

Abstract
Mechanisms for pathogenic metal signaling in airway injury or disease promotion are poorly understood. It is widely believed that one mechanism for pathogenic and possible carcinogenic effects of inhaled chromium (Cr(VI)) is inhibition of inducible gene transactivation. However, we recently reported that Cr(VI) inhibition of Sp1-dependent transactivation required signal transducer and activator of transcription 1 (STAT1)-dependent expression of an inhibitory protein in airway epithelium. Thus, Cr(VI) exposures can induce genes and we hypothesized this induction resulted from Cr(VI) signaling through an innate immune-like STAT1-dependent pathway initiated by Fyn. Exposure of human airway epithelial (BEAS-2B) cells to Cr(VI) selectively transactivated STAT-responsive interferon-stimulated response element (ISRE) and induced ISRE-driven transactivation of interferon regulatory factor 7 (IRF7), without affecting the gamma interferon-activated site (GAS)-driven IRF1 expression. Cr(VI)-induced IRF7 was absent or greatly reduced in cells that lacked STAT1, were treated with the Src family kinase inhibitor, PP2, or lacked Fyn. Expressing Fyn, but not Src, in mouse embryonic fibroblasts cells null for Src, Yes, and Fyn restored Cr(VI)-stimulated STAT1 tyrosine phosphorylation and IRF7 expression. Finally, shRNA knockdown of Fyn in BEAS-2B cells prevented Cr(VI)-activated STAT1 transactivation of IRF7. These data support a novel mechanism through which Cr(VI) stimulates Fyn to initiate interferon-like signaling for STAT1-dependent gene transactivation.
Keywords: Chromium, STAT1, IRF7, HDAC, Fyn
Introduction
Chromium is an environmental pollutant that is known to promote airway diseases. Several million people world-wide are exposed to Cr(VI) or Cr(VI)-containing compounds (1,2) and chronic inhalation of the metal causes pulmonary diseases such as chronic bronchitis, asthma, emphysema, and cancers through poorly defined mechanisms(2–4). Under physiological conditions, Cr(VI) exists as a chromate oxyanion and, unlike Cr(III), can readily enter the cell through anion channels (4,5). Once Cr(VI) enters an airway cell, greater than 90% is trapped following reduction to Cr(III) (1,6–8). This reduction process generates reactive intermediates and these intermediates or the more thermodynamically stable Cr(III) have been implicated as the primary mediators of toxic effects on proteins and DNA (1). These one-way kinetics reduce cellular Cr clearance and thus make the epithelial lining of the airways a main target of inhaled Cr(VI) (9).
Cr(VI) exposures cause a well recognized, but poorly understood, repression of inducible gene transactivation, while failing to affect constitutive gene expression (8,10–12). Proposed mechanisms for this repression include response to DNA damage (10,11) or epigenetic effects resulting from chromium cross-linking of histone deacetylase-1 (HDAC-1) or DNA methyltransferase-1 to chromatin (11). The latter has been reported to repress more than 50 aryl hydrocarbon receptor-driven genes, including those expressing enzymes that metabolize environmental carcinogens (11). In addition to preventing aryl hydrocarbon receptor ligand from inducing genes, Cr(VI) blocks cytokine-induced NF-κB-dependent gene transactivation (12) and metal-induced Sp1-dependent transactivation of repair genes (13). In cells lacking STAT1, however, Cr(VI) both stimulates Sp1 transactivation and fails to block induced Sp1 transactivation (13).
The STAT proteins are a family of latent cytoplasmic transcription factors. There are seven different family members that are activated by exogenous stresses (14–17), including metals (18), to produce diverse biological responses. In the canonical activation scheme, cytoplasmic STAT protein monomers are thought to be activated through phosphorylation on a conserved tyrosine residue in the C-terminus to promote dimerization and translocation into the nucleus (19–21). However, this activation scheme is oversimplified, since there is ample evidence that STAT dimers exist in the cell cytosol and that other post-transcriptional modifications are involved in modulating STAT localization and activity (20). Nonetheless, it is evident that phosphorylation by non-receptor tyrosine kinases, such as Janus kinases (JAKs) and Src family kinases (SFKs), are involved in initiating STAT-dependent signaling (18–20,22,23). For instance, we previously demonstrated that Cr(VI)-activates both STAT1 and STAT3 in airway epithelium and that prolonged STAT3 activation required the SFK, Lck (18).
STAT1 was originally identified as a downstream effector of interferon (IFN) signaling (19) and is required to induce innate immune antiviral and antiproliferative responses (24,25). IFNγ stimulates STAT1 phosphorylation to form homodimers that bind to GAS DNA cis elements to transactivate interferon regulated gene promoters (e.g. interferon regulatory factor 1 (IRF1)) (26). In contrast, type I interferons (e.g IFN-α2) stimulate formation of both STAT homodimers and a heterotrimeric complex, interferon-stimulated gene factor 3 (ISGF3), consisting of STAT1, STAT2 and interferon regulatory factor 9 (IRF9). This heterotrimer binds to interferon-stimulated response elements (ISRE) to transactivate selective innate immune gene promoters (e.g. IRF7) (19,21,27,28). As opposed to its effect on aryl hydrocarbon receptor transactivation, HDAC-1 binding to the ISGF3 complexes and its enzymatic activity are required to recruit RNA polymerase II to the promoters of ISRE-, but not GAS-, driven genes (27,29).
The antiviral and anti-proliferative effects of IFN-α are limited or converted to proliferative responses in cells that lack STAT1 (25,30,31). STAT1 is deleted in a number of cancers and without STAT1 activation, IFN stimulation of STAT3 and 5 or their constitutive activation provides cell survival advantage and can cause transformation (25,30,31). In addition, STAT1 has also been implicated in the pathogenesis of lung diseases (17), including metal (vanadium)-induced airway fibrosis and asthma (32). STAT1 activation was localized to the bronchial epithelial cells in asthmatic patients (33), suggesting that aberrant expression of STAT1 may be a precursor to disease. In contrast and similar to the loss of repression in cancer cells, loss of STAT1 in the airways increases the severity of viral lung infections (34), as well as metal or chemical-induced injury (32,35).
We recently reported that Cr(VI) stimulated STAT1 in human airway epithelial cells to repress Sp1 transactivation of inducible genes (13). In addition, STAT-dependent transcriptional inhibition following Cr(VI) required protein synthesis, suggesting that Cr(VI) stimulates STAT1-dependent gene transactivation. Thus in the current study, we sought to identify the mechanism through which Cr(VI) exposures of airway epithelial cells stimulate STAT1-dependent transactivation of an innate immune response gene, IRF7. A significant finding was that this mechanism is unique to Cr(VI) relative to other lung toxic metals, such as vanadium.
