

Abstract
P190-B RhoGAP (p190-B, also known as ARHGAP5) has been shown to play an essential role in invasion of the terminal end buds (TEBs) into the surrounding fat pad during mammary gland ductal morphogenesis. Here we report that embryos with a homozygous p190-B gene deletion exhibit major defects in embryonic mammary bud development. Overall, p190-B deficient buds were smaller in size, contained fewer cells, and displayed characteristics of impaired mesenchymal proliferation and differentiation. Consistent with the reported effects of p190-B deletion on IGF-1R signaling, IGF-1R deficient embryos also displayed a similar small mammary bud phenotype. However, unlike the p190-B deficient embryos, the IGF-1R deficient embryos exhibited decreased epithelial proliferation and did not display mesenchymal defects. Because both IGF and p190-B signaling affect IRS-1/2, we examined IRS-1/2 double knockout embryonic mammary buds. These embryos displayed major defects similar to the p190-B deficient embryos including smaller bud size. Importantly, like the p190-B deficient buds, proliferation of the IRS-1/2 deficient mesenchyme was impaired. These results indicate that IGF signaling through p190-B and IRS proteins is critical for mammary bud formation and ensuing epithelial-mesenchymal interactions necessary to sustain mammary bud morphogenesis.
Keywords: p190-B, ARHGAP5, IGF-1R, IRS-1, IRS-2, mammary bud, epithelial-mesenchymal
Introduction
Mouse mammary gland development can first be detected as a line of epithelial cells that thicken between the limb buds at embryonic day 10.5–11.5 (E10.5–11.5) (DasGupta and Fuchs, 1999). The line fragments into individual placodes that appear as elevated domes, which then invaginate into the underlying dermal mesenchyme to form bulb-shaped buds at E12.5 (Mailleux et al., 2002). At E13 the bud enters a resting phase, and the mesenchyme of fibroblasts adjacent to the bud condenses into concentric rings that are separated from the bud by basement membrane (Kratochwil, 1969{Kimata, 1985 #122). This process coincides with differentiation of the mesenchyme, which is characterized by upregulation of fibronectin, tenascin-C, and androgen and estrogen receptors. Expression of these proteins distinguishes the mammary mesenchyme from the surrounding dermis and the underlying mesenchyme (Chiquet-Ehrismann et al., 1986; Durnberger and Kratochwil, 1980; Inaguma et al., 1988). This mesenchyme has been shown to be both permissive and instructive in mammary morphogenesis through a series of explant and tissue recombination experiments (Hens et al., 2007; Sakakura et al., 1976; Veltmaat et al., 2003). The cells underlying the mammary mesenchyme condense to form the fat pad precursor at E14 (Sakakura et al., 1982). The fad pad precursor becomes less compact at E15–16 forming a loose connective tissue and producing fatty substances (Sakakura, 1991). During this time the bud undergoes rapid proliferation leading to bud elongation. The distal end of the primary sprout breaks through the mammary mesenchyme and penetrates the fat pad precursor where it branches to give rise to the rudimentary ductal tree that is present at birth. Several studies have identified signaling pathways that are critically involved in regulating embryonic mammary bud development, and these studies highlight the importance of the interactions between the mammary bud and underlying mesenchyme (Hens and Wysolmerski, 2005).
P190-B is a member of the RhoGTPase activating protein (RhoGAP) family, which function as negative regulators of Rho activity (Burbelo et al., 1995). P190-B and Rho are both recruited to the plasma membrane following integrin crosslinking where p190-B functions to inhibit Rho activity by enhancing the intrinsic GTPase activity of Rho converting the active GTP to inactive GDP (Burbelo et al., 1995). Homozygous deletion of p190-B results in central nervous system defects leading to perinatal lethality (Sordella et al., 2002). Interestingly, p190-B deficient embryos and cells are smaller than their wildtype counterparts, which is due to impaired IGF signaling. Importantly, the p190-B and IGF signaling pathways have been shown to directly interact and to be critically involved in regulating both cell growth and differentiation (Sordella et al., 2002; Sordella et al., 2003).
The IGF-1 receptor (IGF-1R) functions through activation of an intrinsic tyrosine kinase in its cytoplasmic domain (Sachev, 2001). Upon ligand binding, the receptor becomes autophosphorylated and recruits intracellular substrates, including insulin receptor substrate (IRS) proteins. The IRS proteins (IRS-1,-2, -3, and -4), are adaptor molecules that organize signaling complexes at sites of receptor activation. IRS-1 and -2 are ubiquitously expressed including in mammary epithelium. IRS-3 and -4 are restricted in their localization, and are predominantly found in adipose tissue and brain, respectively (White, 1998). IRS-1 and IRS-2 play a critical role in IGF signaling as well as insulin, interferon, growth hormone, and integrin signaling.
The IGF signaling axis has been demonstrated to play a critical role in postnatal mammary gland development. Both loss and gain of IGF-IR disrupts postnatal ductal morphogenesis (Bonnette and Hadsell, 2001; Carboni et al., 2005; Jones et al., 2007). Loss of IRS-1 has been shown to reduce mammary fat pad size. IRS-1 does not appear to be critical for normal ductal development or pregnancy induced proliferation although it was hypothesized that IRS-2 might compensate for the loss of IRS-1 expression (Lee et al., 2003). However, despite structural and functional similarities, IRS-1 and IRS-2 are not interchangeable in terms of IGF-stimulated gene expression and cell cycle progression (Bruning et al., 1997).
Previous studies from our laboratory have demonstrated that p190-B interacts with the IGF-1R signaling pathway to regulate postnatal mammary ductal morphogenesis (Chakravarty et al., 2003). Specifically, both deletion of IGF-1R or haploinsufficiency of p190-B resulted in decreased proliferation in the terminal end buds (TEBs) and delayed ductal elongation. Additionally, embryonic mammary transplantation studies from p190-B and IGF-1R deficient mice have shown that these pathways are essential for postnatal mammary gland development, since the deficient transplants fail to grow out (Chakravarty et al., 2003), as is the case with p190-B, or have only limited outgrowth potential, as seen in IGF-1R deficient transplants (Bonnette and Hadsell, 2001). Overexpression of p190-B using a mammary specific tetracycline-inducible system also resulted in impaired TEB and ductal morphogenesis (Vargo-Gogola et al., 2006). In these mice, IGF signaling was altered further indicating a role for interaction of these pathways in regulating postnatal mammary gland development. Although it is clear that the IGF and p190-B pathways interact to regulate postnatal mammary gland development, whether they also contribute to embryonic mammary bud formation remained to be determined.
The studies presented here indicate a critical interaction of p190-B with the IGF signal transduction pathway during embryonic mammary morphogenesis and suggest that these pathways are critical for migration of epithelial progenitors. Furthermore, loss of either p190-B or IRS-1/2 results in major mesenchymal defects implicating this signaling network in establishment of the epithelial-mesenchymal interactions that are necessary to promote mammary bud development. We propose that these embryonic mammary defects underlie the failure of the p190-B deficient mammary glands to develop postnatally in part through modulation of the IGF signaling pathway.
Materials and Methods
Tissue preparation
Heterozygous females (p190-B C57Bl/6; IGF-1R FVB; IRS-1/IRS-2 FVB) were mated with heterozygous males with detection of a plug marked as E1. Pregnant females at E14.5 of pregnancy were injected with BrdU (100 mg/kg) intraperitoneally. Three hours later, embryos were dissected by Cesarean section in PBS and fixed overnight in 4% paraformaldehyde (PFA). Specimens were dehydrated and embedded in paraffin. Serial sections were then taken in the frontal plane at 7 μm on probe on plus slides (Fisher Scientific). Prior to hybridization, the slides were deparaffinized in xylene, rehydrated, and fixed in 4% PFA for 30 min.
In situ hybridization
Riboprobes were labeled with [DIG]-UTP (Boehringer 1277073), using T7 transcription system from Stratagene. To generate antisense riboprobe template, p190B cDNA was amplified from mouse brain by PCR using forward primer 5′-GGATCCTAATACGACTCACTATAGGGAGAATGAGATTTATGTTGTCCCAG and reverse primer 5′-GTAATTACTTTCCCAATTTCT. The sense riboprobe template was generated from the same cDNA by PCR using forward primer 5′-ATGAGATTTATGTTGTCCCAG and reverse primer 5′-GGATCCTAATACGACTCACTATAGGGAGAGTAATTACTTTCCCAATTTCT.
