

Abstract
Airway hyperresponsiveness is the hallmark of allergic asthma and caused by multiple factors. Nerve growth factor (NGF), a neurotrophin, is originally known for regulation of neural circuit development and function. Recent studies indicated that NGF contributes to airway hyperresponsiveness and pathogenesis of asthma. The objective of this study is to develop a small interfering RNA against NGF to attenuate airway hyperresponsiveness and further elucidate the underlying mechanism. In a murine model of allergic asthma, the ovalbumin-sensitized mice were intratracheally delivered small interfering RNA against NGF or administered an inhibitor targeting NGF receptor, tropomyosin-related kinase A, as a positive treatment control. In this study, knockdown NGF derived from pulmonary epithelium significantly reduced airway resistance in vivo. The levels of NGF, proinflammatory cytokines and infiltrated eosinophils in airway were decreased in small interfering RNA against NGF group but not in tropomyosin-related kinase A inhibitor and mock siRNA group. Furthermore, induction of neuropeptide (substance P) and airway innervation were mediated by NGF/tropomyosin-related kinase A pathway. These findings suggested that NGF targeting treatment holds the potential therapy for antigen-induced airway hyperresponsiveness via attenuation of airway innervation and inflammation in asthma.
Keywords: siRNA, asthma, lentivirus, nerve growth factor
Introduction
Airway hyperresponsiveness (AHR) is an asthmatic feature and regulated by multiple factors such as immune responses and structural changes.1 Inflammatory response increases the sensitivity and reactivity of airway nerve, thus, many investigations have been focused on studying the immunologic incidents which resulted in an allergic airway inflammation. However, AHR might also occur in absence of allergic inflammation.2 These evidences suggested that the interactive mechanisms of AHR especially in abnormal neural modulation are complicated and still unclear.
Nerve growth factor (NGF), a neurotrophin, plays a major role in the development and maintenance of nervous function via its receptors (tropomyosin-related kinase A (TrkA) and p75NTR receptor).3,4 NGF contributed to the sensitivity of nerves5,6 and enhanced expressions of neuropeptides (such as substance P (SP)) which were stored in the terminals of sensory afferent nerves and induced neurogenic inflammatory responses after allergen exposure.7,8 In contrast, overwhelming evidences suggested that NGF is increased in inflamed airway9,10 and involved in airway inflammation11 and remodeling.12,13 These studies suggested that NGF might be extremely important in AHR by various effects of participation. In fact, numerous studies carried out approaches to show the significant role of NGF/TrkA pathway in AHR. NGF administration induced AHR in the absence of airway inflammation in mice.14 In NGF-Tg mice, AHR was induced by overexpression of NGF and accompanied by increase of substance P and airway innervation.15 Administering anti-NGF antibodies or an inhibitor of TrkA receptor diminished the progression of AHR in mice.8,16 These studies demonstrated that NGF contributed to AHR. However, it is difficult to characterize the major source for NGF-induced AHR.
Although many NGF-producing cells are involved in pathogenesis of allergic asthma,16,17 airway epithelium is commonly believed to be the main source and plays a key role in airway inflammation and remodeling by releasing inflammatory mediators and growth factors.17,18 NGF derived from epithelium cells promoted the survival of eosinophils, which initiated and maintained the allergic response.19 Meanwhile, activated eosinophils released inflammatory factors and NGF to aggravate airway inflammation.20
In this study, airway epithelium of ovalbumin (OVA)-sensitized and challenged mice was administrated small interfering RNA (siRNA) targeting NGF which was packaged in lentivirus. The inhibitory effects on AHR, airway inflammation, the substance P level, and airway innervation in siRNA treatment were evaluated and compared to those in TrkA inhibitor treatment. The finding demonstrated that the functional role of NGF on AHR and siRNA treatment is a potential therapy in asthma.
Results
Lentivirus containing constructed-siRNA against NGF reduced NGF production and mRNA level
It is well known that allergic inflammation results in increased NGF level in bronchoalveolar lavage (BAL) fluid of OVA-challenge mice16 and epithelial cells are the major resource of NGF.19 We constructed the siRNA against NGF (siNGF) into the lentiviral expression system and determined the inhibitory efficiency of siNGF treatment on cultured primary lung cells. At the beginning of in vitro system, the cultured lung cells, which were clarified by flow cytometry, were stained with the specific marker of alveolar type II cells, prosurfactant protein C (proSP-C), and marker of clara cells, clara-cell-specific 10 kDa protein (CC10). About 90% cells were positive for surfactant protein C and 5% cells were for CC10 (Figure 1a). First, NGF induction was determined by tumor necrosis factor (TNF)-α with different dosage. NGF levels were significantly increased by TNF-α stimulation (20 ng/ml) at 24 hours, and extremely increased at 48 hours (Figure 1b). Second, we investigated the suppressive efficiency of siNGF in vitro. Lentivirus-containing mock siRNA or three segments of siNGF (siNGF-1 to siNGF-3) was delivered to cell cultures for 48 hours before TNF-α stimulation. In order to enhance the infective efficiency, lentivirus was added with polybrene, a cationic polymer. The level of NGF in siNGF treatment was significantly lower than that of mock siRNA treatment and the reduction was noticeable in siNGF-2 group (Figure 1c). Compared with NGF gene expression in mock siRNA and polybrene treatment, the targeting knockdown resulted in about 50% reduction in siNGF treatment (Figure 1c). The similar reduction was shown in protein levels (Figure 1d). These results showed that lentivirus containing siNGF was successfully constructed and ready to be applied to study the functional roles of NGF in airway inflammation and AHR in a murine asthmatic model.
Figure 1.