Materials and Methods
Cell Culture
Human bronchial epithelial cells (BEAS-2B) (ATCC, Manassas, VA) and the stable BEAS-2B cell lines expressing random (shNC) or STAT1 (shSTAT1) shRNA were cultured on a matrix of 0.01 mg/ml of human fibronectin (Invitrogen, Carlsbad, CA), 0.029 mg/ml Vitrogen 100 (COHESION Inc, Palo Alto, CA), and 0.01 mg/ml bovine serum albumin (Invitrogen) in LHC-9 medium (Invitrogen). The parental BEAS-2B cells were cultured in LHC-9 medium and the stably transfected cell lines were cultured in the same medium supplemented with 75 μg/ml G418 (Sigma-Aldrich, St. Louis, MO). The cells were maintained at 37°C under an atmosphere of 5% CO2, as described previously (13,18). Experiments were performed on 1-day post-confluent cells unless otherwise indicated. Medium was changed 12–16 h prior to all experiments. Under these conditions, chromium stimulation of STAT1 and STAT3 was qualitatively identical in BEAS-2B cells and primary human bronchial epithelial cells grown in air/liquid interface cultures (18). SV-40 immortalized Janus kinase (MEF) (ATCC) were grown at 37°C under an atmosphere of 5% CO2 in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Pittsburgh, PA), 4 mM L-glutamine (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). The media was changed 12–16 h prior to the experiment to media containing 0.1% bovine serum albumin (BSA) in replacement of FBS to arrest cell growth. SYF MEF cells (ATCC) were derived from mouse embryos with functionally null mutations in Src, Yes, and Fyn (36). Src++ MEF cells (ATCC) were deficient in Yes and Fyn, but expressed endogenous Src (36). Wild-type MEF cells were used as the control cells in these experiments.
Treatments
Cr(VI) solutions (2.5 mM) were prepared fresh from potassium dichromate (Sigma-Aldrich, St. Louis, MO) and post-confluent cells were treated with 1 ml of stock solution per ml of LHC-9 culture medium for the indicated times (final Cr(VI) concentration = 5 μM). This exposure level is relevant to occupational exposures and is based on our previous demonstration of effective increases in cell signaling for gene expression changes without cytotoxicity in 76 h of exposure (18). Cells were pretreated with sodium butyrate (NaB) (Sigma-Aldrich) for 16 h to inhibit HDAC activity prior to addition of Cr(VI). Neutralizing antibody (1μg/ml) to IFN α/β receptor chain 2 (CD118) that block the proliferative response of all type 1 interferons (37) (Clone: MMHAR-2)PBL Biomedical Laboratories, New Brunswick, NJ) was administered 30 min prior to Cr(VI) exposures. IFN-α2 (100 U/ml) (PBL Biomedical Laboratories) was used as a positive control.
Construction of the Fyn Expression Vector (Fyn-myc)
The c-Fyn cDNA from pRK5 c-Fyn (Addgene, Inc, Cambridge, MA) was subcloned into pcDNA 3.1/myc-His(−) (Invitrogen) using BamHI and HindIII sites. The resultant clones were sequenced at the Genomics and Proteomics Core Laboratories at the University of Pittsburgh to verify the presence of c-Fyn cDNA.
Transient Transfections
Cells were transiently transfected at 70–80% confluence according to the Lipofectamine PLUS protocol (Invitrogen). For the luciferase reporter constructs, cells were transfected with ISRE-luc (BD Biosciences, San Jose, CA). After 24 h, cells were exposed to Cr(VI) or IFN-α2. For the MEF experiments, cells were transfected with either control vector, pcDNA™ 3.1/mycHis(−)C, or the Fyn-myc expressing plasmid. After 48 h, cells were exposed to Cr(VI).
Luciferase Assays
Cells were harvested and luciferase activity was measured as previously described (12,38). Relative light units (RLU) were determined for 20 sec in a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA). Data are presented as mean ± SEM of fold control.
RNA Isolation and RT-PCR
Total RNA was isolated using TRIzol® reagent (Invitrogen) and quantified by measuring absorbance (260 nm). Total RNA was reverse transcribed and IRF7 and Irf7 cDNA were amplified by RT-PCR (MJ Research PTC-100 Thermocycler (BioRad Laboratories, Hercules, CA)), as described previously (39). IRF1, β-actin, and ribosomal protein L13A (RPL13A) cDNA was amplified by real-time PCR (MJ Research Opticon 2 (BioRad Laboratories), as described previously (13). The following primers sets were used: IRF7 (forward 5′-TGTGGACACCTGTGACACCT-3′; reverse 5′-GTTATCTCGCAGCATCACGA-3′), IRF1 (forward 5′-TGCTAAGAGCAAGGCCAAGAG-3′; reverse 5′-CGACTGCTCCAAGAGCTTCAT- 3′), Irf7 (forward 5′-CAGTTGATCCGCATAAGGTGT-3′; reverse 5′-CTCGTAAACACGGTCTTGCTC-3′), ribosomal protein L13A (RPL13A) (forward 5′-cgaggttggctggaagtacc-3′; reverse 5′-attccagggcaacaatggag-3′), and β-actin (forward 5′-GGGACCTGACCGACTACCTC-3′; reverse 5′-GGGCGATGATCTTGATCTTC-3′). PCR products were separated on 2% agarose gels stained with ethidium bromide. The intensity of the bands was measured using Image J. IRF7 and IRF1 mRNA levels were normalized to RPL13A. Irf7 mRNA levels were normalized to β-actin. Data are presented as mean ± SEM of fold control.
Total protein isolation
Cells were rinsed twice in stop buffer (10 mM Tris-HCl, pH 7.4, 10 mM EDTA, 5 mM EGTA, 0.1 M NaF, 0.2 M sucrose, 100 μM sodium orthovanadate, 5 mM pyrophosphate) and scraped in boiling lysis buffer (20 mM Tris, pH 7.5, 1% SDS, 100 μM sodium orthovanadate, and supplemented with protease inhibitors (EMD Biosciences, San Diego, CA)). The lysates were boiled for 5 min. Protein concentrations were determined by the absobance at 595nm after addition of Coomassie blue dye (Pierce Biotechnology, Inc., Rockford, IL) using BSA as a reference standard.
