CREM protein in all three subsets (Fig. 3b and Table 1). Unexpectedly, expression was highest in IFN-γ+IL-10− cells, which continued to express IL-2 (Table 1). However, immunoblot analysis of cytoplasmic and nuclear fractions showed consistent nuclear translocation of ICER/CREM only after 48 h of activation with anti-CD3 and anti-CD46 (Fig. 3c) or with anti-CD3, anti-CD28 and anti-CD46 (data not shown), coincident with the appearance of IL-10 in the culture medium (Fig. 1c). Activation with anti-CD3 alone or with anti-CD3 and anti-CD28 did not lead to measurable nuclear translocation of ICER/CREM at any time point (data not shown). To determine if ICER/CREM bound ‘preferentially’ to the IL2 promoter in IL-10+ cells, we used chromatin-immunoprecipitation analysis, with an ICER/CREM-specific mAb, of IFN-γ+IL-10− and IFN-γ−IL-10+ cells purified 36 h after activation with anti-CD3 and anti-CD46. IL2 promoter–specific DNA sequences were precipitated only from IL-10+ cells and not from IFN-γ+ T cells (Fig. 3d). Thus, activation with anti-CD3 and anti-CD46 induces considerable ICER/CREM expression in TH1 cells, but this is inactive in IFN-γ+ cells that continue to produce IL-2. Instead, active nuclear translocation occurs ‘preferentially’ in IL-10+ cells, consistent with their lower IL-2 production. IL-2 production is not attenuated secondarily via IL-10 production, because neither IL-10 neutralization during activation with anti-CD3 and anti-CD46 nor the addition of recombinant human IL-10 during CD3–IL-2 stimulation affected ICER/CREM translocation or IL-2 production (data not shown).
CD46 CYT-1 domain promotes IL-10 via the kinase SPAK
As outlined above, CD46 is commonly expressed as four isoforms with two possible cytoplasmic domains, CYT-1 and CYT-2 (ref. 25; Supplementary Fig. 2). To determine whether CYT-1 or CYT-2 affect IL-10 switching differently, we generated Jurkat T cells stably expressing either the BC–CYT-1 isoform (BC1) or BC–CYT-2 isoform (BC2). Untransfected Jurkat cells expressed only the C2 isoform and did not produce IFN-γ or IL-10 after activation with anti-CD3 and anti-CD46 in the presence of IL-2 (Fig. 4a and data not shown). In contrast, this property was acquired by BC1-transfected cells but, notably, not by BC2-transfected cells (Fig. 4a).
Figure 4.

The intracellular CYT-1 domain of CD46 and SPAK are required for IL-10 production in CD4+ T cells. (a) Secretion of IL-10 by Jurkat T cells stably transfected with the BC1 isoform (Jurkat-BC1) or BC2 isoform (Jurkat-BC2) of CD46 and either not activated or activated for 48 h with anti-CD3 and anti-CD46 or with anti-CD46 alone. (b) Immunoprecipitation (with a CD46-specific mAb or isotype-matched control mAb) of lysates of primary human CD4+ T cells activated for 15 min with various antibodies (above lanes), followed by immunoblot analysis with anti-CD46 (top) or anti-SPAK (bottom). Left margin: CD46, BC1 + BC2, CD46 isoform with BC region and either CYT-1 or CYT-2; CD46, C1 + C2, CD46 isoform with only the C region. Lysate (far left lane), untreated lysates from nonactivated cells (control). (c) IL-10 secretion by purified CD4+ T cells left untransfected (NT) or transfected with SPAK-specfic siRNA, nonspecific siRNA (Control) or buffer alone (Mock) and then either not activated or activated for 36 h with mAb to CD3 alone or mAb to CD3 and mAb to CD46 (key). (d) IL-10 production by Jurkat T cells left untransfected (Jurkat) or stably transfected with wild-type SPAK (Jurkat-SPAK) or a ‘kinase-dead’ version of SPAK (Jurkat-ΔSPAK) and either not activated or stimulated with anti-CD3 and anti-CD46 or with anti-CD46 alone. *P < 0.01 (Student’s t-test). Data are representative of three experiments (mean ± s.d. in a,c,d).
A yeast two-hybrid screen identified the proline-alanine–rich serine-threonine kinase SPAK as an interaction partner with CYT-1 and CYT-2 (data not shown). SPAK is broadly expressed and has been linked to the regulation of mitogen-activated protein kinases and T cell activation38,39. Immunoprecipitation confirmed that SPAK constitutively bound to CD46 in unstimulated primary CD4+ T cells (Fig. 4b). Although activation with anti-CD3 completely disrupted the CD46-SPAK interaction within 15 min, this was partially preserved when CD46 was also engaged (Fig. 4b). SPAK also immuno-precipitated together with CD46 (but with neither CD3 nor CD28) in unstimulated Jurkat T cells (data not shown). When we knocked down SPAK expression in purified CD4+ T cells through the use of small interfering RNA (siRNA), the cells had a lower capacity to produce IL-10 after activation with anti-CD3 and anti-CD46 (Fig. 4c). Consistent with that finding, Jurkat cells stably transfected with SPAK responded to activation with anti-CD3 and anti-CD46 by producing IL-10, whereas cells transfected with a ‘kinase-dead’ SPAK mutant did not produce IL-10 under any of the activation conditions tested (Fig. 4d). In short, the induction of IL-10 by anti-CD46–anti-CD3 activation of human T cells is attributable at least in part to a signaling pathway from the CYT-1–BC1 tail of CD46 via SPAK. That proposal is supported by the finding that knockdown of SPAK protein (Supplementary Fig. 3a) substantially decreased the previously observed CD46-mediated hyperphosphorylation of Erk23,24,32, a key factor in IL-10 upregulation in T cells36 (Supplementary Fig. 3b). The observation that SPAK silencing abrogated both anti-CD3- and anti-CD46-induced phosphorylation of the kinase Jnk suggests that SPAK might also contribute to Jnk activation in a CD46-independent pathway. In contrast, and as a specificity control, phosphorylation of the mitogen-activated kinase p38 in T cells activated with anti-CD3 alone or with anti-CD3 and anti-CD46 remained largely unaffected by knockdown of SPAK protein (Supplementary Fig. 3b).