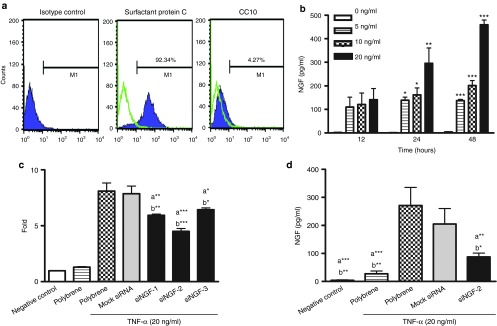
The suppressive effects of siRNA against NGF (siNGF) in vitro. (a) The phenotypes of primary lung cells. (b) NGF inductions were assessed in primary lung culture in the absence or presence of different doses of TNF-α (0, 5, 10, and 20 ng/ml) at different time points (12, 24, and 48 hours). *P < 0.05, **P < 0.01, and ***P < 0.001 versus no TNF-α. (c) siNGF decreased NGF mRNA expression. Primary lung cells were infected by lentvirus for 48 hours and stimulated by TNF-α (20 ng/ml) for 6 hours. (d) The knockdown efficiency of siNGF. Primary lung cells were infected by lentvirus containing siNGF or mock siRNA (MOI: 10) for 48 hours and stimulated by TNF-α (20 ng/ml) for 48 hours. a*P < 0.05, a**P < 0.01, and a***P < 0.001 versus positive control. b*P < 0.05, b**P < 0.01, and b***P < 0.001 versus mock siRNA. Data are shown as mean ± SEM and representative of three independent experiments. MOI, multiplicity of infection; NGF, nerve growth factor; siRNA, small interfering RNA.
siNGF reduced allergic airway inflammation
To evaluate the suppressive effect of siNGF in vivo, OVA-sensitized mice were delivered lentivirus-containing siRNA or TrkA inhibitor which was the positive control into airway before the challenges and the detailed protocol was summarized in Figure 2. The OVA-specific antibodies in sera, cytokine levels in the BAL fluid, and pathological results in lung were measured after sacrifice. NGF widely expressed in many types of cells especially in bronchioles and alveolar epithelium in lung.21 Intratracheal administration with lentivirus containing green fluorescent protein (GFP) presented the main targets of lentivirus therapy including bronchus, alveolar cells, and partially macrophages (Figure 2a). These findings suggested that NGF knockdown in lung mediated by lentivirus treatment was primarily reduced in airway epithelium. There were no significant differences in OVA-specific IgG2a and IgE in OVA-sensitized groups including positive control (PC), siNGF, and TrkA inhibitor and mock siRNA group. However, the level of OVA-specific IgG1 was decreased in siNGF group (Figure 2b). The results of cytokine profile in BAL fluid showed that NGF level was significantly induced by airway inflammation and obviously increased in TrkA inhibitor treatment but significantly decreased in siNGF treatment. IL-5 was increased accompanying with airway inflammation but decreased in siNGF group. The difference in IL-5 level was significantly noted between siNGF and TrkA inhibitor group. However, there was the similar trend in eotaxin but without statistical difference (Figure 2c). IL-4, IL-13, and IFN-γ were also noted with no significant differences (data not shown). In contrast, the results of cytokine levels in lung homogenates showed that not only NGF level but also proinflammatory cytokines including IL-4, IL-5, IL-6, IL-12p70, and IL-17 were significantly decreased in siNGF treatment group. On the other hand, IL-12p70 and IFN-γ concentration were increased in mock siRNA treatment group (Figure 3). It is well known that airway inflammation is the key factor of AHR. These findings suggested that NGF knockdown could affect AHR by decreasing airway inflammation and siNGF treatment might be a useful therapy for asthma.
Figure 2.

siNGF decreased OVA-specfic Abs and cytokine productions in BAL fluid. Lentivirus containing siNGF or mock siRNA (3 × 106 infectious unit) was intratracheally delivered into the OVA-sensitized mice 3 days before the first challenge with OVA. The TrkA inhibitor was intranasally administered 3 hours before OVA challenge. After OVA challenges, sera and BAL fluid from the treated mice were collected and analyzed by ELISA assay. (a) The targeting location of lentivirus which was determined by immunofluorescent stain of anti-GFP expression After intratracheally delivering lentivirus containing GFP for 6 days, BAL cells were collected and lungs were isolated from treated mice following perfusion, fixation, and optimal cutting temperature compound embedding. BAL cells and lung were stained with anti-GFP antibody and secondary antibody conjugated DyLight-594. The sections were analyzed by fluorescence microscope. (b) OVA-specific antibody productions in sera. Data are presented as ELISA unit (E.U.). (c) NGF, IL-5 and eotaxin productions in BAL fluid. a*P < 0.05, a**P < 0.01, and a***P < 0.001 versus PC. b*P < 0.05, b**P < 0.01, and b***P < 0.001 versus mock siRNA. c*P < 0.05, c**P < 0.01, and c***P < 0.001 versus TrkA inhibitor. d***P < 0.001 versus siNGF. N = 6–8 per group. Data are shown as mean ± SEM and representative of five independent experiments. ELISA, enzyme-linked immunosorbent assay; GFP, green fluorescent protein; siNGF, small interfering RNA against nerve growth factor; TrkA, tropomyosin-related kinase A. BAL, bronchoalveolar lavage.
Figure 3.

siNGF decreased proinflammatory cytokines in lung. After sacrifice, lungs were isolated from the treated mice and homogenized by lysis buffer. Protein concentrations of lung homogenates were measured by bicinchoninic acid assay. NGF and cytokine concentrations were determined in 10 μg lung homogenate with ELISA kits. a*P < 0.05, a**P < 0.01, a***P < 0.001 versus positive control (PC). b*P < 0.05, b**P < 0.01, b***P < 0.001 versus mock siRNA. c*P < 0.05, c**P < 0.01, c***P < 0.001 versus TrkA inhibitor. d*P < 0.05, versus negative control. N = 6–8 per group. Data are shown as mean ± SEM and representative of five independent experiments. ELISA, enzyme-linked immunosorbent assay; NGF, nerve growth factor; TrkA, tropomyosin-related kinase A.
NGF-TrkA pathway modulated the progression of AHR
To study the inhibitory effect of siNGF on AHR, we used two systems to measure airway function including whole-body plethysmography and invasive plethysmography in OVA-sensitized mice. After OVA challenges, mice were increased airway constriction with methacholine stimulation in PC and mock siRNA group but reduced in siNGF or TrkA inhibitor group. The relative ratio (50 mg methacholine) in PC group was three times higher than that of siNGF and TrkA inhibitor group. In contrast, the degree of hyperresponsiveness in mock siRNA-treated group was not improved (Figure 4a,b). These data suggested that NGF might play a regulatory role in AHR through the TrkA receptor and decreased NGF resulted in AHR improvement in asthmatic animal model.
Figure 4.