Nuclear protein isolation
Cells were rinsed twice, scraped in stop buffer, and centrifuged at 400g for 10 min at 4°C. The pellet was resuspended in buffer A (10 mM HEPES, pH 7.9, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM KCl, 1 mM DTT, 0.1% NP-40, 100 μM sodium orthovanadate, supplemented with protease inhibitors), incubated on ice for 10 min, and centrifuged at 13,000g for 2 min at 4°C. The supernatant (cytosolic fraction) was transferred to a new tube and the pellet was rinsed in buffer A and centrifuged at 13,000g for 2 min at 4°C. The pellet was resuspended with buffer C (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 100 μM sodium orthovanadate, supplemented with protease inhibitors), vigorously shaken for 15 min at 4°C, and centrifuged at 13,000g for 5 min at 4°C. The supernatant (nuclear fraction) was combined with buffer D (20 mM HEPES, pH 7.9, 20% glycerol, 0.1 M KCl, 1 mM EDTA, 0.1 mM EGTA, 0.1% NP-40, 1 mM DTT, 100 μl sodium orthovanadate, supplemented with protease inhibitors) and protein concentrations were determined.
Immunoprecipitatons
Cells were rinsed twice with cold stop buffer and scraped in modified RIPA buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 10 mM NaF, 1% Triton X-100, 0.1% SDS, and supplemented with protease inhibitors and sodium orthovanadate). Lysates were incubated for 30 min on ice and then sonicated 3 times for 5 sec intervals. Lysates were centrifuged at 13,000g for 15 min at 4°C and the supernatants were collected. Equal amounts of protein were incubated with the antibody against total Fyn (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C on a rotating platform. Protein A/G beads (Thermo-Fisher Scientific) were added and incubated for an additional 3 h at 4°C. The beads were collected by centrifugation at 13,000g for 1 min, rinsed 3 times with modified RIPA buffer, resuspended in 2X sample buffer, and boiled for 5 min.
Western analysis
Changes in protein abundance were measured by SDS-PAGE. Proteins were transferred to PVDF membranes (Millipore, Billerica, MA) and blocked in 5% milk or BSA for 1 h before incubation with primary antibodies overnight at 4°C. Primary antibodies included STAT1 (human) (Cell Signaling Technology, Danvers, MA), phospho-Tyr416-Src family kinase (Cell Signaling Technology), phospho-STAT1 (Millipore), STAT1 (mouse) (Santa Cruz Biotechnology), total Fyn, and β-actin (Sigma-Aldrich). Antibody binding was detected with horseradish peroxidase-conjugated antibodies (GE Healthcare, Piscataway, NJ) and enhanced chemiluminescence (PerkinElmer, Boston, MA). Reactive bands were quantified using ImageJ. Data are presented as mean ± SEM of fold control.
Lentiviral shRNA transduction
Lentiviral particles expressing GFP or Fyn shRNA were provided by the University of Pittsburgh Cancer Institute (UPCI) Lentiviral Facility. Virus stocks were generated by co-transfection of the shRNA expression plasmid (pLK0.1; Mission shRNA library from Sigma) into 293-FT cells together with the packaging plasmids pMD2.g(VSVG), pRSV-REV, pMDLg/pRRE. Forty-eight hours post transfection viral particles were collected in the culture supernatant, filtered (0.45 μM) and stored at −80°C or used immediately to transduce BEAS-2B cells seeded at 50% confluence in 6 well plates. Briefly, 1 ml of LHC-9 media was combined with 1 ml of the specific virus (106 infectious units per ml) and 2 μl polybrene and the mixture was added to each well. Cells were incubated overnight and fresh media was given the following day. The cells were analyzed for GFP expression and protein knockdown 3 days after transduction.
Statistics
One-way analysis of variance (ANOVA) was used to determine whether the mean of each treatment was different from the untreated cells (control). Dunnett’s or Tukey’s Multiple Comparisons post hoc tests were used to determine significant differences between the means of each group. Two-way ANOVAs with Bonferroni’s Multiple Comparisons post hoc tests were used when comparing two factors. All statistics were performed using GraphPad Prism version 5 (GraphPad Software, San Diego, CA). Data are represented as mean ± SEM or as fold control.
Results
Cr(VI) selectively activates ISRE-dependent STAT1 signaling
We previously reported that within 1 h of exposing BEAS-2B cells to Cr(VI), STAT1 becomes tyrosine phosphorylated and translocates to the nucleus (18). Upon activation, STAT1 can form homodimers or heterodimers that bind to GAS and ISRE consensus sequences, respectively, to induce gene transcription (27). To determine which type of transcriptional complexes form in response to Cr(VI), luciferase expression was compared in cells transiently transfected with either ISRE or GAS-driven expression constructs. As shown in Fig. 1, Cr(VI) exposure induced ISRE-driven luciferase in the transfected cells, as well as endogenous ISRE-driven IRF7 in non-transfected cells. Cr(VI) had no effect on GAS-driven luciferase (not shown) or expression of endogenous IRF1 (Fig. 1).
Figure 1.
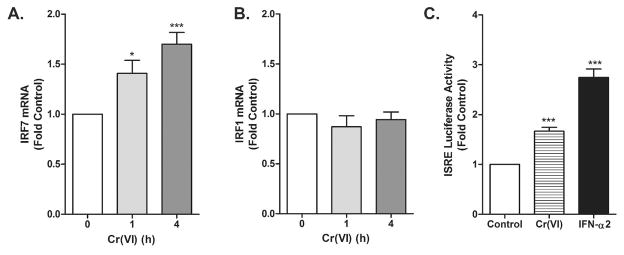
Cr(VI) stimulates ISRE-, but not GAS-dependent transactivation. A–B. BEAS-2B cells were exposed to Cr(VI) for indicated times and total RNA was isolated. IRF7 and IRF1 mRNA levels were measured by RT-PCR and real-time PCR, respectively, and normalized against the housekeeping gene, RPL13A. C. BEAS-2B cells were transfected with the ISRE-luc reporter construct. After 24 h, cells were treated with 5 μM Cr(VI) or 100 U/ml IFN-α2 for 4h. Cell lysates were analyzed for luciferase activity. Data are presented as mean ± SEM of fold control (n=3). * and *** designate p<0.05 and p<0.001, respectively.
STAT1 is required for Cr(VI) activation of ISRE
To confirm the requirement for STAT1 in the Cr(VI)-stimulated ISRE transactivation, BEAS-2B cells stably expressing random (shNC) or STAT1-specific (shSTAT1) shRNA were exposed to Cr(VI). The Western analysis shown in Fig 2A verified the knockdown of STAT1 in the shSTAT1 cells compared to shNC cells. Cr(VI) activated ISRE-driven luciferase and increased IRF7 mRNA expression in the shNC cells. In contrast, these responses were lost in the shSTAT1 cells (Fig. 2B–C) indicating the necessity of STAT1 in gene induction in response to Cr(VI).