For whole mounts
Embryos were dissected and fixed in 4% PFA overnight, washed with 70% alcohol. To reduce the background the embryos were bleached with 4:1 mix of ethanol and 30% H202 for 1 hr and treated with 15 μg/ml proteinase K (PK) for 15 min. Proteinase K treatment was blocked with a short rinse in PBT followed by two washes in freshly prepared 2 mg/ml glycine in PBT. Embryos were re-fixed with freshly prepared 0.2% glutaraldehyde/4%PFA/PBS for 20 min. Fixative was replaced with pre-warmed (68°C) hybridization buffer (DAKO S3304) and rocked gently until the embryos sank (indicating that the formamide had penetrated the embryos). This hybridization buffer was replaced with fresh pre-warmed hybridization buffer containing the probe at a concentration of 1 μg/ml and hybridized overnight at 68°C. The embryos were then washed two times for 30 min each with freshly prepared pre-warmed solution I (50% Formamide, 5XSSC, 1%SDS) at 68°C followed by another wash of 10 min in a prewarmed 1:1 mix of solutions I & solution II (0.5M NaCl, 10mM TrisHCl pH 7.5, 0.1% Tween 20) at 68°C followed by 3 final washes of 5 min each with solution II at RT. Nonspecific hybridization was reduced by incubating the embryos for 30 min in 100ug/ml RNAase A in solution II at 37°C. Excess RNAse A was removed with two 30 min washes with freshly prepared, prewarmed solution III (50% deionized formamide, 2XSSC) at 65°C. Embryos were equilibrated with TBST containing 2mM levamisole and blocked with 10% FCS/2% blocking reagent (Roche 1096176)/2mM levamisole in TBST for at least 1 hr. This was followed by overnight incubation of the blocked embryos at 4°C with 1:1000 anti-digoxigenin-AP antibody (Roche 1093274) supernatant solution diluted 1:1 in 10% FCS/2% blocking reagent/TBST/2mM levamisole. Subsequently they were washed with 2mM levamisole/TBST solution for 5–6 hrs and left overnight in fresh levamisole/TBST at 4°C. Color development was initiated by washing the embryos with freshly prepared NTMT (100mM Nacl, 100mM TrisHCl pH 9.5, 50mM MgCl2, 0.1% Tween 20) +2mM Levamisole twice for 20 min and incubating them in prewarmed BM purple (Roche 1442074) from 30 min to overnight in the dark at room temperature (RT). The stained embryos were post fixed with freshly prepared 4%PFA.
For embryo sections
5 μm paraffin sections of E14.5 embryos were deparaffinized, rehydrated and washed in PBS. Then treated with PK (25 μg/ml) for 1 hr. at 37°C and fixed with 4% PFA for 30 min followed by washing with 2xSSC. Pre-warmed hybridization buffer containing the probe at a concentration of 1 μg/ml was added and hybridized overnight at 55°C. Slides were washed with stringency wash (50% 2xSSC, 50% formamide) for 15 min at 42 °C followed by washes with 2xSSC 20 min at RT and digestion with 40 μg/ml of RNase A for 15 min at 37 °C then washed with 0.1xSSC for 15 min at 42 °C and then 10 min at RT. Slides were then washed with Buffer I (100mM Tris pH 7.5, 150mM NaCl) for 5 min at RT and blocked for 2 hrs in Buffer I with 3% sheep serum and 0.3% triton X-100 at RT. The slides were then incubated overnight with 1:200 anti-digoxigenin-AP at 4 °C. For color development slides were washed with Buffer I for 10 min at RT and then washed with Buffer II (100mM Tris pH9.5, 100mM NaCl, 50mM MgCl) for 2 min at RT and incubating them in prewarmed BM purple (Roche 1442074) from 30 min to overnight in the dark at RT. Slides were counterstained with nuclear fast red, dehydrated, and mounted using Permount (Sigma, St. Louis, MO).
Immunohistochemistry
Five to seven micrometer sections were incubated overnight at 37°C, deparaffinized with xylene, and rehydrated with ethanols. Heat-induced antigen retrieval was performed in 10mM citrate by boiling for 20 min. All washes were performed with PBS unless otherwise stated. Slides were then incubated with the respective antibodies p63 (Neomarkers, MS-1081-P(Ab1): 1:500 in M.O.M diluent buffer (Vector Laboratories); androgen receptor (Upstate, 06-680): 1:500 in 5%BSA, 0.5% blocking buffer; estrogen receptor alpha (sc-542 (MC-20)): 1:200 in 5%BSA, 0.5% blocking buffer; IRS-1 (Upstate, 06-248): 1:800 in 5%BSA, 0.5% blocking buffer; IRS-2 (Upstate 06-506): 1:800 in 5%BSA, 0.5% blocking buffer; Biotin-conjugated BrdU (BD Pharmingen, 550803): 1:10 in 5% BSA, 0.5% blocking buffer all incubated overnight at RT. ER-α was used in place of AR due to non-specific staining in new lot of AR antibody. Sections were washed in PBS and incubated with anti-rabbit (Oncogene Research), or anti-mouse (Oncogene Research) secondary antibodies diluted 1:1000 in 5% BSA, 0.5% blocking buffer for 1 hr at RT. Vectastain Elite ABC and diaminobenzidine (DAB) substrate kits were used to detect immunoperoxidase staining according to the manufacturer’s instructions (Vector Laboratories). As a negative control, slides were incubated with purified rabbit immunoglobulin (The Jackson Laboratory). Detection was achieved by incubation with diaminobenzidine (DAKO) for 10 min or less. Slides were counterstained with hematoxylin for 30 sec, dehydrated, and mounted using Permount. Change in IRS-1 and IRS-2 staining was quantitated using ImagePro software by adding the mean color values within the same size ellipsis in both the wildtype and deficient p190-B embryos and converting the raw number to a percentage followed by inverting this percentage because in brightfield imaging more stain in the sample means that the camera sees less light. pAkt(Ser473) punctate staining was quantified using ImagePro software by subtracting the blue hematoxylin counterstain and then calculating the percentage of brown staining within the same size ellipsis to correct for the change in the area of the bud between the p190-B wildtype and deficient embryonic buds.
Results
p190-B is expressed in the developing mammary anlagen
Previously it was reported that p190-B is ubiquitously expressed in most adult tissues (Burbelo et al., 1995). In contrast, p190-B is developmentally regulated throughout postnatal mammary gland development (Chakravarty et al., 2000). Lack of ductal outgrowths from p190-B deficient transplants indicated that its expression might also be spatiotemporally restricted during embryonic development. We examined this by performing whole mount in situ hybridization for p190-B expression on wildtype embryos. For this analysis, we examined E8.5 embryos, prior to mammary development, and E12.5 embryos, which is the stage in mammary bud development where the spherical mammary placode differentiates into an epithelial bud. Although p190-B mRNA shares only 57% homology with p190-A at the nucleotide level, the specificity of p190-B antisense probe was further ascertained by aligning the probe sequence with that of mouse p190-A. In either case, no significant homology was detected between the two sequences. The sense probe was included as a negative control (Figure 1d) and all hybridizations were performed under highly stringent conditions.
Figure 1. p190-B is expressed throughout the differentiating mammary anlagen.