Regulation of the effector phase of γδ T cells by CD46
T cells expressing the TCR γ-chain variable region 9 and δ-chain variable region 2 (Vγ9Vδ2 T cells), the main γδ T cell subset in human peripheral blood, are biased mainly toward a TH1-like phenotype, but the generation of Tr1-like cells from these cells has not been convincingly demonstrated. If CD46 regulation is a chief means of inducing Tr1-like cells, we reasoned that the scarcity of IL-10-producing Vγ9Vδ2 T cells might be associated with differences in the CD46 pathway. Indeed, although Vγ9Vδ2 T cells expressed CD46, albeit in smaller amounts than did CD4+ T cells (Supplementary Fig. 4a), these different subsets differed considerably in their expression pattern of CD46 variants. The BC2 isoform, which did not confer IL-10 production in Jurkat transfectants stimulated with anti-CD3 and anti-CD46 (Fig. 4a), was unequivocally the main isoform in Vγ9Vδ2 T cells, in contrast to the variable expression pattern commonly observed in CD4+ T cells26 (Fig. 5a). As expected, stimulation of peripheral blood mononuclear cells (PBMCs) from healthy donors with HMBPP (a Vγ9Vδ2 T cell–specific agonist) elicited negligible IL-10 production, and this was not affected by CD46 costimulation, even in high concentrations of IL-2 (Fig. 5b). However, we noted slightly less IFN-γ production (data not shown; discussed below), in contrast to the promotion of IFN-γ by anti-CD3 and anti-CD46 in CD4+ T cells (Fig. 1a,b).
Figure 5.

CD46 directly regulates γδ T cell function. (a) PCR analysis of CD46 isoform use by human γδ T cells (Vγ9Vδ2; middle) and CD4+ T cells (CD4; right) and in Chinese hamster ovary (CHO) cell lines transfected with single isoforms of CD46 (left). (b–d) Production of IL-10 (b) and IFN-γ and tumor necrosis factor (TNF; c) by human γδ T cells among PBMCs stimulated with HMBPP (10 nM) with (+) or without (-) mAb to CD46 in the presence (filled bars) or absence (open bars) of IL-2 (100 U/ml). (d) CD25 expression by Vγ9Vδ2 T cells stimulated with HMBPP (10 nM) or CD4+ T cells stimulated with mAb to CD3 plus mAb to CD28, with or without mAb to CD46 and in the presence or absence of IL-2 (as in b,c). MFI, mean fluorescence intensity. (e) Flow cytometry of purified cultures of CD4+ or Vγ9Vδ2 T cells (both >97% pure) activated with mAb to CD3 or HMBPP, respectively, in the presence of mAb to CD46 and IL-2 (100 U/ml). Numbers in plots indicate percent IFN-γ+IL-10− cells (top left), IFN-γ+IL-10+ cells (top right) or IFN-γ−IL-10+ cells (bottom right). (f) Production of IFN-γ by purified Vγ9Vδ2 T cells with or without anti-CD46 treatment in the presence or absence of IL-2 (indicated as in b,c). D1–D8, donors 1–8. Data are representative of three experiments (a–d) or one experiment each with six (e) or two (f) different donors.