siNGF and TrkA inhibitor could reduce AHR. After OVA challenges, airway function of the treated mice was measured by (a) whole-body plethysmography or (b) invasive plethysmography. Results were expressed as the ratio of the baseline Penh value a or as the airway resistance value b. N = 6–8 per group. a*P < 0.05, a**P < 0.01, a***P < 0.001 versus PC. b*P < 0.05, b**P < 0.01, b***P < 0.001 versus mock siRNA. Data are shown as mean ± SEM and representative of five independent experiments. AHR, airway hyperresponsiveness; siNGF, small interfering RNA against nerve growth factor; siRNA, small interfering RNA; TrkA, tropomyosin-related kinase A.
NGF knockdown decreased eosinophil recruitment in lungs
Since NGF plays an important role in promoting survival of eosinophils, we assessed the effects of siNGF on the infiltration of inflammatory cell by counting the cells from BAL fluid and classifying the cell population in airway. The eosinophil infiltration was detected and that in TrkA inhibitor group was higher than that in siNGF or negative control (NC) groups, but not in the PC group. In contrast, the macrophage population was higher in NC and siNGF group (Figure 5a). The inflammatory infiltration was reduced in siNGF group but not in TrkA inhibitor, mock siRNA, and PC group (Figure 5b). The similar findings were noted in pathological results of lung section, the inflammatory infiltration in TrkA inhibitor, mock siRNA, and PC group were much more severe than that in NC and siNGF group (Figure 6a,b). These data suggested that NGF modulated the recruitment of inflammatory cells in airway and also showed the differences between receptor blocker and siRNA strategy.
Figure 5.
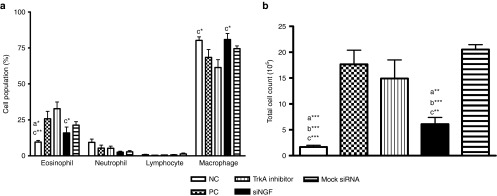
TrkA inhibitor enhanced airway eosinophil infiltration. The cells from BAL fluid were stained with Liu staining after using cytospin. A total of 300 cells were counted under microscopy for the (a) cell population or (b) total cell number. N = 6–8 per group. a*P < 0.05, a**P < 0.01, a***P < 0.001 versus PC. b*P < 0.05, b**P < 0.01, b***P < 0.001 v.s mock siRNA. c*P < 0.05, c**P < 0.01, c***P < 0.001 versus TrkA inhibitor. Data are shown as mean ± SEM and representative of five independent experiments. siRNA, small interfering RNA; TrkA, tropomyosin-related kinase A. BAL, bronchoalveolar lavage.
Figure 6.

siNGF decreased cell infiltration in lung. (a) Lung sections of the treated mice were prepared and stained with H&E staining for measurement of inflammatory cells around the airway. Bar = 100 µm (magnification: ×100). (b) The quantification of infiltration percentage in lung was analyzed by Image J software. a**P < 0.01 versus PC. b**P < 0.01 versus mock siRNA. c**P < 0.01 versus TrkA inhibitor. N = 6–8 per group. Data are shown as mean ± SEM and representative of five independent experiments. siNGF, small interfering RNA against nerve growth factor; siRNA, small interfering RNA; TrkA, tropomyosin-related kinase A.
siNGF decreased substance P production in lungs
As previously described, substance P induces neurogenic inflammation and is induced by NGF/TrkA pathway. To test whether knockdown NGF from airway epithelium could affect neurogenic inflammation via modulating substance P release, total substance P level was determined in lung homogenates during airway challenges. The targeting knockdown carried out a significantly decreased NGF level than that in other OVA-treated groups (Figure 3). In siNGF and TrkA inhibitor group, attenuated NGF/TrkA pathway resulted in decrease of substance P compared to that of PC group (Figure 7a). This result suggested that NGF/TrkA pathway affected AHR mediated by regulating substance P that plays an important role in neurogenic inflammation.
Figure 7.

siNGF reduced neuropeptide production and reduced airway innervation. (a) Lungs were isolated from the treated mice and homogenized by lysis buffer. Protein concentrations of lung homogenate were measured by bicinchoninic acid assay. Substance P concentrations were determined in 10 μg lung homogenate. (b) After OCT embedding, section and immunofluorescent staining, nerve fibers in lung sections were stained with nerve marker (PGP9.5). Bar = 100 µm. a*P < 0.05, a**P < 0.01, a***P < 0.001 versus positive control (PC). b*P < 0.05, b**P < 0.01, b***P < 0.001 versus mock siRNA. c*P < 0.05, c**P < 0.01, c***P < 0.001 versus TrkA inhibitor. N = 6–8 per group. Data are shown as mean ± SEM and representative of five independent experiments. siNGF, small interfering RNA against nerve growth factor; TrkA, tropomyosin-related kinase A.
NGF played an important role in hyperinnervation in airways
Immunofluorescent staining for nerve fibers was performed to examine whether NGF was a key factor in airway innervation during asthma progression. We measured the expression of PGP9.5 (the marker of nerve fibers) in airways. Innervation was increased after airway inflammation but the expressions of PGP9.5 were significantly reduced in TrkA inhibitor and siNGF group (Figure 7b). These findings suggested NGF promoted hyperinnervation during the progression of asthma and further increased AHR by modulating sensitivity of airway nerve.
Discussion
This study showed that the main producer of NGF in the airway was alveolar epithelial cells during allergen exposure, and NGF played a significant role in AHR via regulating airway inflammation, neuropeptide production, and airway innervations. In this study, effective therapeutic outcomes with siRNA technology and receptor inhibitor might provide new thoughts for asthmatic therapy.
It is well known that NGF is a mediator derived from multiple sources. Airway epithelium is the barrier and sensor to protect airway from damages that are induced by microbes and exogenous particles. Increasing evidences suggested epithelial cells skew the immune response to Th2 response via cytokine production including IL-33, IL-25, and thymic stromal lymphopoietin.22 However, airway epithelium could also spontaneously secrete or induce NGF expression by stimuli of proinflammatory cytokines or allergens.17,18,23 Bronchial epithelium increased NGF expression after allergen exposure and promoted survival of lung eosinophil via Trk receptor.19 Furthermore, local overexpression of NGF in the airway after OVA sensitization and challenges resulted in increase of OVA-specific IgE antibody, augmentation of eosinophilic airway inflammation, and increase of IL-5 production in BAL fluids.24 In this study, knockdown NGF in airway resulted in significantly decrease in Th2 cytokines and eosinophilic recruitment. These results showed that NGF released from epithelial cells was critical in development of Th2 response.