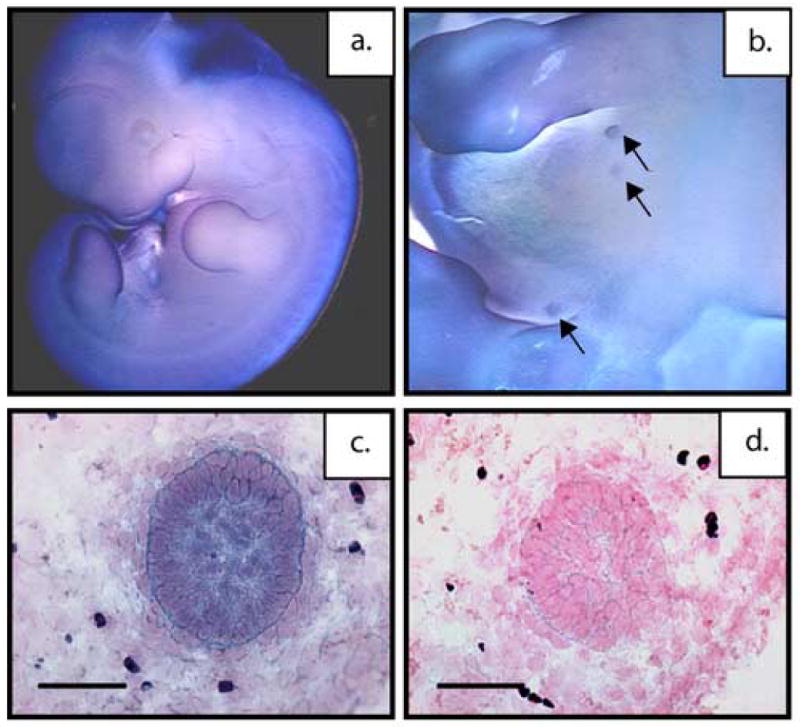
Whole-mount in situ hybridization of wildtype, E14.5 embryos with p190-B antisense riboprobe showing strong transcript expression in the in the developing mammary anlagen (a) low magnification (b) high magnification. Spatial localization of p190-B mRNA in E14.5 mammary buds of wildtype mice using DIG-labeled antisense riboprobe. Shown are representative antisense (c) and sense (d) images with strong transcript expression in the epithelial compartment of the mammary bud and lower expression in the mesenchyme. Scale bar 50 μm (c,d).
Ubiquitous expression of p190-B was detectable as early as E8.5 (data not shown). By E12.5, strong expression was detected in the brain, spinal cord, skin, and the limbs (Figure 1a). At E12.5, the p190-B transcript is detected throughout the mammary epithelial bud compartment (Figure 1b). This was further confirmed by in situ hybridization in tissue sections at E14.5 where expression of p190-B is present in the epithelium and at a lower level in the surrounding mesenchyme of wildtype embryonic mammary buds (Figure 1c) as compared to the sense control (Figure 1d). This expression pattern suggested p190-B might play an essential role in mammary placode formation and differentiation.
Loss of p190-B results in a smaller mammary bud size with a disorganized mesenchyme
While a number of signaling molecules have been shown to be expressed within the epithelium or mesenchyme of the developing bud, few have been shown to play a functional role in development of the bud. Because loss of p190-B resulted in complete failure of postnatal ductal development we examined whether p190-B deficiency also impacted formation and differentiation of the mammary anlagen. For this analysis, wildtype, heterozygous, and deficient E14.5 embryos were isolated and the histology of hematoxylin and eosin (H&E) stained sections was analyzed. Because the buds are known to form at different rates a bud-to-bud comparison was performed (Veltmaat et al., 2003). The wildtype buds (Figure 2a) had an organized epithelial center surrounded by a dense mesenchyme. The heterozygous buds displayed a variable intermediate phenotype. Some buds were comparable to the wildtype, while in others, the epithelial compartment was smaller and the surrounding mesenchyme appeared disorganized. In contrast, the buds from deficient embryos exhibited markedly fewer epithelial cells and the mesenchyme surrounding the epithelium appeared to be diminished and disorganized (Figure 2c). Bud size was determined by quantifying the number of sections through which the bud is detected, and for this analysis 3 buds were counted from 3 independent animals. A significant decrease in bud size is observed in the heterozygous (p<.001) and deficient (p<.0001) embryos as compared to the wildtype animals(Figure 2d).
Figure 2. p190-B−/− mice do possess distinct embryonic mammary buds but have reduced epithelial content and exhibit marked reduction of the mammary mesenchyme.
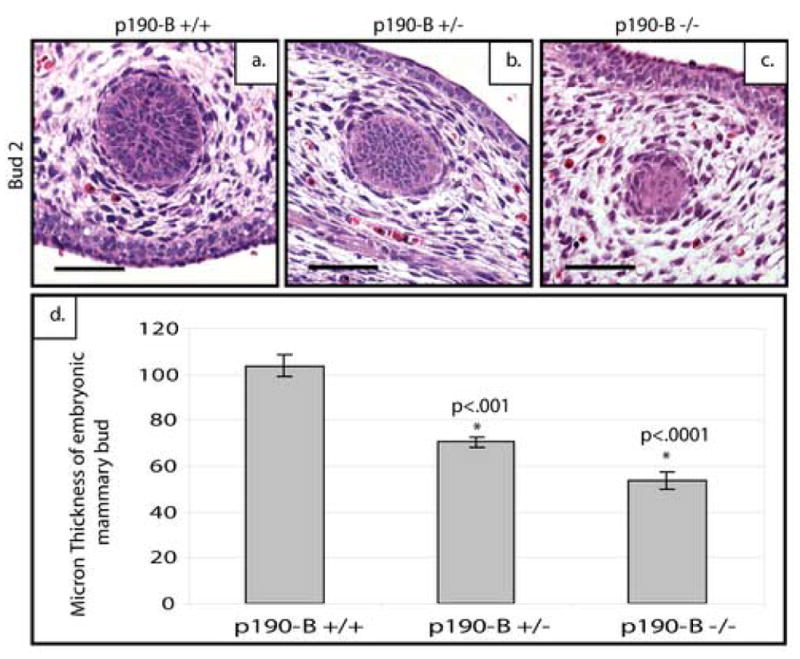
Sagittal sections of E14.5 embryonic mammary buds were stained with hematoxylin and eosin (H&E) to demonstrate a bud to bud comparison of the reduced number of epithelial cells and loss of a well-defined condensed mesenchyme around the p190-B-deficient (c) and heterozygous (b) buds compared to wildtype (a). Bud size is significantly decreased as shown by quantitation (d). Scale bar 50 μm.
To gain further insight into the role of p190-B signaling in placode formation, we examined the expression of markers of mammary epithelium and mesenchyme in tissues from the three genotypes (n=6). To evaluate possible alterations in progenitor epithelial content, the expression of p63 was compared in wildtype, heterozygous and p190-B-deficient E14.5 mammary anlagen. p63 is a member of the p53 gene family that has been demonstrated to be critical for the regulation of proliferation and differentiation in epithelial progenitor cells (Koster et al., 2004). p63 staining was seen in the epithelium of the mammary anlagen in wildtype mice (Figure 3a). Interestingly, a pronounced reduction in the number of p63-positive cells (p<.009) was detected in the p190-B deficient embryos suggesting that fewer p63-positive epithelial progenitors had migrated into the bud from the overlying epidermis (Figure 3c). As seen in Figure 3b, no statistically significant change was seen in the heterozygous group of embryos (Figure 3g).
Figure 3. Loss of p190-B results in reduced epithelial content and a marked reduction in mammary mesenchyme.
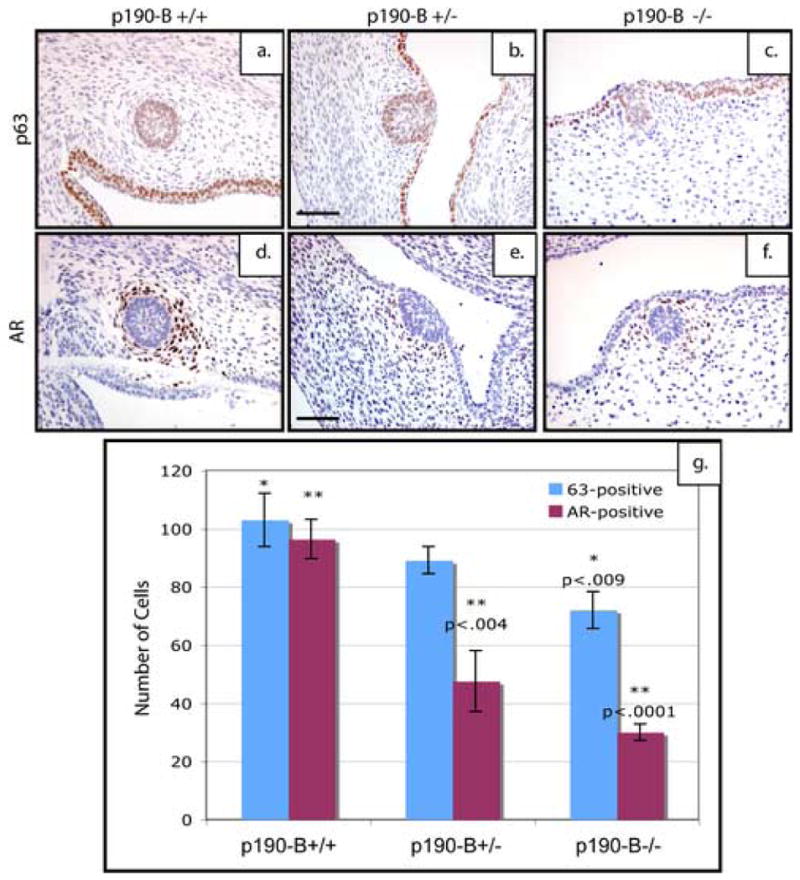
p63 expression in the epithelium of E14.5 mammary buds of (a)p190-B wildtype, (b)p190-B heterozygous & (c)p190-B deficient mice. Androgen receptor (AR) is expressed in the condensed mesenchyme surrounding the epithelial buds in (d) wildtype and (e) heterozygotes, however, the number of cells expressing AR are dramatically reduced in p190-B-deficient embryonic mammary buds (f). Quantitation of these staining patterns (g) show a statistically significant decrease in the number of p63-positive cells within the bud of the p190-B deficient embryos as well as a statistically significant decrease in the number of AR-positive cells within both the p190-B heterozygous and deficient anlagen as compared to the wildtype. Scale bar 100 μm.