To further investigate the more direct immunosuppressive role of CD46 described above, we stimulated PBMCs from three additional donors with HMBPP with or without IL-2 and crosslinking of CD46. Again, anti-CD46 did not induce IL-10 (data not shown) but consistently decreased production of IFN-γ and secretion of tumor necrosis factor (Fig. 5c). This effect was not due to anti-CD46-induced cell death, because the proportion of apoptotic (annexin V–positive) and necrotic (annexin V–positive and propidium iodide–positive) Vγ9Vδ2 T cells and the overall proportion of Vγ9Vδ2 T cells were each similar in PBMCs stimulated with HMBPP and IL-2 in the presence or absence of anti-CD46 (Supplementary Fig. 4b). A direct immunosuppressive effect of CD46 on Vγ9Vδ2 cells was also evident in downregulation of CD25 (IL-2 receptor α-chain), especially in the absence of IL-2, in contrast to the response of CD4+ T cells (Fig. 5d). To preclude potential interfering effects of other subpopulations, we sorted Vγ9Vδ2 T cells (>95% pure) from two donors by flow cytometry and stimulated the cells with HMBPP, and separately activated CD4+ T cells from the same donors with anti-CD3 and anti-CD28. Although costimulation with anti-CD46 during these activation conditions allowed us to detect IFN-γ+IL-10+ and IFN-γ−IL-10+ CD4+ T cells, we did not detect such subpopulations among Vγ9Vδ2 T cells (Fig. 5e), and IFN-γ production was again generally lower in high concentrations of IL-2 (Fig. 5f). Finally, we investigated the possibility that IL-10 could be induced by restimulation of Vγ9Vδ2 T cells with anti-CD46 (as observed for naive CD4+ T cells; Supplementary Fig. 2). Thus, we obtained a polyclonal T cell line (>75% Vγ9Vδ2+) after incubating PBMCs with HMBPP and IL-2 and stimulated it with HMBPP and/or IL-2 and anti-CD46. Again, anti-CD46 decreased the secretion of both tumor necrosis factor and IFN-γ (Supplementary Fig. 4c) but could not induce substantial IL-10 production (data not shown). Although these results confirm the finding that the BC1 isoform is needed to switch cells to IL-10 producers, they also show that activation of the BC2 isoform of CD46 directly inhibits TH1-like responses by a principal subset of unconventional T cells linked to the early phases of immune responses. Thus, CD46 seems to use two distinct mechanisms to regulate the production of pro-inflammatory cytokines.
Possible contribution of defective CD46 signals to autoimmunity
We obtained further evidence that CD46 actively regulates IL-10 in human pathophysiology by comparing the responses of CD4+ T cells from three healthy donors and three adult patients with rheumatoid arthritis to activation with anti-CD3 and anti-CD46 in the presence of IL-2 at a concentration of 25 U/ml. The baseline expression of CD3, CD46 and CD25 was similar among all T cell samples (data not shown), yet at 36 h after activation, cultures from patients with rheumatoid arthritis had higher percentages of IFN-γ+IL-10− and IFN-γ+IL-10+ T cells but very few IFN-γ−IL-10+ T cells (Fig. 6a). In addition, whereas the amount of IFN-γ and IL-10 in the supernatants of T cells from healthy donors was roughly similar, T cells from patients with rheumatoid arthritis had tenfold or more greater production of IFN-γ than IL-10 (Fig. 6b), and supernatants derived from these cells had no suppressive activity (data not shown). Because chronic inflammatory conditions such as rheumatoid arthritis are often attributed to inappropriate responses to persistent antigen, we examined cytokine expression after restimulating T cells with anti-CD3 and anti-CD46. Cultures of T cells from healthy donors showed more switching to the IFN-γ−IL-10+ state and contained approximately equal distributions of IFN-γ-producing T cells (IFN-γ+IL-10− and IFN-γ+IL-10+) and IL-10-secreting T cells (IFN-γ+IL-10+ and IFN-γ−IL-10+; Fig. 6c), with a ratio of 1:1 for IFN-γ and IL-10 secreted into the culture media (data not shown). In contrast, restimulated cultures from patients with rheumatoid arthritis lacked a substantial IFN-γ−IL-10+ population, with a diminution even in IFN-γ+IL-10+ cell frequencies relative to the cells’ initial stimulation (Fig. 6c). Thus, the ratio of IFN-γ to IL-10 in the culture media was ~20:1 at that point (data not shown). Notably, T cells from normal donors and patients with rheumatoid arthritis responded similarly to activation with anti-CD3 and anti-CD28 (Supplementary Fig. 5a–c) and TH1- and TH2-driving cytokines (Supplementary Fig. 5d), which showed that the observed deregulation in the expression of IFN-γ versus IL-10 in rheumatoid arthritis relates to a defect in CD46 responsiveness.
Figure 6.
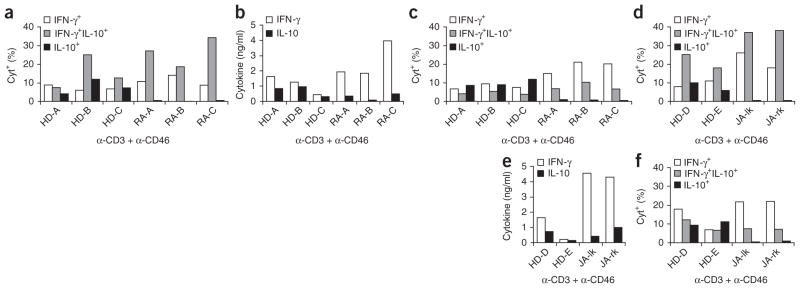
CD4+ T cells from patients with rheumatoid arthritis are defective in the IFN-γ–IL-10 switch induced by anti-CD3 and anti-CD46. (a) Frequency of cytokine-secreting cells among blood-derived T cells obtained from three healthy donors (HD-A–HD-C) and three patients with rheumatoid arthritis (RA-A–RA-C) and activated for 36 h with anti-CD3 and anti-CD46 plus IL-2 (50 U/ml), presented as the percentage of cytokine-positive cells (key; middle value of two data points). (b) IFN-γ and IL-10 in supernatants of T cells activated as described in a and then maintained in culture for 5 d with IL-2 (50 U/ml). (c) Frequency of cytokine-secreting cells among cells treated as described in b, then restimulated for 8 h with anti-CD3 and anti-CD46 plus IL-2 (50 U/ml); results presented as in a. (d) Frequency of cytokine-secreting cells among T cells from the synovial fluid of the left knee (JA-lk) and right knee (JA-rk) of a patient with juvenile arthritis and blood-derived T cells from two healthy donors (HD-D and HD-E) after primary stimulation with anti-CD3 and anti-CD46; results presented as in a. (e) Secretion of IFN-γ and IL-10 by the cells in d during population expansion as described in b. (f) Frequency of cytokine-secreting cells among the cells in d after secondary stimulation as in c. Data are representative of two experiments (a–f).