It has been suggested that the parasympathetic nervous system plays a key role in bronchial constriction and mucus production by acetylcholine reactivity.25 The effects of acetylcholine on airway smooth muscle tissue are regulated by tachykinins (e.g., substance P and neurokinin A) which are released from nonmyelinated sensory C-fibers in the airways.26 Many studies have suggested that NGF modulated neural sensitivity by increasing the expression of TRPV1 receptor,6 which received allergen and chemical challenges, and enhancing the production of substance P.27 Furthermore, NGF increased the NK-1 expression in airways.28 The biological activities of SP in airways include microvascular hyper-permeability, bronchoconstriction, hyperplasia of fibroblast and smooth muscle cells, and immune responses.29,30 NK-1 receptor is involved in AHR induced by NGF treatment31 and the TrkA receptor engages in activation of NGF-induced SP.8,31 These studies showed that NGF might play an important role in AHR through neural control. In our findings, decreased NGF production further reduced airway inflammation, AHR, and diminished airway hyperinnervation. Substance P levels in TrkA inhibitor group and siNGF group were notably decreased. Moreover, eosinophil infiltration was not different among the PC, mock siRNA, and siNGF groups. These data demonstrated that decreased NGF levels in airway inflammation and downregulated NGF-derived hyperinnervation by blocking NGF-TrkA pathway played a key role in reducing development of AHR.
There are a variety of therapeutic approaches in allergen-induced AHR and inflammation. Accumulating evidences suggested that NGF/TrkA pathway played a key role but the results in those studies were not very consistent. One of the possible explanations was the approach of administration. de Vries et al.8 showed that intranasal administration of TrkA antagonist, K252a, did not decrease the eosinophil recruitment in an asthmatic model but reversed AHR. Braun et al.16 found that intranasal delivery of anti-NGF antibody reduced AHR and inflammatory cytokines in BAL fluid in OVA-inducing asthma model. Ogawa et al.32 assessed that repeated siNGF sequence treatment inhibited AHR by decreasing hyperinnervation in chronic asthma model induced by house-dust mite. In this study, siRNA technology and TrkA inhibitor were applied and presented diverse findings. The ways to block NGF/TrkA signal included the neutralization of substrates such as an anti-NGF antibody and the inhibition of receptor activation such as an antagonist or the TrkA inhibitor. Once NGF-producing cells were active, NGF and its receptor such as TrkA receptors could be widely expressed on many kinds of cells to extend immune responses around the reactive location. Therefore, treatment with anti-NGF antibody was usually given before airway challenges to attenuate NGF-induced responses.12,16 Administration with antagonists or TrkA inhibitors before airway challenges or after challenges might block NGF-induced responses.8,33 Therefore, the dosage and timing might be the key points for the successful treatment.
K252a is the inhibitor of protein kinases by acting the competition with adenosine triphosphate-binding sites and could be a specific inhibitor of Trk receptor. However, K252a inhibits activation of TrkA receptor but it also blocks other signals that are triggered by protein kinase A and so on. Therefore, it was difficult to characterize the functional role of TrkA receptor in NGF-induced response. In this study, the specific TrkA inhibitor was used before airway challenges to decrease the progression of AHR. In addition to reducing degree of AHR, production of substance P and nerve hyperinnervation, TrkA inhibitor treatment led to NGF accumulation (Figure 2c), enhanced the eosinophil infiltration (Figure 5a), sustained airway inflammation (Figure 3) but reduced AHR. Compared with the findings of de Vries et al.8, treatment with K252a or TrkA inhibitor (AG879) reduced AHR and expression of substance P but did not affect eosinophil infiltration in airway in allergenic model. The possible explanation was distribution of receptor expression. The TrkA-positive tissues mainly include fibers-innervated skin,34 muscle35, and bone36; therefore, TrkA inhibitor approach might significantly reduce AHR via diminishing airway constriction. Furthermore, TrkA receptor was also expressed on a variety of inflammatory cells. Some other critical factors including dosage of inhibitor, NGF-inducing TrkA expression,37 and activation of p75 receptor might affect the NGF-induced process.
Further, our results also suggested that the blocking receptor might result in the compensatory NGF accumulation in airways to enhance eosinophil recruitment. Local treatment targeting TrkA accumulated NGF production.38 Hahn et al.19 confirmed that upregulation of NGF in epithelial cell after allergic inflammation promoted the survival of tissue eosinophil. Eosinophil is the main source of IL-5, and IL-5 induces NGF expression in bronchial epithelial cell. There is a positive feedback between NGF and IL-5 in airways. These finding suggested that administration of receptor blockage in attenuating NGF-induced response might be more considerable and cautious.
These results, which were administrated with anti-NGF antibody, suggested that neutralization of NGF significantly assuaged the progression of airway inflammation and AHR. However, some dilemmas existed in antibody therapy including penetration in physiological structure, immunogenic response induced by repeated injection, and expensive cost. Thus, we used another approach, siRNA technique, to suppress NGF production in airway and further to prevent NGF-induced immune responses in asthmatic animal model.
The siRNA has been used for gene silencing by specific degradation of complementary mRNA. This technique is widely used in both experimental and clinical approaches.39 However, it could not be denied some challenges of siRNA therapeutics including efficient delivery system and off-targeting effect were needed to overcome. Ogawa et al.32 used the stealth-siRNA sequences in chronic model of asthma and found that decreased NGF production from bronchial epithelium after allergen exposure affected AHR by reducing hyperinnervation but maintained airway inflammation. However, treatment with lentivirus expressing siRNA could reduce either AHR or airway inflammation in this study. One possible explanation was the original characterization of siRNA. Though these modified siRNA sequences were more stable and effective than naked siRNA sequences for knockdown, degradation and consumption could not be continually and spontaneously supplied during allergen-induced process. Therefore, the repeated procedure was similar with administration of anti-NGF antibody. In the past several years, our team has successfully developed siRNA strategies for treatment of airway inflammation and AHR in an asthmatic model40,41,42,43,44 Compared to administration of antibody, receptor antagonist or inhibitor, which was treated before every challenge, lentivirus-containing siRNA was just treated once and exerted long term and stable efficiency on target cells. For the reason, we suggested virus-expressing siRNA against NGF was a potential tool for asthmatic therapy.