To further evaluate the functional status of the mesenchyme, expression of Androgen Receptor (AR), a marker of mesenchymal differentiation, was examined in tissues from the three genotypes. In the wildtype embryonic anlagen, a large number of AR-positive cells formed the condensed mesenchyme (Figure 3d). However, the mammary anlagen from the p190-B heterozygous (p<.004) and deficient (p<.0001) mice consistently exhibited a reduction in the number of cells staining positive for AR in the mesenchyme (Figure 3e, f) as shown by the quantitation (Figure 3g). Taken together, these results suggest that loss of p190-B function impairs mesenchymal condensation and differentiation.
p190-B may interact with IGF-1R to affect migration of epithelial progenitors
Disruption of the IGF-1R gene has also been shown to retard postnatal mammary development (Bonnette and Hadsell, 2001). In particular, it has been reported that IGF-1R deficient embryonic mammary buds display reduced growth potential when transplanted into syngeneic hosts. This phenotype is reminiscent of p190-B heterozygous transplants. Furthermore, our previous studies implicated interaction of these pathways in the developing postnatal mammary gland (Chakravarty et al., 2003; Vargo-Gogola et al., 2006). We, therefore, explored the possibility that IGF-1R might also inhibit mammary anlagen formation and differentiation. To test this hypothesis, we analyzed mammary buds from IGF-1R wildtype and deficient embryos both histologically and through expression analysis of markers of epithelial and mesenchymal differentiation.
As shown in Figure 4, the H&E stained sagittal sections from E14.5 IGF-1R deficient embryonic mammary buds indicated that the smaller mammary bud phenotype of p190-B deficient mice was similarly observed in IGF-1R deficient mice (Figure 4b). The wildtype buds (Figure 4a) had an organized epithelial center surrounded by a dense mesenchyme whereas the deficient buds displayed very little epithelium (n=9). However, unlike the p190-B deficient mice, these buds did not show any apparent mesenchymal defects. These observations were further confirmed by staining tissue sections from all three genotypes with both p63 and Estrogen Receptor-α (ER) antibodies, another marker of mesencymal differentiation (n=9). The mammary buds from IGF-1R deficient mice had a significantly reduced number of p63-positive cells (Figure 4c, d), but did not appear to exhibit defects in the number of ER-positive cells (Figure 4e, f). A significant decrease in bud size is observed in the IGF-1R deficient (p<1×10−7) as compared to the wildtype (Figure 4g). Quantification of marker staining (Figure 4h) shows a decrease in the number of p63-positive cells within the IGF-1R deficient buds (p<3×10−6) as compared to the wildtype, but no significant difference in the number of intensely staining ER-positive cells. These results suggests that IGF-1R signaling is required for the recruitment of p63-positive progenitors into the mammary bud, but that loss of IGF-1R does not effect the epithelial-mesenchymal interactions that facilitate mesenchymal condensation and differentiation.
Figure 4. IGF-1R deficient mice phenocopy the small mammary bud phenotype of p190-B−/− mice, but not the mesenchymal defect.
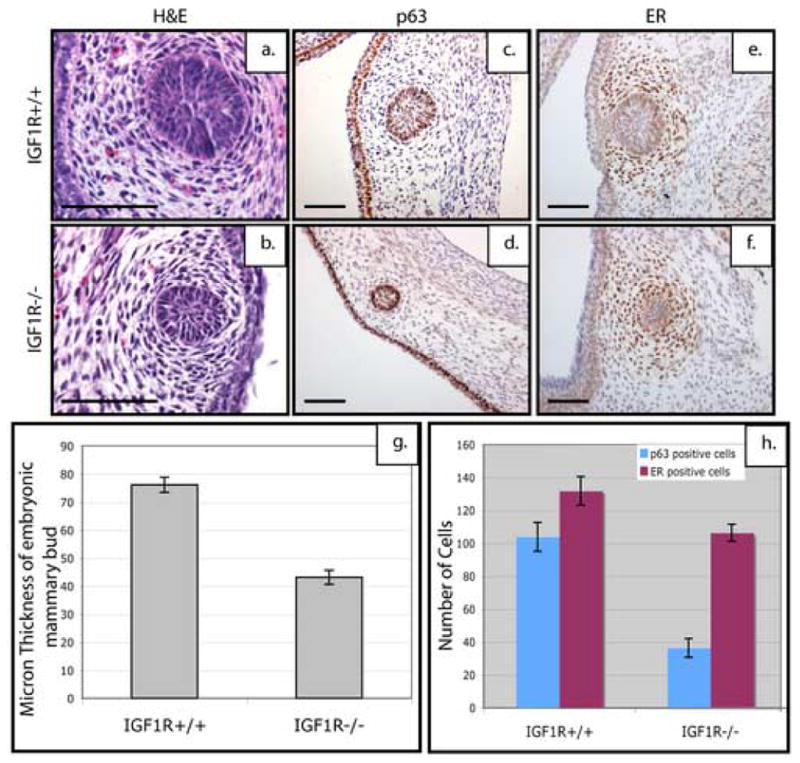
Shown here are the sagittal sections from wildtype and IGF-1R deficient E14.5 embryonic mammary buds stained with H&E. Note the smaller mammary buds in the deficient embryos (b) as compared to the wildtype (a) littermates. p63 expression in the epithelium of E14.5 mammary buds of (c) IGF-1R wildtype & (d) IGF-1R deficient mice. ER expression can be detected in the condensed mesenchyme of (e) wildtype and (f) IGF-1R deficient embryonic mammary buds. In panel (g) Bud size is significantly decreased as shown by quantitation. Quantitation of markers of each compartment (h) show a statistically significant decrease in the number of p63-positive cells within the bud of the IGF-1R deficient embryos but no statistically significant difference in the number of intense staining ER-positive cells between the IGF-1R deficient anaglen as compared to the wildtype. Scale bar 100 μm (a–f).
IRS-1/IRS-2 expression is decreased within the p190-B deficient mammary buds resulting in inhibition of downstream signaling
We previously reported that IRS-1 and -2 expression was decreased in TEBs of p190-B heterozygous mice (Chakravarty et al., 2003). Fibroblasts from p190-B deficient embryos exhibited an increase in Rho kinase activity, which resulted in inhibition of IGF/insulin signaling as well as the activity of several downstream effectors including p38, JNK, and Akt (Sordella et al., 2002). This modulation of IGF signaling was shown to be due to phosphylation of IRS-1 on Ser612, which targets IRS-1 for degradation (Sordella et al., 2002). To examine if the downstream effectors of the IGF signaling pathway are affected in the p190-B deficient mammary buds, we examined the level of IRS-1 and IRS-2 (n=3). Quantitation of this staining (Figure 5e) demonstrated that the intensity of IRS-1 and IRS-2 staining was decreased (p<.05) within the epithelial compartment of the p190-B deficient (Figure 5b, d) as compared to the wildtype mammary bud (Figure 5a, c). Interestingly the IRS-1 expression is also decreased in the mesenchyme. These results are consistent with previous reports that loss of p190-B alters the IRS proteins.
Figure 5. Decrease in IRS protein expression and the downstream IGF signaling component phospho-Akt in p190-B deficient mammary buds.