To further investigate IL-10 switching in arthritis, we isolated CD4+ T cells from the synovial fluid of both inflamed knee joints of a patient with juvenile arthritis. Of note, primary T cells from synovial fluid mostly had an activated CD25hiCD45RO+ phenotype, although expression of CD3 and CD46 was equivalent (data not shown). Similar to the results obtained with blood-derived CD4+ T cells from patients with rheumatoid arthritis, these synovial T cells did not switch to the IFN-γ−IL-10+ state after an initial stimulation with anti-CD3 and anti-CD46 (Fig. 6d); produced much more IFN-γ than IL-10 during population expansion (Fig. 6e); were mostly locked into the IFN-γ+IL-10− state after restimulation (Fig. 6f); and secreted ~30 times more IFN-γ than IL-10 into the culture medium after restimulation (data not shown). These data suggest that the anti-CD3–anti-CD46–IL-2–mediated switch of the proinflammatory IFN-γ+ (TH1) phenotype to an immunoregulatory phenotype of secreting only IL-10 is dysfunctional in patients with rheumatoid arthritis and juvenile arthritis. Moreover, the defect seems to lie mainly in the shutdown of IFN-γ production, as IL-10 secretion itself by T cells from patients with rheumatoid arthritis is induced almost normally after CD46 engagement.
IL-10 secretion driven by local complement production
Evidence underpinning CD46 as a key regulatory axis was provided by the finding that highly purified (antigen-presenting cell–free) anti-CD3-activated CD4+ T cell cultures contained the CD46 ligand C3b (data not shown) and had considerable deposition of C3b onto their cell surfaces (Fig. 7a). C3b generation was increased by costimulation with either anti-CD28 or anti-CD46. The prospect that this functionally engages CD46 was supported by the finding that small amounts of IL-10 production induced by activation of CD4+ T cells with anti-CD3 or with anti-CD3 and anti-CD28 was decreased to background production by the addition of soluble anti-CD46 (Fig. 7b). Accordingly, the presence of immobilized C3b increased IL-10 production by cells activated with anti-CD3 alone or with anti-CD3 and anti-CD28 (data not shown).
Figure 7.

Engagement of CD46 by locally produced C3b drives IL-10 expression in CD4+ T cells stimulated with anti-CD3 and anti-CD28. (a) Flow cytometry of C3b surface deposition on purified CD4+ T cells either not activated or activated for 12 h with immobilized mAbs (key). Data are representative of four experiments. (b) Production of IL-10 by purified T cells either not activated or activated for 36 h with mAb to CD3 alone or mAb to CD3 plus mAb to CD28 (horizontal axis) plus media alone or media plus soluble CD46 (sCD46; key). NS, not significant; *P < 0.01 (Student’s t-test). Data are representative of three experiments.
DISCUSSION
Irrefutable evidence of the importance of IL-10 in suppressing immunopathology has now been extended to humans, in whom it has both biological and clinical implications40. As a result, there is much interest in IL-10-producing cells. This area of study has progressed from the idea of an IL-10 lineage to one that embraces cells that switch to IL-10 production from an earlier incarnation as IFN-γ-producing TH1 cells. Cultures that coexpress IL-10 and IFN-γ have been documented11–14, and such a switch would have the seeming advantage of inducing a regulatory cytokine and simultaneously suppressing an effector cytokine based on recognition of the same antigen. Nonetheless, how such a switch might naturally occur, particularly in humans, has not been examined in detail before, to our knowledge40. The results presented in our study here make the case that human TH1 cells are promoted by coengagement of the TCR and CD46 but that as environmental IL-2 increases, as would be the case in a flourishing effector response, a switch to regulatory IL-10 production occurs, concomitant with diminished endogenous IL-2 production.
Attesting to the importance of the mechanism outlined above, we found it to be defective in patients with rheumatoid arthritis. Moreover, it is striking that this switching also did not function in the main subset of human γδ-cells that are known to express IL-10 only very rarely33. Anti-CD46-induced IL-10 production required expression of CYT-1 of CD46; CYT-1-bearing isoforms were expressed in CD4+ T cells but were undetectable in γδ T cells. Together these findings would be consistent with the idea that the CD46-TCR activation regime is a chief means of peripheral IL-10 induction tightly regulated by the expression patterns of the respective CD46 isoforms. Notably, the analysis of γδ cells did show an effect of CD46-TCR coengagement: instead of switching to IL-10 production, the cells showed substantial suppression of the secretion of IFN-γ and tumor necrosis factor as well as a decrease in CD25 expression. These data permit us to propose that CD46 uses distinct mechanisms to regulate the immune response from its beginning to its close: in the early stages it may promote IFN-γ in conventional T cells while controlling the rapidly activated effector functions of unconventional T cells; in the later stages, it switches conventional T cells to Treg cells.