Figure 2.

Cr(VI) induction of IRF7 mRNA requires STAT1. A. Total protein was isolated from BEAS-2B cell lines stably expressing either random (shNC) or STAT1 (shSTAT1) shRNA and total STAT1 and β-actin protein levels were determined by western analysis. B. shNC (open bars) and shSTAT1 (closed bars) cells were exposed to 5 μM Cr(VI) for 0, 1, and 4 h. Total RNA was isolated and IRF7 mRNA was measured by RT-PCR and normalized to the housekeeping gene, RPL13A. C. shNC (open bars) and shSTAT1 (close bars) cells were transiently transfected with the ISRE-luc reporter construct. After 24 h, cells were exposed to 5 μM Cr(VI) for 4 h and luciferase activity was measured. Data are presented as mean ± SEM of fold control (n=3). * and *** designate p<0.05 and p<0.001, respectively, compared to untreated (control) cells. ^ and ^^^ designate p<0.05 and p<0.001, respectively, compared to the shNC cells at the same time point.
HDAC activity is required for Cr(VI) induction of IRF7
Cr(VI) inhibits aryl hydrocarbon receptor-induced gene induction by retaining HDAC in their proximal promoters (8,11). In contrast, HDAC activity is required to recruit RNA polymerase to the promoters of ISRE-driven genes (27,29). To examine the role of HDAC in Cr(VI) induction of IRF7, BEAS-2B cells were pretreated with the global HDAC inhibitor, sodium butyrate (NaB), before exposure to Cr(VI). Cr(VI) had no effect on ISRE transactivation of the luciferase construct or endogenous IRF7 in cells pretreated with NaB compared to vehicle-treated cells (Fig. 3). These data suggested that, as in type I IFN-stimulated gene induction, HDAC activity was essential for Cr(VI)-induced gene transactivation.
Figure 3.
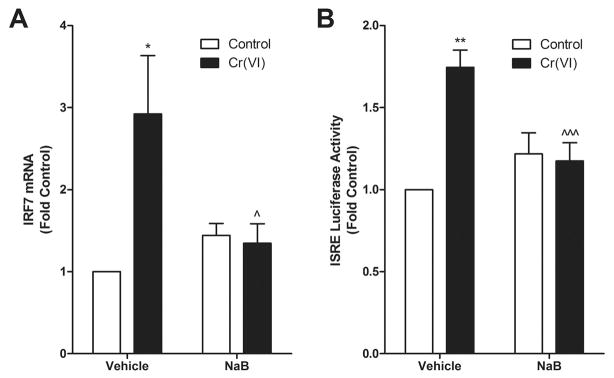
HDAC activity is necessary for Cr(VI)-stimulated ISRE transactivation and IRF7 induction. A. BEAS-2B cells were exposed to 5 μM Cr(VI) for 4 h after a 16 h pretreatment with 2 mM NaB and total RNA was isolated. IRF7 mRNA levels were measured by RT-PCR and normalized against the housekeeping gene, RPL13A. B. BEAS-2B cells were transfected with the ISRE-luc reporter construct. After 24 h, cells were pretreated with 2 mM sodium butyrate for 16 h followed by treatment with 5 μM Cr(VI) for 4 h. Cell lysates were analyzed for luciferase activity. Data is presented as mean ± SEM of fold control (n=3). * and ** designate p<0.05 and p<0.01, respectively, compared to untreated (control) cells. ^ and ^^^ designate p<0.05 and p<0.001, respectively, compared to cells treated with Cr(VI) alone.
Cr(VI) transactivation of ISRE is independent of type I interferon signaling
Type I IFNs bind to the interferon α/β receptors (IFNAR) to stimulate STAT1 and STAT2 tyrosine phosphorylation and subsequent dimerization (21,40). Another lung toxic metal, vanadium pentoxide (V2O5), causes a slow, oxidant dependent increase in IFN-b protein to activate STAT1 signaling pathways (32). To determine if Cr(VI) stimulated IFN signaling in a similar manner, a neutralizing antibody against CD118 that blocks the proliferative response of all Type 1 interferons (37) was added to cells prior to Cr(VI) treatment. IFN-α2 induction of IRF7 was abrogated in cells treated with the neutralizing antibody, but the antibody had no effect on Cr(VI)-stimulated induction of the gene (Fig. 4). These data indicate that Cr(VI) signaling for IRF7 induction is independent of IFNAR signaling. They also indicate that Cr(VI) and V2O5 differ in mechanisms for activating STAT1.
Figure 4.

Cr(VI) induction of IRF7 mRNA is independent of type I IFN signaling. BEAS-2B cells were treated with IFNAR2, a neutralizing antibody for the IFN-α/β receptor, for 30 min prior to treatment with 5 μM Cr(VI) or 100 U/ml IFN-α2 for 4 h. Total RNA was isolated and IRF7 mRNA levels were measured by RT-PCR and normalized against the housekeeping gene, RPL13A. Data is presented as means ± SEM of fold control. ** and *** designate p<0.01 and p<0.001, respectively, compared to the respective untreated (control) cells. ^^^ designates p<0.001 compared to cells treated with IFN-α2 alone.
Cr(VI)-activated STAT1 signaling requires Fyn
Incubating the BEAS-2B cells with the global SFK inhibitor PP2 blocked Cr(VI)-activated STAT1 and IRF7 induction (Fig. 5A–B) indicating an essential role for a SFK in the STAT1-dependent response. We previously reported that Cr(VI) activates both Fyn and Lck, but not Src or Yes in A549 airway adenocarcinoma cells and that this activation resulted from a direct effect of Cr(VI) on the kinases (41). In addition, we demonstrated that Cr(VI)-stimulated STAT3 translocation and induction of interleukin-6 (IL-6) mRNA transcription required Lck activity in BEAS-2B and primary bronchiolar epithelial cells (18). The identity of the essential SFK mediating Cr(VI) stimulation of STAT1 signaling was determined in genetically modified MEF cells. STAT1 was activated by Cr(VI) in wildtype, but not in SYF −/− or Src++ MEF cells (Fig. 5C–E). In contrast, transient expression of human Fyn in SYF −/− cells restored Cr(VI)-stimulated STAT1 signaling (Fig. 6). The overexpression of Fyn was verified by Western analysis with an antibody that recognized human Fyn (Fig. 6A). As in the A549 cells, Cr(VI) activated Fyn in BEAS-2B cells within 15 min of exposure (Fig. 7A–B). In reciprocal experiments, BEAS-2B cells were transduced with lentiviral constructs expressing eGFP (control) or Fyn shRNA. Knockdown of Fyn was verified by Western analysis (Fig. 7C) and inhibited both Cr(VI)-activated STAT1 and IRF7, relative to responses in control shRNA cells (Fig. 7D–F). These data indicate that Cr(VI)-activated Fyn mediates the phosphorylation of STAT1 and induction of IRF7 mRNA expression.