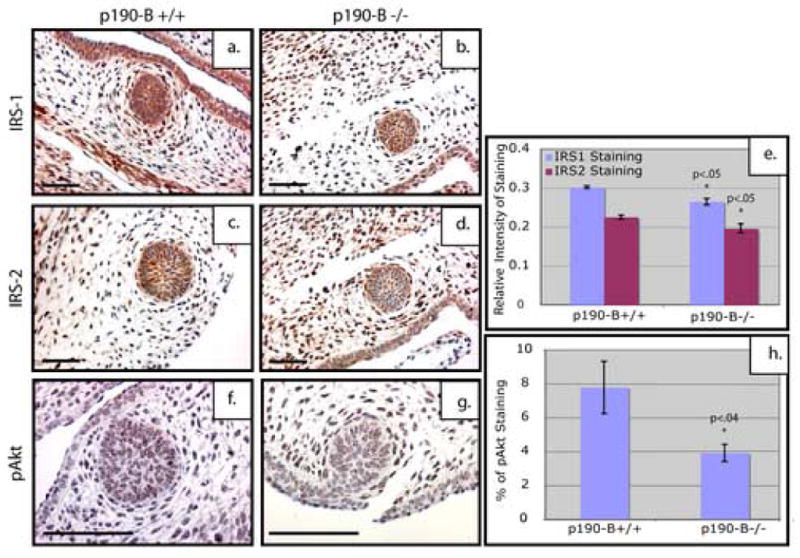
Sagittal sections of E14.5 embryonic mammary buds stained with IRS-1 (a, b); IRS-2 (c, d); pAkt(Ser473) (f, g). Note the obvious reduction in both IRS-1 and IRS-2 as well as the decrease in pAkt in the p190-B deficient buds (b, d, g) as compared to the wildtype (a, c, f). Quantitation of IRS-1 and IRS-2 staining (e) using ImagePro software shows a statistically significant decrease in intensity of brown staining in the p190-B deficient embryos as compared to the wildtype. Quantitation of the pAkt staining (h) shows a statistically significant decrease in pAkt within the mammary bud of the p190-B-deficient embryos as compared to the wildtype. Scale bar 100 μm.
To further examine the mechanism by which p190-B loss leads to a decrease in IRS-1 and affects downstream signaling, we assessed the activation of Akt by performing immunostaining to detect phospho-Akt (Ser473) (n=3). Punctate nuclear staining for phospho-Akt staining was detected in the p190-B wildtype buds (Figure 5f) and was decreased in the p190-B deficient buds (Figure 5g). Quantitative analysis shows a decrease in pAkt within the p190-B-deficient epithelium (p<.04) as compared to the wildtype (Figure 5h). These results suggest that loss of p190-B disrupts signaling downstream of IGF-1R, which may affect formation of the mammary bud.
Loss of IRS-1/2 phenocopies loss of p190-B affecting migration of epithelial progenitors and condensation of mammary mesenchyme
Because p190-B has been shown to regulate the level of IRS proteins both during postnatal ductal development and within the mammary bud, we hypothesized that loss of IRS-1 and IRS-2 may phenocopy the loss of p190-B (Chakravarty et al., 2003). To test this, we analyzed mammary buds from IRS-1/2 double deficient embryos both histologically and through expression analysis of markers of epithelial and mesenchymal differentiation (n=6). As shown in Figure 6, the E14.5 IRS-1/2 double deficient embryos appeared to exhibit a similar smaller mammary bud phenotype seen in the IGF-1R deficient mice and the p190-B deficient mice. The wildtype buds (Figure 6a) displayed an organized epithelial center surrounded by a dense mesenchyme, whereas the double-deficient buds displayed very little epithelium (Figure 6c), and the double heterozygous buds displayed a variable phenotype. This was quantitated in the same manner as the p190-B deficient buds and a similar decrease in size was seen in the IRS-1/2 heterozygous (p<.02) and deficient (p<.001) mammary buds (Figure 6d). However, unlike the IGF-1R deficient mice, the mesenchyme surrounding the epithelium appeared to be diminished and disorganized much like the p190-B deficient embryos. These observations were further confirmed by staining tissue sections of all three genotypes with both p63 and AR antibodies. The mammary buds from both the IRS-1/2 heterozygous and deficient mice exhibited a decrease in p63-positive cells (Figure 7c), as well as a marked reduction in the number of cells staining positive for AR in the mesenchyme (Figure 7f). Quantification of these staining patterns (Figure 7g) showed a statistically significant decrease in the number of p63-positive cells within the buds of the IRS-1/2 heterozygous (p<.003) and deficient (p<.0006) embryos as well as a statistically significant decrease in the number of AR-positive cells within the heterozygous (p<.001) and deficient (p<4×10−7) anlagen as compared to the wildtype. Taken together, these results demonstrate that the IRS-1/2 deficient mammary buds phenocopy the p190-B deficient buds.
Figure 6. Defective epithelial and mesenchymal differentiation in IRS-1/2-deficient mice.
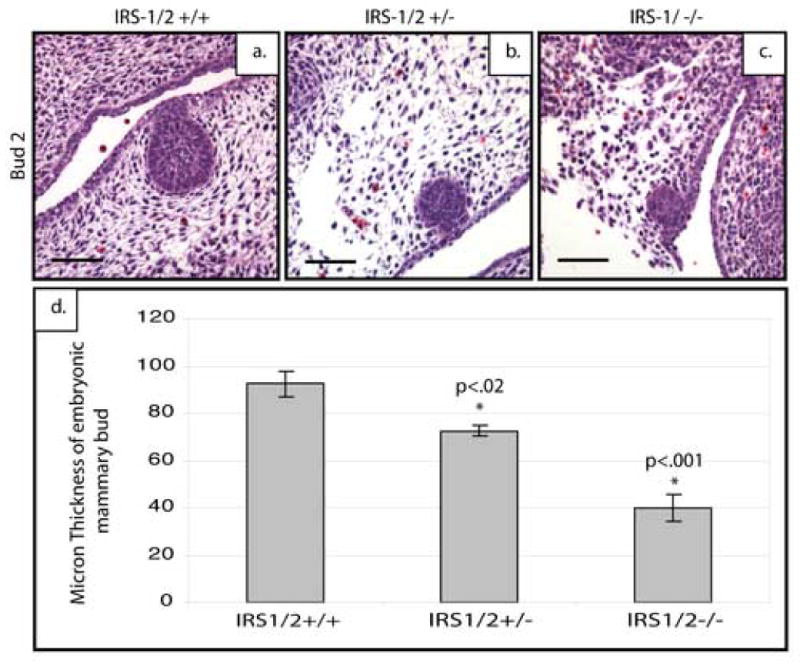
Sagittal sections of E14.5 embryonic mammary buds stained with H&E to demonstrate a bud to bud comparison of the reduced number of epithelial cells and loss of a well defined condensed mesenchyme around the IRS-1/2-deficient (c) and heterozygous (b) buds compared to wildtype (a). Bud size is significantly decreased as shown by quantitation (d). Scale bar 100 μm.
Figure 7. Loss of IRS-1/2 expression results in reduced bud size and disrupted mesenchyme at E14.5.