In conclusion, we demonstrated that epithelium played a key role in the development of allergen-induced AHR which was mediated by NGF-TrkA pathway and suggested the therapeutic potential of siRNA strategy in asthma.
Materials and methods
Preparation and phonotypical determination of primary cultures. Lung cells which were isolated from BALB/c mice were cut into 2–3 mm3 and ground in MEM-α complete medium (Thermo Fisher Scientific, Waltham, MA) which were supplemented with 10% fetal bovine serum, 2 mmol/l l-glutamine, 100 U/ml penicillin, 1 mg/ml streptomycin, and 0.25 mg/ml amphotericin (Biological industries, Kibbutz Beit Haemek, Israel) and 25 mmol/l hydroxyethyl piperazine-1-ethane sulfonic acid (pH 7.2). After blood cell deletion, cells were incubated for 10–14 days. The primary cells were harvested and prepared for assay.
Phenotypes of primary cells were clarified by immunofluorescence staining. After fixation and permeabilization, cells were incubated with polyclonal rabbit antimouse prosurfactant protein C (proSP-C; dilution: 1:1,000, Millipore, Billerica, MA) and AQP5 (dilution: 1:100, Abcam, Cambridge, UK) or polyclonal goat antimouse clara-cell-specific 10 kDa protein (CC10; dilution: 1:100, Santa Cruz, Dallas, TX) for 1 hour at room temperature. After washing twice with phosphate-buffered saline (PBS) plus 0.05% Tween-20, cells were incubated with secondary antibody including Alexa 488 goat antirabbit IgG (Invitrogen, Carlsbad, CA) and FITC donkey antigoat IgG (Santa Cruz) for 30 minutes. Phenotype determination was performed by flow cytometry.
Protocol of NGF induction. Primary cells were seeded into 24-well plate (5 × 104/well) for cytokine assay. Before TNF-α stimulation, cells were starved overnight in fetal bovine serum-free medium. NGF production was induced by different doses of TNF-α (0, 5, 10, 20 ng/ml, Peprotech, Rocky hill, NJ) in fresh fetal bovine serum-free medium at different timing (12, 24, and 48 hours) after TNF-α stimulation. NGF levels in culture medium were harvested and determined by enzyme-linked immunosorbent assay (ELISA; Promega, Madison, WI).
Construction and preparation of short hairpin RNA (shRNA)-expressing lentiviruses. The targeting sequence of mouse NGF gene was siNGF-1: AAGCCAGTGAAATTAGGCTCC; siNGF-2: AAGGCGTTGACAACAGATGAG; and siNGF-3: AAGGACGCAGCTTTCTATACT. The control sequence of mock shRNA was GTCAGACTGTGCCATGACTG. The control shRNA was provided by the shRNA expression cassette kit and it was no homologous gene in any mouse gene. These shRNAs were synthesized by polymerase chain reaction and ligated into pTY-linker vector for the lentivirus delivering system. The plasmids for lentivirus (pHP-dl.Nde/Ase, pCEP4-tat, pHEF-VSV-G, and pTY-linker) were provided by Dr Lih-Hwa Hwang (Graduate Institute of Microbiology, National Yang-Ming University, Taiwan). The 293T cells were seeded before plasmid transfection. The lentivirus plasmids and pTY-linker containing targeting shRNA, GFP, or control shRNA were cotransfected into 293T cells at ratios of 3:2:1:3, respectively. The supernatant containing lentivirus was collected daily for 72 hours and followed by deletion of cellular debris and filtration with 0.45 μm filer. The viral supernatant was concentrated with 100 kDa centrifuge tube (Merck Millpore, Billerica, MA) and purified by ultracentrifugation with 20% sucrose buffer. The virus pellets were resuspended in PBS and stored at −80 °C.
Lentvirus titration was measured by qPCR lentivirus litration kit (Applied Biological Materials, Richmond, Canada) and the viral titer was indicated as the infectious unit/ml. 293T cells were infected by mixture containing 8 μg/ml polybrene (Sigma, St Louis, MO) and lentivirus packaging GFP for 48 hours. The GFP expression on 293T cells was assayed by FACS Calibur (BD, Franklin Lakes, NJ) and as the multiplicity of infection.
Determination of suppressed efficiency of shRNA. For protein assay, the primary cells were harvested and seeded into and 24-well plate (5 × 104/well). The cells were infected with mixture including polybrene and lentiviruses containing siNGF, or mock respectively (multiplicity of infection: 10) for 48 hours. Infected cells were stimulated by TNF-α (20 ng/ml) for 48 hours. The supernatants were collected and NGF levels in the supernatants were measured by ELISA assay. For RNA assay, cells were seeded into six-well plate (2 × 105/well). After 48 hours infection, infected cells were stimulated with TNF-α (20 ng/ml) for 6 hours. The RNA extractions were collected. Total RNA was performed with random hexamer primers and Super-Script III RNase H- reverse transcriptase (Invitrogen) to convert to cDNA. Quantitative real-time polymerase chain reaction was using the mouse NGF TaqMan gene expression assays (Applied Biosystems, Carlsbad, CA) in ABI PRISM 7400 Sequence Detector (Applied Biosystems, Foster City, CA). All reported mRNA levels were normalized by glyceraldehyde-3-phosphate dehydrogenase mRNA level.