Figure 5.

Cr(VI) activation of STAT1 requires Src family kinases. A. BEAS-2B cells were pretreated with PP2 for 30 min prior to the addition of 5 μM Cr(VI) for 1 h. pYSTAT1, STAT1, and β-actin levels were determined by western analysis. Density of the protein bands from three separate experiments were quantified with ImageJ software. B. BEAS-2B cells were pretreated with PP2 for 30 min prior to the addition of 5 μM Cr(VI) for 4 h. Total RNA was isolated and IRF7 mRNA levels were measured by RT-PCR and normalized against the housekeeping gene, RPL13A. C. Wild-type (WT), SYF, and Src++ MEF cells were incubated with 5 μM Cr(VI) for the indicated times and nuclear protein was isolated. pYSTAT1, total STAT1, and β-actin levels were determined by western analysis and a representative blot from a single experiment is shown. D. Density of the protein bands from three separate experiments were quantified with ImageJ software. E. WT, SYF, and Src++ MEF cells were exposed to 5 μM Cr(VI) for the indicated times and total RNA was isolated. Irf7 mRNA levels were measured by RT-PCR and normalized to the housekeeping gene, β-actin. Data is presented as mean ± SEM of fold control (n=3). * and *** designate p<0.05 and p<0.001, respectively, compared to untreated (control) cells. ^ and ^^^ designate p<0.05 and p<0.001, respectively, compared to cells treated with Cr(VI) alone.
Figure 6.

Fyn mediates Cr(VI)-activated STAT1 signaling in MEF cells. Wildtype and SYF MEF cells were transiently transfected with either the control vector or the Fyn-myc expression vector. A. Total protein was isolated and Fyn and β-actin protein levels were determined by western analysis. B. Cells were treated with 5 μM Cr(VI) for 2 h and nuclear protein was isolated. pYSTAT1, total STAT1, and β-actin levels were determined by western analysis and a representative blot from a single experiment is shown. C. Density of the protein bands from three separate experiments were quantified with ImageJ software. D. Cells were treated with 5 μM Cr(VI) for 4 h and total RNA was isolated and Irf7 mRNA levels were measured by RT-PCR and normalized to the housekeeping gene, β-actin. Data is presented as mean ± SEM of fold control. * designates p<0.05 compared to respective untreated (control) cells.
Figure 7.

Fyn is required for Cr(VI) activation of STAT1 in BEAS-2B cells. A. BEAS-2B cells were exposed to 5 μM Cr(VI) and total Fyn was immunoprecipitated from whole cell lysates and immunoblotted for pFyn and total Fyn. A representative blot from a single experiment is shown. B. Density of the protein bands from three separate experiments were quantified with ImageJ software. C–F. BEAS-2B cells were transduced with GFP-expression control or Fyn shRNA. C. Total protein was isolated and Fyn and β-actin protein levels were determined by western analysis. D. Cells were exposed to 5 μM Cr(VI) for 1 h and nuclear protein was isolated. pYSTAT1, total STAT1, and β-actin levels were determined by western analysis and a representative blot from a single experiment is shown. E. Density of the protein bands from three separate experiments were quantified with ImageJ software. F. Cells were exposed to 5 μM Cr(VI) for 4 h and total RNA was isolated and Irf7 mRNA levels were measured by RT-PCR and normalized to the housekeeping gene, RPL13A. Data is presented as mean ± SEM of fold control. *, **, and *** designate p<0.05, p<0.01, and p<0.001, respectively, compared to respective untreated (control) cells. ^^ designates p<0.01 compared to GFP-expression control-transfected cells.
Discussion
The data presented here address a mechanism responsible for Cr(VI)-stimulated gene induction in target cells of Cr(VI)-promoted lung diseases. The data demonstrated that a non-cytotoxic exposure to Cr(VI) rapidly activated Fyn to initiate transient STAT1 signaling and gene transactivation. These data contrast with the long standing belief that Cr(VI) inhibits inducible genes, but is consistent with our previous observation of Cr(VI) inducing a transcriptional repressor protein through a STAT1-dependent pathway (13). This repressive pathway is similar to innate immune type I IFN antiviral and antiproliferative responses regulated through NRTKs (20,27,28). Since airway epithelial STAT1 activation can contribute to the development of pulmonary inflammatory diseases (17,32,33), these novel findings may reveal mechanistic understanding of the molecular pathogenesis of Cr(VI)-induced lung diseases. In addition, STAT1 has been implicated as an important factor in modulating airway injuries, especially fibrotic changes, stimulated by other inhaled metals, such as V2O5 (32). However, it is apparent the two metals activate STAT1 signaling through two separate, but perhaps complementary pathways.
The current data and our previous reports (13,18) indicate that exposing airway epithelial cells to Cr(VI) increases DNA binding and promoter transactivation by members of the STAT family, including STAT1 and STAT3. STATs are activated through the phosphorylation of a conserved tyrosine residue in the C-terminus by JAKs or SFKs (18,21–23). We previously demonstrated that prolonged Cr(VI) stimulation of STAT3 in BEAS-2B and primary human airway epithelial cells was mediated by the SFK, Lck (18). In the current studies, pharmacological and genetic studies were used to demonstrate that Fyn is the Cr(VI)-stimulated SFK upstream of STAT1 activation. The role of Fyn was confirmed by demonstrating that Cr(VI) failed to activate STAT1 and transactivate ISRE-driven luciferase or IRF7 promoters in MEF (Fig. 5C–D) and BEAS-2B cells that lacked Fyn (Fig. 7D–F). Restoring Fyn, but not Src to SYF cells, provided a fully functional STAT1 response to Cr(VI) (Fig. 6), which is consistent with our earlier observation in A549 cells that Cr(VI) stimulated Fyn, and not Src or Yes (41). It is interesting to note that overexpression of Fyn in wild type cells (Fig. 6) did not lead to enhanced Cr(VI) stimulation of STAT1 phosphorylation. Fyn signaling is highly contextual and often requires adapter proteins to facilitate substrate phosphorylation (42,43). It is possible that the normal pool of Fyn that mediates STAT1 phosphorylation is saturated in the wild type cells or that an adapter protein is rate limiting and thus adding more Fyn would not increase the amount of STAT1 phosphorylated in response to Cr(VI).