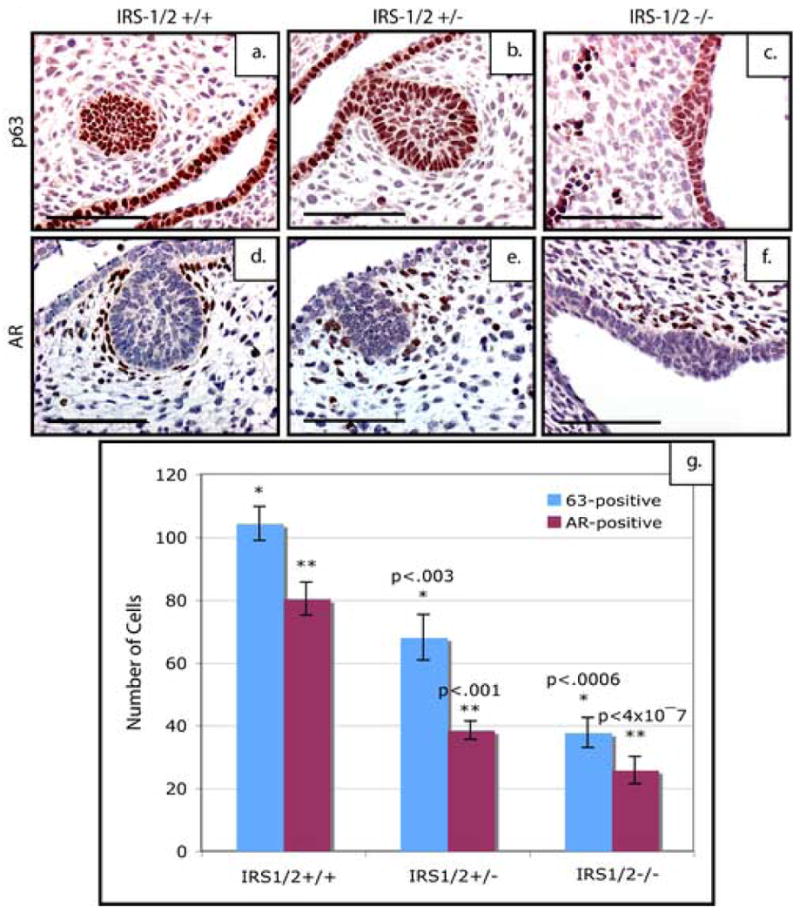
p63 expression in the epithelium of E14.5 mammary buds of (a)IRS-1/2+/+, (b) IRS-1/2+/−, (c) IRS-1/2−/− mice. Note the dramatic reduction in the number of epithelial cells that stain for p63 both in the heterozygous (b) and IRS-1/2-deficient mammary placodes (c). Note the dramatic reduction of AR-positive cells within the condensed mesenchyme surrounding the epithelial buds in (e) heterozygous and (f) deficient anlagen as compared to the wildtype. Quantitation of these staining patterns (g) show a statistically significant decrease in the number of p63-positive cells with in bud of the IRS-1/2 heterozygous and deficient embryos as well as a statistically significant decrease in the number of AR-positive cells within the p190-B heterozygous and deficient anlagen as compared to the wildtype. (f). Scale bar 100 μm.
Loss of either p190-B or IRS-1/2 leads to a defect in mesenchymal proliferation at E14.5
It is possible that p190-B affects the mesenchyme by either interrupting pathways necessary for the mesenchymal differentiation or affecting signaling pathways necessary for the proliferation. Previous studies have pointed to a role of IGF-1R in proliferation and migration of mammary epithelial and breast cancer cells (Bonnette and Hadsell, 2001; Zhang et al., 2004; Zhang et al., 2005). To investigate whether p190-B is required for mesenchymal proliferation, we examined cell proliferation by quantitating the number of BrdU-positive cells associated with the mammary bud at day E14.5 of p190-B (Figure 8a–c) and IRS-1/2 (Figure 8d–f) embryos (n=6). As shown in Figure 8g, loss of p190-B resulted in a decrease in proliferation of the mesenchymal cells surrounding the E14.5 mammary bud (p<.01). This decrease was calculated by determining the percentage of BrdU-positive cells that also stained positive for AR in paired serial sections. The IRS-1/2 deficient E14.5 mammary buds exhibited a similar decrease in the level of mesenchymal proliferation (p<.01) (Figure 8h). A significant decrease was also detected in the heterozygous IRS-1/2 mammary buds (p<.03). Importantly, there were no cleaved-caspase 3-positive cells detected in either the p190-B or IRS-1/2 mammary buds in any of the genotypes tested (data not shown), suggesting that the smaller bud size is not due to increased apoptosis. Only a few of the epithelial cells within the E14.5 bud were proliferating, and no difference was observed in the epithelial proliferation between wildtype and p190-B or IRS-1/2 deficient mammary buds as calculated by determining the percentage of BrdU-positive cells within the mammary epithelium. Interestingly, in the IGF-1R embryonic buds stained with BrdU (Figure 9a–c) there was no statistically significant change in the proliferation within the mammary mesenchyme as determined by the percentage of BrdU-positive cells that also stained positive for ER in a serial section (n=6). However there was a significant decrease in the proliferation within the mammary epithelium in both the heterozygous (p<.05) as well as the deficient (P<3×10−5) buds as compared to the wildtype. Furthermore, there was no change in the proliferation, ranging from 25–35%, of the overlying epidermis in any of the genotypes including the p190-B, IRS-1/2, or IGF-1R embryos (data not shown). These results coupled with the altered expression of mesenchymal markers indicate that p190-B and the IRS proteins play a critical role in regulating the mesenchymal compartment within the developing mammary anlagen and may play a role in migration of the mammary epithelial cells from the overlying epiderimis. However, our findings suggest that IGF-1R is critical for proliferation of the mammary bud, but is not critical for mesenchymal proliferation.
Figure 8. Loss of either p190-B or IRS-1/2 leads to a defect in mesenchymal proliferation at E14.5.
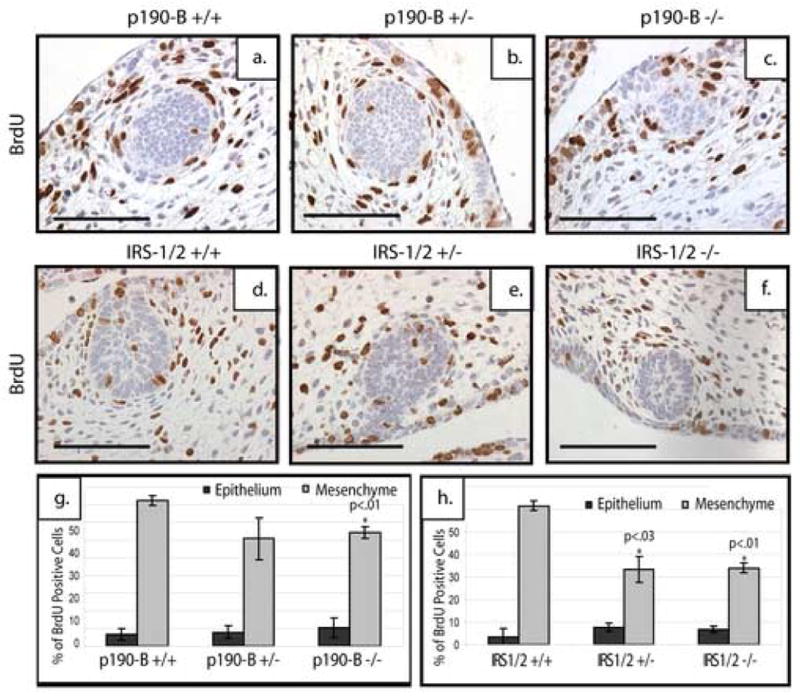
Saggital sections of E14.5 mammary buds stained for BrdU. Loss of p190-B (c) leads to a reduced number of BrdU-positive cells in the mesenchyme as compared to wildtype (a) and heterozygous (b) mice. Loss of one (e) or two copies (f) of IRS-1/2 also leads to a reduced number of BrdU-positive cells within the mammary mesenchyme as compared to the wildtype (d). Quantitation of BrdU postitive staining revealed loss of p190-B (g) or IRS-1/2 (h) leads to a decrease in the percentage of proliferating mesenchymal cells, but no change in epithelial proliferation. Scale bar 100 μm.
Figure 9. Loss of IGF-1R leads to a defect in epithelial proliferation at E14.5 but no significant change in mesenchymal proliferation.
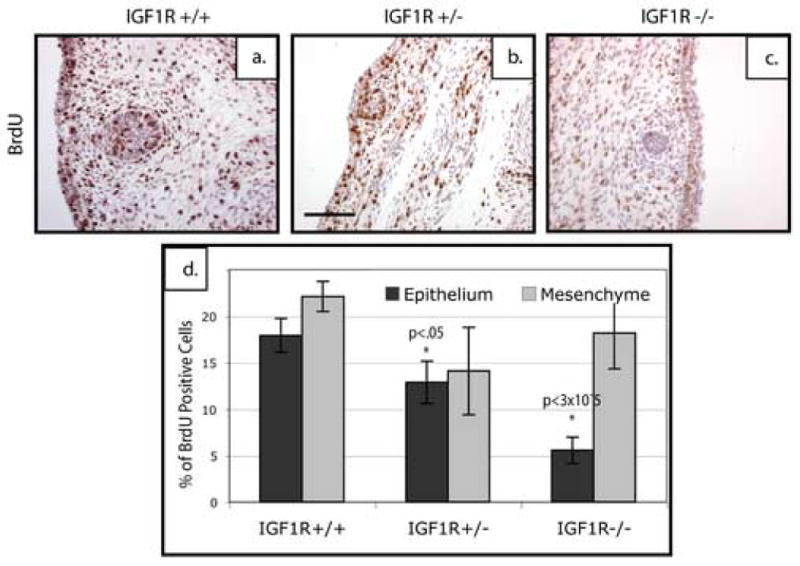
Saggital sections of E14.5 mammary buds stained for BrdU. Loss of one (b) or two copies (c) of IGF-1R leads to a reduced number of BrdU-positive cells in the epithelium as compared to wildtype controls (a). Quantitation of this proliferation shows a significant decrease in proliferation within the epithelial compartment of the heterozygous and p190-B-deficient mammary buds as compared to the wildtype, but no significant change in the mesenchymal proliferation. Scale bar 100 μm.