It is now widely acknowledged that the complement system functions well beyond simple danger recognition and microbe clearance and participates actively in adaptive immune responses41. Two paradigms regarding complement functions have renewed interest in this evolutionary old innate system: first, complement has a role not only in the induction of T cell responses but also in their contraction and thus consequently in immune homeostasis20,42,43; second, the local production of complement components by immunocompetent cells participates decisively in shaping adaptive immune responses31,41,44. The latter observation, derived mostly from mouse models, is in agreement with our observation that C3b was produced early after activation of human CD4+ T cells with anti-CD3 and was increased after costimulation. Thus, our data allow us to suggest a model in which presentation of antigen and TCR engagement induces the subsequent generation of CD46 ligands, which provides a means for CD46 activation early during T cell activation. Such activated cells are now poised to integrate the third signal—high environmental concentrations of IL-2, reflecting a successfully expanded TH1 response—and switch appropriately into the Treg cell or contraction phase.
The scenario described above then raises a question about the hierarchy of CD46 activation (specifically in relation to CD28 costimulation) in IL-10 induction by TH1 cells. The complete inhibition of CD3-CD28 costimulation–induced IL-10 production by interference with ‘intrinsic’ CD46 activation suggests a dominant role for CD46 in IL-10 expression by TH1 cells. Moreover, such ‘physiological’ CD46 engagement through C3b or C4b produced by activated T cells might provide the molecular mechanisms by which IL-10-secreting Tr1 cells are induced in vitro by other groups. The generation of ‘classic’ adaptive IL-10-producing (and IFN-γ-producing) Tr1 cells in culture requires minimally repetitive activation with anti-CD3 and anti-CD28 (or exposure to dendritic cells) of CD4+ T cells in the presence of high IL-2 concentrations7,45, which are conditions conducive for CD46 engagement and CD46-induced IL-10 production.
Testing the idea that CD46 is important in regulating CD4+ and γδ T cell responses in a small animal model is hampered at present by the fact that rodents lack CD46 expression on somatic cells and that a murine molecule recapitulating CD46’s role in TCR–IL-2–dependent IL-10 induction has not yet been identified20. A published study of humans, however, has connected substantially lower IL-10 expression after CD46 activation of CD4+ T cells with multiple sclerosis46. Although the switch from the TH1 to Tr1 state was not examined in that study46, low IL-10 production by T cells from patients with multiple sclerosis correlate with an abnormal increase in CYT-2 expression, consistent with our finding that CYT-1 was required for IL-10 expression whereas CYT-2 was inefficient in inducing this. Those data combine with our observation that T cells from patients with rheumatoid arthritis were unable to promote the CD46-mediated TH1-Tr1 switch to link defects in this pathway in clinically important autoimmune and/or inflammatory conditions. We speculate that a high intrinsic signal threshold of T cells to switch from the TH1 phenotype to the Tr1 phenotype after IL-2-dependent CD46 activation might be a risk factor for autoimmunity. Consistent with that, certain CD25 polymorphisms have been correlated with multiple sclerosis, rheumatoid arthritis and type 1 diabetes47. Conversely, a low intrinsic threshold for CD46–IL-2–mediated IL-10 production might protect from autoimmunity but possibly at the price of a greater risk of chronic infection.
Notably, the induction of IL-10 from naive CD4+ T cells through TCR–CD46–IL-2 activation required repetitive stimulation of cells. This is reminiscent of the observation that the tolerance-inducing switch of TH1 cells into an IL-10+IFN-γ+ state in mice requires persistence of (self) antigen18. Similarly, only continuous exposure of nonallergic beekeepers to high doses of bee venom induces the switch from IFN-γ-secreting TH1 cells to IL-10-producing Tr1-like cells indicated by a decrease in T cell–mediated cutaneous swelling48. After antigen withdrawal, bee venom–specific T cells again produce only large amounts of IFN-γ and no IL-10 after the first reactivation and switch to IL-10 production only after repetitive stimulation. Anti-CD46-activated TH1 or Tr1 cells followed a similar scheme: sorted IFN-γ−IL-10+ T cells switched back to initial IFN-γ production after population expansion and restimulation (data not shown). These data are consistent with the idea that IL-10-secreting cells might not necessarily represent a lineage but instead represent the endpoint of a successful effector T cell response. Indeed, they comply with the facts that TH1 cells, TH2 cells and IL-17-producing helper T cells can produce IL-10 and that so far no Tr1-specific lineage marker has been identified. Consequently, organs such as the gut, skin and lungs, in which there is chronic interaction between the host and the environment, might provide a local milieu, through continuous stimulation, that locks TH1 cells into a regulatory state, potentially explaining the abundance of IL-10-secreting Tr1 cells in these sites49.
Although such T cell plasticity ensures the important flexibility to respond to microenvironmental signals appropriately, it poses a considerable obstacle in the therapeutic use of IL-10-secreting TH1 suppressor cells. Tr1 cells generated in a controlled in vitro environment might reacquire a proinflammatory TH1 phenotype after injection into autoimmune or transplant patients. Identifying the molecular signature that characterizes the effector and regulatory phases of CD46-induced TH1 and Tr1 cells might provide a means for actively inducing and locking these cells into the desired functional state. The ability to identify specific signaling events required for IL-10 induction in TH1 cells (for example, the CD46–CYT-1–mediated activation of SPAK, as well as the expression and nuclear translocation of ICER/CREM) could be a useful step in creating a platform to monitor and potentially manipulate these pathways in the future.