Cr(VI) stimulates Fyn substrate phosphorylation through direct effect on the enzyme (41). This was demonstrated that Fyn substrate phosphorylation was increased by adding Cr(VI) to a solution containing only affinity purified enzyme, enolase as a substrate, and ATP (41). The effect of Cr(VI) was not inhibited in the presence of catalase, but was prevented by addition of NAC to the reaction mixture. This suggested that either that Cr(VI) reacts with a thiol in the kinase or needs to be reduced to activate substrate phosphorylation. Cr(VI) causes similar activation of recombinant Src under the same reaction conditions, but not in intact cells (41). The thiol dependence for activation of purified Fyn or Src is consistent with structural studies that identified a metal-responsive motif in a carboxyl regulatory domain of the SFK enzymes that contains two critical cysteines separated by ten amino acids (44). Conformational change in this motif during activation was previously recognized to increase substrate affinity (45). Whether Cr(VI) binds these cysteines or binds to other amino acids in the regulatory domain in intact cells or in vivo remains to be determined. However, the fact that Cr(VI) exposure of intact cells prevents Src activation (13,41) suggests that specificity of the Cr(VI) in vivo is more complex. Also, Fyn may not directly phosphorylate STAT1, since Fyn can phosphorylate another NRTK, c-Abl (46), which has been demonstrated to phosphorylate STAT1 (47,48) and is an indirect target of PP2. Nonetheless, despite multiple potential intracellular targets of the metal, The findings indicate that Cr(VI) selectively activates Fyn-dependent signaling for induction of type 1 IFN responsive genes.
Direct Cr(VI) activation of Fyn to stimulate STAT1 signaling contrasts with the oxidant dependent activation of SFK in response to arsenic exposure (49) and the mechanism for STAT1 activation following exposure to V2O5 (32,50). Arsenic activates SFK only in intact cells and this activation is inhibited by the antioxidant enzyme catalase or thiol protective N-acetyl cysteine (NAC) (49). In a similar fashion, V2O5-stimulated STAT1 signaling is also attenuated by catalase or NAC (32,50). Oxidized vanadium increases the tyrosine phosphorylation state of signaling proteins by binding a critical cysteine in the active site of tyrosine phosphatases (51,52). This enhances tyrosine phosphorylation, but either basal or stimulated kinase activity is required to initiate the phosphorylation (53). As indicated above, Cr(VI) stimulation of Fyn activity is not inhibited by catalase (41). While this finding do not rule out a dependence on another oxygen-centered radical species, it is consistent with the insensitivity of Fyn to H2O2 relative to Cr(VI) (41) and the thiol dependent mechanism for Fyn discussed above.
In addition to the differential requirement for H2O2, mechanisms for Cr(VI) and V2O5 stimulation of STAT1 signal are differentiated by temporal and concentration dependence. Low micromolar concentrations of Cr(VI) increase nuclear localization of STAT1 within 1 h of exposure and this increase resolves between 4 and 8 h (18). In contrast, V2O5 is 100 times less potent than Cr(VI) and causes a slow, protein synthesis-dependent STAT1 activation (32,50). Cr(VI) stimulation of STAT1 is not inhibited by cycloheximide, but Cr(VI)-induced, STAT1-dependent gene suppression was absent when protein synthesis was blocked (13). The STAT1 activating proteins induced by oxidants and V2O5 are IFNs (32). IFN-mediated signaling is the most characterized pathway for STAT1 activation and type I IFNs, such as IFN-α2, are well known to bind cell surface type I IFN receptors to activate kinases that phosphorylate STAT1 (21,40). However, the signaling events activated by Cr(VI) are not mediated by induction of IFN and are independent of IFNAR signaling, since neutralizing antibody that blocks all type 1 IFNs from binding their receptors did not affect Cr(VI)-induced IRF7 expression, but did prevent the IFN-α2 induction of the gene (Fig. 4). Thus, while the two metals both stimulate STAT1 signaling, they differ substantially in both mechanism of action and in the type of STAT1 signaling produced.
Activated STAT1 forms homodimers or heterotrimers with STAT2 and IRF9 in the ISGF3 complex to bind to GAS or ISRE consensus sequences, respectively (19,27,28). Cr(VI) selectively activated ISRE-driven transcription and had no effect on GAS transactivation (Fig. 1). Cr(VI)-stimulated ISRE transactivation and IRF7 mRNA induction was attenuated in BEAS-2B cells stably expressing STAT1 shRNA (Fig. 2), thus confirming the requirement for STAT1. However, the data cannot rule out a Cr(VI) effects on STAT2 or IRF9 in the ISFG3 complex. Tyrosine phosphorylated STAT2 was observed in BEAS-2B cells treated with Cr(VI) (data not shown) and deletion of any of the three proteins in the heterotrimer results in loss of IFN induction of a protein repressor of Sp1 and NF-κB transcriptional activity (28). Since Cr(VI) induces a protein to inhibit Sp1 in cells with intact STAT1 signaling (13), further investigation of its full effects on the ISGF3 complex formation and transactivation of the unknown repressive gene are warranted.
Cr(VI) effects on HDAC-1 activity and Cr(VI) cross-linking of HDAC-1 with DNA methytransferase 1 and chromatin has been demonstrated to repress aryl hydrocarbon receptor mediated transactivation of CYP1A (8,11). Although histone deacetylation is most often correlated with gene repression, HDAC-1 activity plays an essential role in induction of ISRE-driven genes by forming complexes with STAT1-containing ISGF3 complexes and facilitating the recruitment of RNA polymerase II to interferon-stimulated gene promoters(27,29). Since the HDAC inhibitor, NaB, prevents Cr(VI) from both repressing CYP1A1 (8,11) and inducing IRF7 (Fig. 3), it is possible that Cr(VI) cross-linking of HDAC-1 to chromatin is a common mechanism in both gene repression and induction. However, it is evident that extra-nuclear signaling events are necessary for Cr(VI) to stimulate formation of the ISGF3 transactivation complexes. Thus, Cr(VI) appears to signal for gene repression through both direct actions on chromatin and DNA (10,11), as well as indirect induction of gene repressors.
In summary, the present study demonstrated that Cr(VI) activates STAT1 to form ISGF3 complexes that induce IRF7 transcript expression. This activation is independent of type I IFN signaling, but is mediated by Fyn. The date support a novel pathway through which Cr(VI) stimulates rather than represses gene induction. Fyn has been implicated in mediating cell transformation (54) and STAT1 both regulates antiproliferative and apoptotic responses (24,25) and plays a role in the pathogenesis of inflammatory and fibrotic lung diseases (32–35). Thus, these findings may provide critical insight into pathogenic mechanisms for development of Cr(VI)-related pulmonary diseases. However, much more study is needed to reveal the full impact of Cr(VI) stimulation of these signaling pathways in the molecular etiology of Cr(VI)-promoted pathologies.