Discussion
Previous genetic studies have implicated several signaling pathways such as fgf/fgfr, Lef-1/Tcf, Tbx-3, PTHrP/PTHrPR, and Eda/Edar in various stages of bud development. Most of these pathways have been shown to play a role in epithelial-mesenchymal interactions where the ligand is expressed in one compartment and the receptor is expressed in the reciprocal compartment (Veltmaat et al., 2003). Here we report that p190-B RhoGAP is another essential gene for mammary bud formation. P190-B mRNA was detected in both the epithelium and mesenchyme of the bud suggesting that it may regulate both compartments. Accordingly, our results propose that p190-B mediates two key processes in mammary bud formation including the migration of mammary progenitor cells from the epidermis into the mammary bud as well as mesenchymal proliferation and condensation. Importantly, these data indicate that defects in embryonic mammary bud development underlie the failure of p190-B deficient mammary glands to develop postnatally.
The IGF signaling axis has been identified as a crucial interacting pathway with p190-B. As evidence of this, p190-B deficient embryos and fibroblasts exhibit impaired cell growth and differentiation as a result of altered IGF signaling (Sordella et al., 2002; Sordella et al., 2003). Previously we demonstrated that both loss and gain of p190-B function affects IGF signaling in the developing postnatal mammary gland (Chakravarty et al., 2003; Vargo-Gogola et al., 2006). Furthermore, we have demonstrated by immunohistochemical analysis that IRS-1 and IRS-2 expression as well as activation of the downstream effector Akt are decreased in the p190-B deficient mammary buds.
To further investigate the role of IGF and p190-B signaling interactions in mammary bud development, we examined the effects of loss of either IGF-1R or IRS-1/2 expression on mammary bud development. Strikingly, IRS-1/2 deficient mammary buds phenocopy the small bud size and aberrant mesechyme observed in the p190-B deficient buds. Although IGF-1R loss resulted in a similar small bud phenotype, the mesenchyme appeared unaffected suggesting that IGF-IR is not required for proper formation of the mesenchyme. In support of this, loss of p190-B or IRS-1/2 significantly inhibited mesenchymal proliferation whereas IGF-1R loss had no effect. These data complement our previous loss and gain of function studies, which suggested that cross-talk between the p190-B and IGF signaling pathways plays a critical role in postnatal mammary gland development.
An important question arising from these studies is what is the mechanistic basis for the small bud phenotype detected in the absence of p190-B, IRS-1/2, or IGF-IR. Furthermore, why does IRS-1/2 loss phenocopy the loss of p190-B, whereas IGF-1R loss only partially recapitulates this phenotype? Our data suggest that there are distinct mechanisms that affect bud size in the absence of p190-B and IRS-1/2 as compared to IGF-1R. It is well known that mesenchymal condensation and differentiation is an early and essential process in mammary bud formation. The mesenchyme is both permissive and instructive and plays a critical role in directing mammary epithelial cell fate (Hens and Wysolmerski, 2005). We have demonstrated that the absence of either p190-B or IRS-1/2 expression in the developing bud profoundly affects mesenchymal proliferation and condensation. We suggest that this aberrant mesenchyme is not capable of directing migration of the p63-positive mammary progenitor cells from the overlying epidermis into the developing bud resulting in the small bud phenotype. In the case of IGF-1R loss, our data indicate that the small bud size results from decreased proliferation in the epithelial compartment of the bud, although we cannot rule out the possibility of a compounding migratory defect. We suggest that in these buds, signaling from the mesenchyme is intact, but that the epithelium is not competent to respond. It is likely that the lack of any apparent proliferation defects in the epithelial compartment of the p190-B or IRS-1/2 deficient buds is obscured by the mesenchymal defects. Importantly, there was no change in proliferation of the overlying epidermis in any of the models examined, supporting the idea of a migratory defect. Interestingly, mice overexpressing p190-B also showed impaired epithelial-mesenchymal interactions (Vargo-Gogola et al., 2006).
The mesechyme may form properly in the IGF-1R deficient mammary anlagen through a variety of compensatory mechanisms. Signaling to IRS-1 and IRS-2 via other growth factor receptors including other IGF-R family members, insulin receptor, or through epidermal growth factor receptor (EGFR) are possible mechanisms, which may explain the observed effects on cell migration. EGF has been shown to increase expression of both IRS-1 and IRS-2 in breast cancer cell lines with IRS-2 being required for cell migration (Cui et al., 2006). Many studies have also shown a link between integrins and IRS-1 and IRS-2. However, there may also be redundancy in this pathway as mammary glands from either α3- or α6-integrin-deficient epithelium are fully developed and functional (Klinowska et al., 2001). Loss of β1 integrin leads to defects in alveolargenesis and lactation in the mammary gland, while loss in the epidermis leads to defects in assembly of basement membrane proteins resulting in defects in hair follicle differentiation though altered proliferation as well as impaired cytoskeletal dynamics leading to reduced migration (Brakebusch et al., 2000; Grose et al., 2002; Naylor et al., 2005; Raghavan et al., 2003).
The studies reported here enhance a growing understanding of the molecular mechanisms that regulate embryonic mammary gland formation. The roles of the p190 RhoGAPs are diverse and include involvement in proliferation (Chakravarty et al., 2003; Su et al., 2003), cell transformation (Ellis et al., 1990), EMT (Ozdamar et al., 2005), gene transcription (Jiang et al., 2005), integrin signaling (Burbelo et al., 1995), cytoskeletal reorganization (Hall, 1998), vesicular trafficking and development (Bernards and Settleman, 2005). Many of these contrasting functions may be attributed to their large molecular weight, the presence of multiple protein motifs, their ability to act as a scaffold, and their capacity to interact with various chromatin-remodeling complexes through its GAP domain (Nagaraja and Kandpal, 2004). Further studies including in vivo mammary bud specific gene arrays and pathway specific inhibitor studies will yield more insight into the roles of p190-B and IRS-1/2 in embryonic mammary bud development.