Establishment of murine model of asthma. Female BALB/c mice (6-weeks old, N = 6–8 mice/group) were obtained from the Animal Center of the College of Medicine, National Taiwan University. The mice were sensitized intraperitoneally by injection of 2 mg of aluminum hydroxide containing 50 µg of OVA (Sigma) in a total volume of 200 μl PBS on day 0, and boosted with 25 µg OVA in the same dosage of adjuvant on days 14 and 28. Finally, the mice were challenged with OVA (100 µg) by intranasal administration for 3 days. Lentivirus-containing shRNA against NGF (3 × 106 infectious unit) were intratracheally delivered into the anesthetized animals 3 days before the first challenge with OVA. Lentivirus-containing GFP, mock siRNA virus or PBS (positive group) was used as the controls. The TrkA inhibitor (Merck, Darmstadt, Germany), an oxindole compound that selectively targeted the adenosine triphosphate-binding pocket of TrkA, was intranasally inhaled 3 hours before OVA challenge.33
Determination of cytokine production. Following sacrifice, IL-4, IL-5, IL-6, IL-12p70, IL-13, IL-17, IFN-γ, and eotaxin levels in BAL fluid and lung homogenates were determined by the ELISA assays (R&D, Minneapolis, MN) according to the manufacturer-recommended protocols. The right lungs were frozen and samples were solubilized by homogenization in lysis buffer (Cell Signaling Technology, Danvers, MA).45 Protein concentrations was measured by bicinchoninic acid kit (Thermo scientific, Rockford, IL). Substance P level in lung homogenates was determined by enzyme immunoassay kit (Cayman chemical, Ann Arbor, MI).
Determination of the airway resistance and function. Airway resistance was assessed as an increase in pulmonary resistance after challenge with aerosolized β-methacholine. Mice were anesthetized and tracheostomized. The breath rate was mechanically maintained at 150 breaths/minute with a tidal volume of 0.3 ml, and a positive end-expiratory pressure of 3–4 cm H2O. PE-50 tubing was inserted into the esophagus to the level of the thorax, coupled with a pressure transducer (LDS GOULD, Valley View, OH). Flow, pressure and volume change were measured by electronic differentiation of volume signal. Pulmonary resistance was calculated by a software program (Model PNM-PCT100W, LDS PONEMAH Physiology Platform, LDS GOULD, Valley View, OH). β-methacholine aerosol was generated with a nebulizer and administrated directly through the ventilator. The resistance of the orotracheal tube (0.45 cm H2O seconds/ml) was subtracted from all airway resistance measurements. Data were expressed as the pulmonary resistance (RL) of three independent experiments.
Airway responsiveness was measured in unrestrained animals by barometric whole-body plethysmography (Buxco, Troy, NY). Briefly, mice were placed in the main chamber and baseline readings were taken and averaged for 3 minutes. Aerosolized PBS or β-methacholine in increasing concentrations (3.125–50 mg/ml) were for 3 minutes. Airway reactivity was expressed as enhanced pause (Penh), and readings were taken and averaged for 3 minutes after each nebulization. Data were expressed as the ratio of Penhβ-methacholine values compared to PenhPBS from three independent experiments.
Determination of airway inflammation and histological examination. BAL fluid was harvested by gentle aspiration three times with 1 ml Hank's Balanced Salt Solution and then centrifuged. Differential cell counts were distinguished on cytologic preparations. The cells slides were stained with Liu staining after using cytospin and total of 300 cells were counted under microscopy. BAL fluid supernatants were assayed by ELISA assay. The lungs were fixed with 10% formalin-PBS buffer and sections were prepared and stained with hematoxylin and eosin to quantify the number of infiltrating inflammatory cells under microscopy. The images of cell infiltration were quantified by Image J software (NIH, v1.45). To calculate the infiltration percentage in the mouse lung, the number of pixels within the defined area of cell particles was measured as A value. The total number of pixels within the field of view was measured as T value. The percentage was calculated according to the following formula:
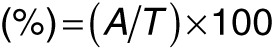 |
The average of the four fields (100× magnification) of view was used to determine cell infiltration in each lung analyzed.
Determination of antigen-specific antibody. Serum anti-OVA antibody titers were determined by ELISA assay. In brief, 96-well microtiter plates were precoated with 1 µg/well OVA in NaHCO3 coating buffer (pH 9.6). After overnight incubation at 4 °C, plates were blocked with 1% bovine serum albumin in PBS solution for 2 hours at room temperature. Serum samples were added for 2 hours at room temperature. Biotin conjugated antimouse IgE, IgG1, and IgG2a (1:500, PharMingen, San Diego, CA) were spectacularly diluted in 1% bovine serum albumin–PBS buffer for 45 min at room temperature. Avidin-conjugated horseradish peroxidase (1:500; Pierce Biotechnology, Rockford, IL) was then added for 30 minutes at room temperature. Finally, the reaction was developed by tetramethylbenzidine substrate, and stopped by 2N H2SO4 solution. The absorbance was determined at 490 nm in a microplate reader (Molecular Devices, Sunnyvale, CA). The titers of OVA-specific antibody were compared to standard serum that were generated in laboratory and the OD490nm reading of IgG1, IgG2a, and IgE in standard serum was allocated 1 ELISA unit (E.U.)/ml.
Immunofluorescence staining. After BAL fluid collection, lungs were perfused by PBS, fixed by 4% paraformaldehyde and embedded in optimal cutting temperature compound. The 5 μm thickness of frozen sections were cut and prepared for staining. The slides were worm for room temperature and washed by PBS following methanol fixation for 10 minutes at −20 °C and rinsed by PBS. Slides were incubated in blocking solution (Biogenex, Fremont, CA) for 2 hours and permeabilized by 0.3% Triton X-100 in PBS for 30 minutes at room temperature. During incubation with goat biotinylated anti-GFP (Dilution: 1:200, Vector Laboratories, Burlingame, CA) or monoclonal rabbit antimouse PGP9.5 (dilution: 1:100, Epitomics, Burlingame, CA) in a humid chamber overnight at 4 °C, the antigen detection was carried out with streptavdin conjugated DyLight-594 (Vector Laboratories) or Alexa 488 goat antirabbit IgG (Invitrogen) for 1 hour. Negative control sections were processed by omitting the specific primary antibody. The sections were added mounting media with 4',6-diamidino-2-phenylindole (Vector Laboratories) and coverslip.
BAL cells were stained with goat biotinylated anti-GFP for overnight at 4 °C after cytospin, fixation with methanol at −20 °C for 10 minutes and blockade with blocking solution plus 0.3% Triton X-100 at room temperature for 1 hour. During twice wash, slides were incubated with secondary antibody conjugated DyLight-594 for 1 hour at room temperature. The sections were added mounting media and coverslip. The Immunofluorescence labelings were analyzed using fluorescence microscope (Zeiss Axio Imager, Zeiss, Oberkochen, Germany) and were visualized with Axio Vision.