Acknowledgments
This work was supported by Public Health Service grants ES10638 and HL06569 from the National Institute of Environmental Health Sciences and the National Heart Lung and Blood Institute.
ABREVIATIONS
- GAS
gamma interferon-activated site
- HDAC
histone deacetylase
- IFN
interferon
- ISRE
interferon-stimulated response element
- IRF 1
interferon regulatory factor 1
- IRF7
interferon regulatory factor 7
- JAK
Janus kinase
- MEF
mouse embryonic fibroblasts
- NRTK
non-receptor tyrosine kinase
- SFK
Src family kinase
- STAT
signal transducer and activator of transcription
Reference List
- 1.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21(1):28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agency for Toxic Substances and Disease Registry. Toxicological profile for chromium. 2005. [PubMed] [Google Scholar]
- 3.Bright P, Burge PS, O’Hickey SP, Gannon PFG, Robertson AS, Boran A. Occupational asthma due to chrome and nickel electroplating. Thorax. 1997;52:28–32. doi: 10.1136/thx.52.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takemoto K, Kawai H, Kuwahara T, Nishina M, Adachi S. Metal concentrations in human lung tissue, with special reference to age, sex, cause of death, emphysema and contamination of lung tissue. Int Arch Occup Environ Health. 1991;62(8):579–586. doi: 10.1007/BF00381111. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533(1–2):3–36. doi: 10.1016/j.mrfmmm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Alcedo JA, Wetterhahn KE. Chromium toxicity and carcinogenesis. Int Rev Exp Pathol. 1990;31:85–108. doi: 10.1016/b978-0-12-364931-7.50008-2. [DOI] [PubMed] [Google Scholar]
- 7.De Flora S, Morelli A, Basso C, Romano M, Serra D, De Flora A. Prominent role of DT-diaphorase as a cellular mechanism reducing chromium(VI) and reverting its mutagenicity. Cancer Res. 1985;45(7):3188–3196. [PubMed] [Google Scholar]
- 8.Wei YD, Tepperman K, Huang MY, Sartor MA, Puga A. Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J Biol Chem. 2004;279(6):4110–4119. doi: 10.1074/jbc.M310800200. [DOI] [PubMed] [Google Scholar]
- 9.De Flora S. Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis. 2000;21(4):533–541. doi: 10.1093/carcin/21.4.533. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton JW, Wetterhahn KE. Differential effects of chromium(VI) on constitutive and inducible gene expression in chick embryo liver in vivo and correlation with chromium(VI)-induced DNA damage. Mol Carcinog. 1989;2:274–286. doi: 10.1002/mc.2940020508. [DOI] [PubMed] [Google Scholar]
- 11.Schnekenburger M, Talaska G, Puga A. Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol Cell Biol. 2007;27 (20):7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shumilla JA, Broderick RJ, Wang Y, Barchowsky A. Chromium(VI) inhibits the transcriptinal activity of NF-κB by decreasing the interaction of p65 with CBP. J Biol Chem. 1999;274:36207–36212. doi: 10.1074/jbc.274.51.36207. [DOI] [PubMed] [Google Scholar]
- 13.Nemec AA, Barchowsky A. Signal Transducer and Activator of Transcription 1 (STAT1) is Essential for Chromium Silencing of Gene Induction in Human Airway Epithelial Cells. Toxicol Sci. 2009;110(1):212–223. doi: 10.1093/toxsci/kfp084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dudley AC, Thomas D, Best J, Jenkins A. The STATs in cell stress-type responses. Cell Commun Signal. 2004;2(1):8. doi: 10.1186/1478-811X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laskin DL, Fakhrzadeh L, Heck DE, Gerecke D, Laskin JD. Upregulation of phosphoinositide 3-kinase and protein kinase B in alveolar macrophages following ozone inhalation. Role of NF-kappaB and STAT-1 in ozone-induced nitric oxide production and toxicity. Mol Cell Biochem. 2002;234–235(1–2):91–98. [PubMed] [Google Scholar]
- 16.Stephanou A. Role of STAT-1 and STAT-3 in ischaemia/reperfusion injury. J Cell Mol Med. 2004;8(4):519–525. doi: 10.1111/j.1582-4934.2004.tb00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286(6):L1282–L1292. doi: 10.1152/ajplung.00349.2003. [DOI] [PubMed] [Google Scholar]
- 18.O’Hara KA, Vaghjiani RJ, Nemec AA, Klei LR, Barchowsky A. Chromium(VI)-stimulated STAT3 tyrosine phosphorylation and nuclear translocation in human airway epithelial cells requires Lck. Biochem J. 2007;402(2):261–269. doi: 10.1042/BJ20061427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 20.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19(4):329–340. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darnell JE., Jr STATs and gene regulation. Science. 1997;277(5332):1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 22.Xi S, Zhang Q, Dyer KF, Lerner EC, Smithgall TE, Gooding WE, Kamens J, Grandis JR. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278(34):31574–31583. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 23.Yu CL, Jove R, Burakoff SJ. Constitutive activation of the Janus kinase-STAT pathway in T lymphoma overexpressing the Lck protein tyrosine kinase. J Immunol. 1997;159(11):5206–5210. [PubMed] [Google Scholar]
- 24.Gimeno R, Lee CK, Schindler C, Levy DE. Stat1 and stat2 but not stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T lymphocytes. Mol Cell Biol. 2005;25(13):5456–5465. doi: 10.1128/MCB.25.13.5456-5465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M. Cutting edge: role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J Immunol. 2005;174(2):609–613. doi: 10.4049/jimmunol.174.2.609. [DOI] [PubMed] [Google Scholar]
- 26.Deb DK, Sassano A, Lekmine F, Majchrzak B, Verma A, Kambhampati S, Uddin S, Rahman A, Fish EN, Platanias LC. Activation of Protein Kinase Cdelta by IFN-gamma. J Immunol. 2003;171(1):267–273. doi: 10.4049/jimmunol.171.1.267. [DOI] [PubMed] [Google Scholar]
- 27.Sakamoto S, Potla R, Larner AC. Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J Biol Chem. 2004;279(39):40362–40367. doi: 10.1074/jbc.M406400200. [DOI] [PubMed] [Google Scholar]