ONLINE METHODS
Donor and blood samples
Purified T cells were obtained from buffy coats (National Blood Service) or blood samples from healthy volunteers. Informed consent was obtained from all subjects included in the study, and blood or synovial fluid was collected and processed with the approval of and in accordance with King’s College Ethics Committee guidelines (06/Q0705/20). Adult patients with inflammatory arthritis (including rheumatoid arthritis and juvenile idiopathic arthritis) were recruited for the study. All patients had active disease with a disease activity score for 28 joints of over 5.1, representing moderately severe activity, despite therapy with a combination of the disease-modifying antirheumatic drugs methotrexate, hydroxychloroquine and sulfasalazine. Synovial fluid was obtained during therapeutic arthrocentesis of knee joints.
Cells, antibodies and recombinant proteins
T cells were maintained as described19. Chinese hamster ovary cells and Jurkat T cells were cultured according to the manufacturer’s protocol (American Tissue Culture Collection). Cell-stimulating antibodies were from BD Biosciences (anti-CD28; CD28.2), were purified from a specific hybridoma line (anti-CD3; OKT-3) or were generated in-house (anti-CD46; TRA-2-10)26. Polyclonal rabbit SPAK antiserum (2281) was from Cell Signaling Technology, and antibodies to phosphorylated and nonphosphorylated Erk1/2 (20A and MK12), Jnk (37/pan-JNK/SAPK1 and pT183/pY185) and p38 (36/p38 and 27/p38α) were from BD Biosciences. Neutralizing mAbs to human IL-2 (MQ1-17H12), IL-4 (MP4-25D2), IL-10 (JES3-19F1) and IFN-γ (4S.B3), and recombinant human IL-4, IL-10, IL-12 and IFN-γ for in vitro TH1 or TH2 induction, as well as fluorochrome-conjugated antibodies to phosphorylated STAT4 (558249), STAT5 (612567) and STAT6 (612701) were from BD Biosciences (and were used with fixing and staining buffers suggested by the manufacturer). Fluorochrome-conjugated mAbs to human CD4 (RPA-T4), CD25 (M-A251), CD45RA (HI100), CD45RO (UCHL1), CCR7 (2D12) and Foxp3 (259D/C7) and allophycocyanin–annexin V were from BD Pharmingen. Phycoerythrin- conjugated antibody to mouse and/or human T-bet (4B10) was from eBiosciences. Fluorescein isothiocyanate–labeled antibody to human ‘pan’ γδTCR (IM157U) and anti–human Vδ2 TCR (IM1464) were from Beckman Coulter. The mAb to CD46 (TRA-2-10) and mAb to human ICER/CREM (ab54625; Abcam) were labeled with phycoerythrin using a Zenon Mouse R-phycoerythrin Mouse IgG1 Labeling kit (Invitrogen). HMBPP (hydroxymethyl-butenyl-pyrophosphate), a phosphorylated antigen that specifically activates human Vγ9Vδ2 T cells, was provided by H. Jomaa. Recombinant C3b was from Complement Technologies. Soluble CD46 was generated by cloning of cDNA encoding short consensus repeats 1–4 of human CD46 into the pET15-b vector. BL21(DE3) bacteria were transfected with the construct and recombinant soluble CD46 was then purified from the inclusion bodies and refolded according to a published method50. Functional activity of soluble CD46 was monitored by C3b-binding and cofactor assays.
T cell isolation, the γδ T cell line and the generation TH1 and TH2 cells
CD4+ T lymphocytes were isolated from PBMCs or synovial fluid with CD4 MicroBeads (Miltenyi Biotec). Where indicated, T cells were isolated by cell sorting after appropriate surface staining (CD4+, CD45RA+, CD45RO−, CD25−, CD127− and CCR7+ for naive CD4+ T cells and ‘pan’ anti-γδTCR for γδ T cells). The purity of isolated lymphocyte fractions was typically >97%. The polyclonal γδ T cell line (>75% TCR Vδ2+) was obtained after stimulation of PBMCs with HMBPP (10 nM) and IL-2 (100 U/ml). Fresh medium plus IL-2 was added every 2–3 d and cells were used for functional assays at day 17. TH1 and TH2 cells were generated by activation and population expansion of sorted naive CD4+ T cells with mAb to CD3 and mAb to CD28 (2 μg/ml each), plus a function-neutralizing mAb to IL-4 (10 μg/ml) and recombinant human IL-12 (10 ng/ml) for TH1 induction or anti-IFN-γ (10 μg/ml) and recombinant human IL-4 (20 ng/ml) for TH2 induction.
T cell activation
Purified CD4+ T cells were activated in 48-well culture plates (2.5 × 105 to 3.5 × 105 cells per well) coated with mAbs to CD3, CD28 and/or CD46 (2.0 μg/ml each). Functional assays with γδ T cells used the same protocol except that HMBPP (10 nM) was used for stimulation.
Cytokine measurement
Cytokines were measured with the TH1/TH2 Cytometric Bead Array kit (BD Biosciences) or the human IFN-γ, IL-10 or IL-2 Cytokine Secretion Assay kit (Miltenyi Biotec) in combination according to the manufacturer’s protocol.