Statistical analysis. All values referred to mean ± SEM using one-way analysis of variance followed by the Fisher protected least significant difference test. The minimal level of significance was a P value <0.05.
Acknowledgments
This project was supported by a grant from College of Medicine, National Taiwan University and China Medical University jointed grant, 99FB008-302 and 100F008-41 and a grant from National Science Council (20100312). We also thank the staff of the imaging core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance.
References
- Hargreave FE, Dolovich J, O'Byrne PM, Ramsdale EH, Daniel EE. The origin of airway hyperresponsiveness. J Allergy Clin Immunol. 1986;78 5 Pt 1:825–832. doi: 10.1016/0091-6749(86)90226-5. [DOI] [PubMed] [Google Scholar]
- Baroffio M, Barisione G, Crimi E, Brusasco V. Noninflammatory mechanisms of airway hyper-responsiveness in bronchial asthma: an overview. Ther Adv Respir Dis. 2009;3:163–174. doi: 10.1177/1753465809343595. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond, B, Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykissas MG, Batistatou AK, Charalabopoulos KA, Beris AE. The role of neurotrophins in axonal growth, guidance, and regeneration. Curr Neurovasc Res. 2007;4:143–151. doi: 10.2174/156720207780637216. [DOI] [PubMed] [Google Scholar]
- Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and calcitonin gene-related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. 1992;49:693–698. doi: 10.1016/0306-4522(92)90237-v. [DOI] [PubMed] [Google Scholar]
- El-Hashim AZ, Jaffal SM. Nerve growth factor enhances cough and airway obstruction via TrkA receptor- and TRPV1-dependent mechanisms. Thorax. 2009;64:791–797. doi: 10.1136/thx.2009.113183. [DOI] [PubMed] [Google Scholar]
- Weigand LA, Undem BJ. Allergen-induced neuromodulation in the respiratory tract. Chem Immunol Allergy. 2012;98:142–162. doi: 10.1159/000336508. [DOI] [PubMed] [Google Scholar]
- de Vries A, Engels F, Henricks PA, Leusink-Muis T, McGregor GP, Braun A, et al. Airway hyper-responsiveness in allergic asthma in guinea-pigs is mediated by nerve growth factor via the induction of substance P: a potential role for trkA. Clin Exp Allergy. 2006;36:1192–1200. doi: 10.1111/j.1365-2222.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- Virchow JC, Julius P, Lommatzsch M, Luttmann W, Renz H, Braun A. Neurotrophins are increased in bronchoalveolar lavage fluid after segmental allergen provocation. Am J Respir Crit Care Med. 1998;158:2002–2005. doi: 10.1164/ajrccm.158.6.9803023. [DOI] [PubMed] [Google Scholar]
- Tortorolo L, Langer A, Polidori G, Vento G, Stampachiacchere B, Aloe L, et al. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2005;172:233–237. doi: 10.1164/rccm.200412-1693OC. [DOI] [PubMed] [Google Scholar]
- Noga O, Englmann C, Hanf G, Grützkau A, Seybold J, Kunkel G. The production, storage and release of the neurotrophins nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 by human peripheral eosinophils in allergics and non-allergics. Clin Exp Allergy. 2003;33:649–654. doi: 10.1046/j.1365-2222.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- Kiliç A, Sonar SS, Yildirim AO, Fehrenbach H, Nockher WA, Renz H. Nerve growth factor induces type III collagen production in chronic allergic airway inflammation. J Allergy Clin Immunol. 2011;128:1058–66.e1. doi: 10.1016/j.jaci.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Freund-Michel V, Frossard N. The nerve growth factor and its receptors in airway inflammatory diseases. Pharmacol Ther. 2008;117:52–76. doi: 10.1016/j.pharmthera.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Braun A, Quarcoo D, Schulte-Herbrüggen O, Lommatzsch M, Hoyle G, Renz H. Nerve growth factor induces airway hyperresponsiveness in mice. Int Arch Allergy Immunol. 2001;124:205–207. doi: 10.1159/000053711. [DOI] [PubMed] [Google Scholar]
- Hoyle GW, Graham RM, Finkelstein JB, Nguyen KP, Gozal D, Friedman M. Hyperinnervation of the airways in transgenic mice overexpressing nerve growth factor. Am J Respir Cell Mol Biol. 1998;18:149–157. doi: 10.1165/ajrcmb.18.2.2803m. [DOI] [PubMed] [Google Scholar]
- Braun A, Appel E, Baruch R, Herz U, Botchkarev V, Paus R, et al. Role of nerve growth factor in a mouse model of allergic airway inflammation and asthma. Eur J Immunol. 1998;28:3240–3251. doi: 10.1002/(SICI)1521-4141(199810)28:10<3240::AID-IMMU3240>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Fox AJ, Patel HJ, Barnes PJ, Belvisi MG. Release of nerve growth factor by human pulmonary epithelial cells: role in airway inflammatory diseases. Eur J Pharmacol. 2001;424:159–162. doi: 10.1016/s0014-2999(01)01138-4. [DOI] [PubMed] [Google Scholar]
- Pons F, Freund V, Kuissu H, Mathieu E, Olgart C, Frossard N. Nerve growth factor secretion by human lung epithelial A549 cells in pro- and anti-inflammatory conditions. Eur J Pharmacol. 2001;428:365–369. doi: 10.1016/s0014-2999(01)01280-8. [DOI] [PubMed] [Google Scholar]
- Hahn C, Islamian AP, Renz H, Nockher WA. Airway epithelial cells produce neurotrophins and promote the survival of eosinophils during allergic airway inflammation. J Allergy Clin Immunol. 2006;117:787–794. doi: 10.1016/j.jaci.2005.12.1339. [DOI] [PubMed] [Google Scholar]