- 28.Laver T, Nozell SE, Benveniste EN. IFN-beta-mediated inhibition of IL-8 expression requires the ISGF3 components Stat1, Stat2, and IRF-9. J Interferon Cytokine Res. 2008;28:13–23. doi: 10.1089/jir.2007.0062. [DOI] [PubMed] [Google Scholar]
- 29.Klampfer L, Huang J, Swaby LA, Augenlicht L. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem. 2004;279(29):30358–30368. doi: 10.1074/jbc.M401359200. [DOI] [PubMed] [Google Scholar]
- 30.Laimer K, Spizzo G, Obrist P, Gastl G, Brunhuber T, Schafer G, Norer B, Rasse M, Haffner MC, Doppler W. STAT1 activation in squamous cell cancer of the oral cavity: a potential predictive marker of response to adjuvant chemotherapy. Cancer. 2007;110(2):326–333. doi: 10.1002/cncr.22813. [DOI] [PubMed] [Google Scholar]
- 31.Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66(7):3649–3657. doi: 10.1158/0008-5472.CAN-05-3612. [DOI] [PubMed] [Google Scholar]
- 32.Antao-Menezes A, Turpin EA, Bost PC, Ryman-Rasmussen JP, Bonner JC. STAT-1 signaling in human lung fibroblasts is induced by vanadium pentoxide through an IFN-beta autocrine loop. J Immunol. 2008;180(6):4200–4207. doi: 10.4049/jimmunol.180.6.4200. [DOI] [PubMed] [Google Scholar]
- 33.Sampath D, Castro M, Look DC, Holtzman MJ. Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J Clin Invest. 1999;103(9):1353–1361. doi: 10.1172/JCI6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shornick LP, Wells AG, Zhang Y, Patel AC, Huang G, Takami K, Sosa M, Shukla NA, Agapov E, Holtzman MJ. Airway epithelial versus immune cell Stat1 function for innate defense against respiratory viral infection. J Immunol. 2008;180(5):3319–3328. doi: 10.4049/jimmunol.180.5.3319. [DOI] [PubMed] [Google Scholar]
- 35.Walters DM, Antao-Menezes A, Ingram JL, Rice AB, Nyska A, Tani Y, Kleeberger SR, Bonner JC. Susceptibility of signal transducer and activator of transcription-1-deficient mice to pulmonary fibrogenesis. Am J Pathol. 2005;167(5):1221–1229. doi: 10.1016/S0002-9440(10)61210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein PL, Vogel H, Soriano P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994;8:1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 37.Colamonici OR, Domanski P. Identification of a novel subunit of the type I interferon receptor localized to human chromosome 21. J Biol Chem. 1993;268(15):10895–10899. [PubMed] [Google Scholar]
- 38.Barchowsky A, Soucy NV, O’Hara KA, Hwa J, Noreault TL, Andrew AS. A Novel Pathway for Nickel-induced Interleukin-8 Expression. J Biol Chem. 2002;277(27):24225–24231. doi: 10.1074/jbc.M202941200. [DOI] [PubMed] [Google Scholar]
- 39.Andrew AS, Barchowsky A. Nickel-induced plasminogen activator inhibitor-1 (PAI-1) expression inhibits the fibrinolytic activity of human airway epithelial cells. Toxicol Appl Pharmacol. 2000;168:50–57. doi: 10.1006/taap.2000.9009. [DOI] [PubMed] [Google Scholar]
- 40.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 41.O’Hara KA, Klei LR, Barchowsky A. Selective activation of Src family kinases and JNK by low levels of chromium(VI) Toxicol Appl Pharmacol. 2003;190(3):214–223. doi: 10.1016/s0041-008x(03)00188-1. [DOI] [PubMed] [Google Scholar]
- 42.Filipp D, Julius M. Lipid rafts: resolution of the “fyn problem”? Mol Immunol. 2004;41(6–7):645–656. doi: 10.1016/j.molimm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase fyn in integrin signaling and anchorage-dependent cell growth. 1998. pp. 625–634. [DOI] [PubMed] [Google Scholar]
- 44.Ahmadibeni Y, Hanley M, White M, Ayrapetov M, Lin X, Sun G, Parang K. Metal-binding properties of a dicysteine-containing motif in protein tyrosine kinases. Chembiochem. 2007;8(13):1592–1605. doi: 10.1002/cbic.200700242. [DOI] [PubMed] [Google Scholar]
- 45.Veillette A, Dumont S, Fournel M. Conserved cysteine residues are critical for the enzymatic function of the lymphocyte-specific tyrosine protein kinase p56lck. J Biol Chem. 1993;268:17547–17553. [PubMed] [Google Scholar]
- 46.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13(18):2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danial NN, Rothman P. JAK-STAT signaling activated by Abl oncogenes. Oncogene. 2000;19(21):2523–2531. doi: 10.1038/sj.onc.1203484. [DOI] [PubMed] [Google Scholar]
- 48.Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183(3):811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low Levels of Arsenic Trioxide Stimulate Proliferative Signals in Primary Vascular Cells without Activating Stress Effector Pathways. Toxicol Appl Pharmacol. 1999;159(1):65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- 50.Wang YZ, Ingram JL, Walters DM, Rice AB, Santos JH, Van HB, Bonner JC. Vanadium-induced STAT-1 activation in lung myofibroblasts requires H2O2 and P38 MAP kinase. Free Radic Biol Med. 2003;35 (8):845–855. doi: 10.1016/s0891-5849(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 51.Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. 2001;13(2):182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 52.Samet JM, Silbajoris R, Wu W, Graves LM. Tyrosine phosphatases as targets in metal-induced signaling in human airway epithelial cells [In Process Citation] Am J Respir Cell Mol Biol. 1999;21(3):357–364. doi: 10.1165/ajrcmb.21.3.3656. [DOI] [PubMed] [Google Scholar]
- 53.Barchowsky A, Munro SR, Morana SJ, Vincenti MP, Treadwell M. Oxidant-sensitive and phosphorylation-dependent activation of NF-kappaB and AP-1 in endothelial cells. Am J Physiol. 1995;269:L829–L836. doi: 10.1152/ajplung.1995.269.6.L829. [DOI] [PubMed] [Google Scholar]
- 54.He Z, Tang F, Ermakova S, Li M, Zhao Q, Cho YY, Ma WY, Choi HS, Bode AM, Yang CS, Dong Z. Fyn is a novel target of (−)-epigallocatechin gallate in the inhibition of JB6 Cl41 cell transformation. Mol Carcinog. 2008;47(3):172–183. doi: 10.1002/mc.20299. [DOI] [PMC free article] [PubMed] [Google Scholar]