Suppression assay
Purified T cells were activated for 36 h with anti-CD3 and anti-CD46 in the presence of recombinant human IL-2 (5 or 50 U/ml). Cells were then sorted by flow cytometry on the basis of their secretion profile (IFN-γ+IL-10−, IFN-γ+IL-10+ or IFN-γ−IL-10+) and were cultured separately for 18 h with a low concentration of IL-2. Supernatants from these cultures were collected and then transferred to freshly purified T cells. T cell–supernatant mixtures were activated for 6 d with mAb to CD3 alone or mAb to CD3 plus mAb to CD28 and cell proliferation was measured with a CellTiter 96 AQueous One Solution Cell Proliferation assay (Promega).
Chromatin-immunoprecipitation analysis
Binding of ICER/CREM to the IL2 promoter was assessed with the MAGnify Chromatin Immunoprecipitation system according to the manufacturer’s protocol (Invitrogen). A mAb to ICER/CREM conjugated to protein G was used for immunoprecipitation of ICER/CREM-DNA complexes.
RNA silencing
The siRNA targeting human SPAK (siRNA identification number 898) and negative control siRNA were from Ambion; siRNA was delivered into primary human CD4+ T cells by electroporation as follows: 2 × 106 cells per ml transfection buffer (Ambion) plus 3 μg/ml of siRNA at 200 V and 325 mF (Gene Pulser; Bio-Rad Laboratories). Transfection efficiency and cell viability were consistently above 80% and 75%, respectively, and protein knockdown peaked at 36–48 h after transfection.
PCR
The IL2 promoter–specific primer pair 5′-TTACAAGAATCCCAAACT-3′ (forward) and 5′-TAGAGGCTTCATTATCAAA-3′ (reverse), between positions +301 and +510 relative to the promoter site, was used for PCR.
RT-PCR
The expression pattern of the CD46 isoform was assessed in CD4+ and γδ T cells with the CD46-specific primers 5′-GTGGTCAAATGTCGATTTCCAGTAGTCG-3′ (forward) and 5′-CAAGCCACATTGCAATATTAGCTAAGCCACA-3′ (reverse)26.
Quantitative real-time RT-PCR
For analysis of GATA-3 expression, RNA isolated from T cells was reverse-transcribed and cDNA specific for GATA-3 was measured with the ABI Prism 7700 Sequence Detection System and a GATA-3-specific primer pair (hs00231122_M1; Applied Biosystems). Results of individual samples were normalized to that of human 18S rRNA (housekeeping gene; Applied Biosystems.
Statistical analysis
Student’s two-tailed t-test (Excel software; Microsoft) was used for statistical analyses.
Supplementary Material
Acknowledgments
We thank H. Jomaa (Justus-Liebig-Universität) for HMBPP; K. Murphy for advice and discussions; and C. Hawrylowicz for help with manuscript revision. Supported by the American Asthma Foundation (formerly Sandler Program for Asthma Research; J.P.A. and C.K.), the Wellcome Trust (A.H. and A.R.), Cancer Research UK (A.H. and P.V.), the US National Institutes of Health (AI037618 to J.P.A.), the Medical Research Council Centre for Transplantation, Guy’s Hospital, King’s College (G.L. and C.K.) and the Department of Health, National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust (G.L.F., A.H and C.K).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
Note: Supplementary information is available on the Nature Immunology website.
AUTHOR CONTRIBUTIONS
J.C., G.L.F., P.V., A.R., A.F., I.J., T.S. and C.K. did the experiments, discussed data and corrected the manuscript; G.L. provided financial support for I.J. and T.S. and aided in data discussion; J.P.A. provided reagents, helped design experiments involving CD46-mediated signaling events, assisted in interpreting the data and revised the manuscript; A.C. designed experiments involving patients with rheumatoid arthritis and provided patient samples; A.H. designed experiments with γδ T cells and revised the manuscript; and C.K. designed the study and wrote the manuscript.