- Solomon A, Aloe L, Pe'er J, Frucht-Pery J, Bonini S, Bonini S, et al. Nerve growth factor is preformed in and activates human peripheral blood eosinophils. J Allergy Clin Immunol. 1998;102:454–460. doi: 10.1016/s0091-6749(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Fujimaki H, Tin-Tin-Win-Shwe, Yamamoto S, Nakajima D, Goto S. The expression of nerve growth factor in mice lung following low-level toluene exposure. Toxicol Lett. 2009;191:240–245. doi: 10.1016/j.toxlet.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Curotto de Lafaille MA, Lafaille JJ, Graca L. Mechanisms of tolerance and allergic sensitization in the airways and the lungs. Curr Opin Immunol. 2010;22:616–622. doi: 10.1016/j.coi.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye YL, Wu HT, Lin CF, Hsieh CY, Wang JY, Liu FH, et al. Dermatophagoides pteronyssinus 2 regulates nerve growth factor release to induce airway inflammation via a reactive oxygen species-dependent pathway. Am J Physiol Lung Cell Mol Physiol. 2011;300:L216–L224. doi: 10.1152/ajplung.00165.2010. [DOI] [PubMed] [Google Scholar]
- Quarcoo D, Schulte-Herbrüggen O, Lommatzsch M, Schierhorn K, Hoyle GW, Renz H, et al. Nerve growth factor induces increased airway inflammation via a neuropeptide-dependent mechanism in a transgenic animal model of allergic airway inflammation. Clin Exp Allergy. 2004;34:1146–1151. doi: 10.1111/j.1365-2222.2004.01993.x. [DOI] [PubMed] [Google Scholar]
- Racké K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol. 2006;533:57–68. doi: 10.1016/j.ejphar.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Myers AC. Transmission in autonomic ganglia. Respir Physiol. 2001;125:99–111. doi: 10.1016/s0034-5687(00)00207-3. [DOI] [PubMed] [Google Scholar]
- Dinh QT, Groneberg DA, Peiser C, Springer J, Joachim RA, Arck PC, et al. Nerve growth factor-induced substance P in capsaicin-insensitive vagal neurons innervating the lower mouse airway. Clin Exp Allergy. 2004;34:1474–1479. doi: 10.1111/j.1365-2222.2004.02066.x. [DOI] [PubMed] [Google Scholar]
- Hu C, Wedde-Beer K, Auais A, Rodriguez MM, Piedimonte G. Nerve growth factor and nerve growth factor receptors in respiratory syncytial virus-infected lungs. Am J Physiol Lung Cell Mol Physiol. 2002;283:L494–L502. doi: 10.1152/ajplung.00414.2001. [DOI] [PubMed] [Google Scholar]
- De Swert KO, Joos GF. Extending the understanding of sensory neuropeptides. Eur J Pharmacol. 2006;533:171–181. doi: 10.1016/j.ejphar.2005.12.066. [DOI] [PubMed] [Google Scholar]
- O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201:167–180. doi: 10.1002/jcp.20061. [DOI] [PubMed] [Google Scholar]
- de Vries A, Dessing MC, Engels F, Henricks PA, Nijkamp FP. Nerve growth factor induces a neurokinin-1 receptor- mediated airway hyperresponsiveness in guinea pigs. Am J Respir Crit Care Med. 1999;159 5 Pt 1:1541–1544. doi: 10.1164/ajrccm.159.5.9808058. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Azuma M, Uehara H, Takahashi T, Nishioka Y, Sone S, et al. Nerve growth factor derived from bronchial epithelium after chronic mite antigen exposure contributes to airway hyperresponsiveness by inducing hyperinnervation, and is inhibited by in vivo siRNA. Clin Exp Allergy. 2012;42:460–470. doi: 10.1111/j.1365-2222.2011.03918.x. [DOI] [PubMed] [Google Scholar]
- Abram M, Wegmann M, Fokuhl V, Sonar S, Luger EO, Kerzel S, et al. Nerve growth factor and neurotrophin-3 mediate survival of pulmonary plasma cells during the allergic airway inflammation. J Immunol. 2009;182:4705–4712. doi: 10.4049/jimmunol.0802814. [DOI] [PubMed] [Google Scholar]
- Lu J, Zhou XF, Rush RA. Small primary sensory neurons innervating epidermis and viscera display differential phenotype in the adult rat. Neurosci Res. 2001;41:355–363. doi: 10.1016/s0168-0102(01)00293-0. [DOI] [PubMed] [Google Scholar]
- Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, et al. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience. 2002;113:155–166. doi: 10.1016/s0306-4522(02)00165-3. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, et al. A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone. 2010;46:306–313. doi: 10.1016/j.bone.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti L, Callegari A, Luin S, Signore G, Viegi A, Beltram F, et al. Ligand signature in the membrane dynamics of single TrkA receptor molecules. J Cell Sci. 2013;126 Pt 19:4445–4456. doi: 10.1242/jcs.129916. [DOI] [PubMed] [Google Scholar]
- Guerios SD, Wang ZY, Boldon K, Bushman W, Bjorling DE. Blockade of NGF and trk receptors inhibits increased peripheral mechanical sensitivity accompanying cystitis in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R111–R122. doi: 10.1152/ajpregu.00728.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Chiang BL. siRNA as a therapy for asthma. Curr Opin Mol Ther. 2009;11:652–663. [PubMed] [Google Scholar]
- Lee CC, Chiang BL. RNA interference: new therapeutics in allergic diseases. Curr Gene Ther. 2008;8:236–246. doi: 10.2174/156652308785160692. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang HY, Chiang BL. Lentiviral-mediated interleukin-4 and interleukin-13 RNA interference decrease airway inflammation and hyperresponsiveness. Hum Gene Ther. 2011;22:577–586. doi: 10.1089/hum.2009.105. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang HY, Chiang BL. Lentiviral-mediated GATA-3 RNAi decreases allergic airway inflammation and hyperresponsiveness. Mol Ther. 2008;16:60–65. doi: 10.1038/sj.mt.6300309. [DOI] [PubMed] [Google Scholar]
- Huang HY, Lee CC, Chiang BL. Short hairpin RNAs against eotaxin or interleukin-5 decrease airway eosinophilia and hyper-responsiveness in a murine model of asthma. J Gene Med. 2009;11:112–118. doi: 10.1002/jgm.1285. [DOI] [PubMed] [Google Scholar]
- Sugita A, Ogawa H, Azuma M, Muto S, Honjo A, Yanagawa H, et al. Antiallergic and anti-inflammatory effects of a novel I kappaB kinase beta inhibitor, IMD-0354, in a mouse model of allergic inflammation. Int Arch Allergy Immunol. 2009;148:186–198. doi: 10.1159/000161579. [DOI] [PubMed] [Google Scholar]