Acknowledgments
These studies were supported through the CA030195-22 (JMR) and CA94118 (AVL). T. V-G. is supported by a Howard Temin Pathway to Independence award (1K99CA127361-01). BMH is supported by a Department of Defense Breast Cancer Program Predoctoral Traineeship Fellowship (DAMD W81XWH-06-1-0704). Thanks to Mr. Walter Olea for technical assistance with the IRS knockout mice and Remigo Lopez from the Breast Center Pathology Core for the embryo sectioning.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernards A, Settleman J. GAPs in growth factor signalling. Growth Factors. 2005;23:143–9. doi: 10.1080/08977190500130480. [DOI] [PubMed] [Google Scholar]
- Bonnette SG, Hadsell DL. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology. 2001 Nov;142(11):4937–45. doi: 10.1210/endo.142.11.8500. [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Grose R, Quondamatteo F, Ramirez A, Jorcano JL, Pirro A, Svensson M, Herken R, Sasaki T, Timpl R, Werner S, Fassler R. Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. Embo J. 2000;19:3990–4003. doi: 10.1093/emboj/19.15.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JC, Winnay J, Cheatham B, Kahn CR. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 1997;17:1513–21. doi: 10.1128/mcb.17.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Miyamoto S, Utani A, Brill S, Yamada KM, Hall A, Yamada Y. p190-B, a new member of the Rho GAP family, and Rho are induced to cluster after integrin cross-linking. J Biol Chem 1995. 1995;270:30919–26. doi: 10.1074/jbc.270.52.30919. [DOI] [PubMed] [Google Scholar]
- Carboni JM, Lee AV, Hadsell DL, Rowley BR, Lee FY, Bol DK, Camuso AE, Gottardis M, Greer AF, Ho CP, Hurlburt W, Li A, Saulnier M, Velaparthi U, Wang C, Wen ML, Westhouse RA, Wittman M, Zimmermann K, Rupnow BA, Wong TW. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005;65:3781–7. doi: 10.1158/0008-5472.CAN-04-4602. [DOI] [PubMed] [Google Scholar]
- Chakravarty G, Hadsell D, Buitrago W, Settleman J, Rosen JM. p190-B RhoGAP regulates mammary ductal morphogenesis. Mol Endocrinol. 2003;17:1054–65. doi: 10.1210/me.2002-0428. [DOI] [PubMed] [Google Scholar]
- Chakravarty G, Roy D, Gonzales M, Gay J, Contreras A, Rosen JM. P190-B, a Rho-GTPase-activating protein, is differentially expressed in terminal end buds and breast cancer. Cell Growth Differ. 2000;200011:343–54. [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Mackie EJ, Pearson CA, Sakakura T. Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell. 1986;47:131–9. doi: 10.1016/0092-8674(86)90374-0. [DOI] [PubMed] [Google Scholar]
- Cui X, Kim HJ, Kuiatse I, Kim H, Brown PH, Lee AV. Epidermal growth factor induces insulin receptor substrate-2 in breast cancer cells via c-Jun NH(2)-terminal kinase/activator protein-1 signaling to regulate cell migration. Cancer Res. 2006;66:5304–13. doi: 10.1158/0008-5472.CAN-05-2858. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–68. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Durnberger H, Kratochwil K. Specificity of tissue interaction and origin of mesenchymal cells in the androgen response of the embryonic mammary gland. Cell. 1980;19:465–71. doi: 10.1016/0092-8674(80)90521-8. [DOI] [PubMed] [Google Scholar]
- Ellis C, Moran M, McCormick F, Pawson T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature. 1990;343:377–81. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- Grose R, Hutter C, Bloch W, Thorey I, Watt FM, Fassler R, Brakebusch C, Werner S. A crucial role of beta 1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development. 2002;129:2303–15. doi: 10.1242/dev.129.9.2303. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science 1998. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hens JR, Dann P, Zhang JP, Harris S, Robinson GW, Wysolmerski J. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction. Development. 2007;134:1221–30. doi: 10.1242/dev.000182. [DOI] [PubMed] [Google Scholar]
- Hens JR, Wysolmerski JJ. Key stages of mammary gland development: molecular mechanisms involved in the formation of the embryonic mammary gland. Breast Cancer Res. 2005;7:220–4. doi: 10.1186/bcr1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaguma Y, Kusakabe M, Mackie EJ, Pearson CA, Chiquet-Ehrismann R, Sakakura T. Epithelial induction of stromal tenascin in the mouse mammary gland: from embryogenesis to carcinogenesis. Dev Biol. 1988;128:245–55. doi: 10.1016/0012-1606(88)90288-6. [DOI] [PubMed] [Google Scholar]
- Jiang W, Sordella R, Chen GC, Hakre S, Roy AL, Settleman J. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Mol Cell. 2005;17:23–35. doi: 10.1016/j.molcel.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, Moorehead RA. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26:1636–44. doi: 10.1038/sj.onc.1209955. [DOI] [PubMed] [Google Scholar]
- Klinowska TC, Alexander CM, Georges-Labouesse E, Van der Neut R, Kreidberg JA, Jones CJ, Sonnenberg A, Streuli CH. Epithelial development and differentiation in the mammary gland is not dependent on alpha 3 or alpha 6 integrin subunits. Dev Biol. 2001;233:449–67. doi: 10.1006/dbio.2001.0204. [DOI] [PubMed] [Google Scholar]
- Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18:126–31. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil K. Organ specificity in mesenchymal induction demonstrated in the embryonic development of the mammary gland of the mouse. Dev Biol. 1969;20:46–71. doi: 10.1016/0012-1606(69)90004-9. [DOI] [PubMed] [Google Scholar]
- Lee AV, Zhang P, Ivanova M, Bonnette S, Oesterreich S, Rosen JM, Grimm S, Hovey RC, Vonderhaar BK, Kahn CR, Torres D, George J, Mohsin S, Allred DC, Hadsell DL. Developmental and hormonal signals dramatically alter the localization and abundance of insulin receptor substrate proteins in the mammary gland. Endocrinology. 2003;144:2683–94. doi: 10.1210/en.2002-221103. [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Spencer-Dene B, Dillon C, Ndiaye D, Savona-Baron C, Itoh N, Kato S, Dickson C, Thiery JP, Bellusci S. Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development. 2002 Jan;129(1):53–60. doi: 10.1242/dev.129.1.53. [DOI] [PubMed] [Google Scholar]
- Nagaraja GM, Kandpal RP. Chromosome 13q12 encoded Rho GTPase activating protein suppresses growth of breast carcinoma cells, and yeast two-hybrid screen shows its interaction with several proteins. Biochem Biophys Res Commun. 2004;313:654–65. doi: 10.1016/j.bbrc.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, Wang P, Schatzmann F, Wintermantel T, Schuetz G, Clarke AR, Mueller U, Hynes NE, Streuli CH. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol. 2005;171:717–28. doi: 10.1083/jcb.200503144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–9. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Raghavan S, Vaezi A, Fuchs E. A role for alphabeta1 integrins in focal adhesion function and polarized cytoskeletal dynamics. Dev Cell. 2003;5:415–27. doi: 10.1016/s1534-5807(03)00261-2. [DOI] [PubMed] [Google Scholar]
- Sachev DaDY. The IGF System and breast cancer. Endocr Relat Cancer. 2001;8:197–209. doi: 10.1677/erc.0.0080197. [DOI] [PubMed] [Google Scholar]
- Sakakura T. New aspects of stroma-parenchyma relations in mammary gland differentiation. Int Rev Cytol. 1991;125:165–202. doi: 10.1016/s0074-7696(08)61219-x. [DOI] [PubMed] [Google Scholar]
- Sakakura T, Nishizuka Y, Dawe CJ. Mesenchyme-dependent morphogenesis and epithelium-specific cytodifferentiation in mouse mammary gland. Science. 1976 Dec 24;194(4272):1439–41. doi: 10.1126/science.827022. [DOI] [PubMed] [Google Scholar]
- Sakakura T, Sakagami Y, Nishizuka Y. Dual origin of mesenchymal tissues participating in mouse mammary gland embryogenesis. Dev Biol. 1982;91:202–7. doi: 10.1016/0012-1606(82)90024-0. [DOI] [PubMed] [Google Scholar]
- Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, Le Z, Takami H, Yamada Y, Settleman J. Modulation of CREB Activity by the Rho GTPase Regulates Cell and Organism Size during Mouse Embryonic Development. Dev Cell. 2002;2(5):553–65. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
- Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113:147–58. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- Su L, Agati JM, Parsons SJ. p190RhoGAP is cell cycle regulated and affects cytokinesis. J Cell Biol. 2003;163:571–82. doi: 10.1083/jcb.200308007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargo-Gogola T, Heckman BM, Gunther EJ, Chodosh LA, Rosen JM. P190-B Rho GTPase-activating protein overexpression disrupts ductal morphogenesis and induces hyperplastic lesions in the developing mammary gland. Mol Endocrinol. 2006;20:1391–405. doi: 10.1210/me.2005-0426. [DOI] [PubMed] [Google Scholar]
- Veltmaat JM, Mailleux AA, Thiery JP, Bellusci S. Mouse embryonic mammogenesis as a model for the molecular regulation of pattern formation. Differentiation. 2003;71:1–17. doi: 10.1046/j.1432-0436.2003.700601.x. [DOI] [PubMed] [Google Scholar]
- White MF. The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Recent Prog Horm Res. 1998;53:119–38. [PubMed] [Google Scholar]
- Zhang X, Kamaraju S, Hakuno F, Kabuta T, Takahashi S, Sachdev D, Yee D. Motility response to insulin-like growth factor-I (IGF-I) in MCF-7 cells is associated with IRS-2 activation and integrin expression. Breast Cancer Res Treat. 2004;83:161–70. doi: 10.1023/b:brea.0000010709.31256.c6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lin M, van Golen KL, Yoshioka K, Itoh K, Yee D. Multiple signaling pathways are activated during insulin-like growth factor-I (IGF-I) stimulated breast cancer cell migration. Breast Cancer Res Treat. 2005;93:159–68. doi: 10.1007/s10549-005-4626-8. [DOI] [PubMed] [Google Scholar]