References
- 1.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 2.Gazzinelli RT, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ and TNF-α. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 3.O’Garra A, Vieira P. TH1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 4.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 5.Franke A, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 6.Hunter CA, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. 1997;158:3311–3316. [PubMed] [Google Scholar]
- 7.Groux H, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 8.Murphy KM, et al. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–494. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 10.McGeachy MJ, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 11.Del Prete G, et al. Human IL-10 is produced by both type 1 helper (TH1) and type 2 helper (TH2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150:353–360. [PubMed] [Google Scholar]
- 12.Assenmacher M, Schmitz J, Radbruch A. Flow cytometric determination of cytokines in activated murine T helper lymphocytes: expression of interleukin-10 in interferon-γ and in interleukin-4-expressing cells. Eur J Immunol. 1994;24:1097–1101. doi: 10.1002/eji.1830240513. [DOI] [PubMed] [Google Scholar]
- 13.Windhagen A, Anderson DE, Carrizosa A, Williams RE, Hafler DA. IL-12 induces human T cells secreting IL-10 with IFN-γ. J Immunol. 1996;157:1127–1131. [PubMed] [Google Scholar]
- 14.Gerosa F, et al. Interleukin-12 primes human CD4 and CD8 T cell clones for high production of both interferon-γ and interleukin-10. J Exp Med. 1996;183:2559–2569. doi: 10.1084/jem.183.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerosa F, et al. CD4+ T cell clones producing both interferon-γ and interleukin-10 predominate in bronchoalveolar lavages of active pulmonary tuberculosis patients. Clin Immunol. 1999;92:224–234. doi: 10.1006/clim.1999.4752. [DOI] [PubMed] [Google Scholar]
- 16.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4+CD25−Foxp3− TH1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jankovic D, et al. Conventional T-bet+Foxp3− TH1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrysova L, et al. Negative feedback control of the autoimmune response through antigen-induced differentiation of IL-10-secreting TH1 cells. J Exp Med. 2009;206:1755–1767. doi: 10.1084/jem.20082118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemper C, et al. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 20.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 21.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Seya T, Turner JR, Atkinson JP. Purification and characterization of a membrane protein (gp45–70) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837–855. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Astier A, Trescol-Biemont MC, Azocar O, Lamouille B, Rabourdin-Combe C. Cutting edge: CD46, a new costimulatory molecule for T cells, that induces p120CBL and LAT phosphorylation. J Immunol. 2000;164:6091–6095. doi: 10.4049/jimmunol.164.12.6091. [DOI] [PubMed] [Google Scholar]
- 24.Zaffran Y, et al. CD46/CD3 costimulation induces morphological changes of human T cells and activation of Vav, Rac, and extracellular signal-regulated kinase mitogen-activated protein kinase. J Immunol. 2001;167:6780–6785. doi: 10.4049/jimmunol.167.12.6780. [DOI] [PubMed] [Google Scholar]
- 25.Liszewski MK, Post TW, Atkinson JP. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu Rev Immunol. 1991;9:431–455. doi: 10.1146/annurev.iy.09.040191.002243. [DOI] [PubMed] [Google Scholar]
- 26.Wang G, Liszewski MK, Chan AC, Atkinson JP. Membrane cofactor protein (MCP; CD46): isoform-specific tyrosine phosphorylation. J Immunol. 2000;164:1839–1846. doi: 10.4049/jimmunol.164.4.1839. [DOI] [PubMed] [Google Scholar]
- 27.Astier AL, et al. RNA interference screen in primary human T cells reveals FLT3 as a modulator of IL-10 levels. J Immunol. 2010;184:685–693. doi: 10.4049/jimmunol.0902443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–577. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heeger PS, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201:1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price JD, et al. Induction of a regulatory phenotype in human CD4+ T cells by streptococcal M protein. J Immunol. 2005;175:677–684. doi: 10.4049/jimmunol.175.2.677. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez A, Feito MJ, Rojo JM. CD46-mediated costimulation induces a TH1-biased response and enhances early TCR/CD3 signaling in human CD4+ T lymphocytes. Eur J Immunol. 2004;34:2439–2448. doi: 10.1002/eji.200324259. [DOI] [PubMed] [Google Scholar]
- 33.Pennington DJ, et al. Early events in the thymus affect the balance of effector and regulatory T cells. Nature. 2006;444:1073–1077. doi: 10.1038/nature06051. [DOI] [PubMed] [Google Scholar]
- 34.Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- 35.Stockinger B, Veldhoen M. Differentiation and function of TH17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 36.Saraiva M, et al. Interleukin-10 production by TH1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31:209–219. doi: 10.1016/j.immuni.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powell JD, Lerner CG, Ewoldt GR, Schwartz RH. The -180 site of the IL-2 promoter is the target of CREB/CREM binding in T cell anergy. J Immunol. 1999;163:6631–6639. [PubMed] [Google Scholar]
- 38.Li Y, et al. SPAK kinase is a substrate and target of PKCθ in T-cell receptor-induced AP-1 activation pathway. EMBO J. 2004;23:1112–1122. doi: 10.1038/sj.emboj.7600125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delpire E, Gagnon KB. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J. 2008;409:321–331. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Liu XS. Development and function of IL-10 IFN-γ-secreting CD4+ T cells. J Leukoc Biol. 2009;86:1305–1310. doi: 10.1189/jlb.0609406. [DOI] [PubMed] [Google Scholar]
- 41.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 42.Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM. Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett. 2005;97:171–179. doi: 10.1016/j.imlet.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Friec GL, Kemper C. Complement: coming full circle. Arch Immunol Ther Exp (Warsz) 2009;57:393–407. doi: 10.1007/s00005-009-0047-4. [DOI] [PubMed] [Google Scholar]
- 44.Kerekes K, Prechl J, Bajtay Z, Jozsi M, Erdei A. A further link between innate and adaptive immunity: C3 deposition on antigen-presenting cells enhances the proliferation of antigen-specific T cells. Int Immunol. 1998;10:1923–1930. doi: 10.1093/intimm/10.12.1923. [DOI] [PubMed] [Google Scholar]
- 45.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest. 2006;116:3252–3257. doi: 10.1172/JCI29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins M. Tipping the balance in autoimmune disease. Genome Biol. 2007;8:317. doi: 10.1186/gb-2007-8-10-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meiler F, et al. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J Exp Med. 2008;205:2887–2898. doi: 10.1084/jem.20080193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 50.White J, et al. Biological activity, membrane-targeting modification, and crystallization of soluble human decay accelerating factor expressed in E. coli. Protein Sci. 2004;13:2406–2415. doi: 10.1110/ps.03455604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.