

Abstract
Duchenne muscular dystrophy (DMD) is caused by lack of functional dystrophin and results in progressive myofiber damage and degeneration. In addition, impaired muscle regeneration and fibrosis contribute to the progressive pathology of DMD. Importantly, transforming growth factor-β (TGF-β) is implicated in DMD pathology and is known to stimulate fibrosis and inhibit muscle regeneration. In this study, we present a new strategy to target TGF-β signaling cascades by specifically inhibiting the expression of TGF-β type I receptor TGFBR1 (ALK5). Antisense oligonucleotides (AONs) were designed to specifically induce exon skipping of mouse ALK5 transcripts. AON-induced exon skipping of ALK5 resulted in specific downregulation of full-length receptor transcripts in vitro in different cell types, repression of TGF-β activity, and enhanced C2C12 myoblast differentiation. To determine the effect of these AONs in dystrophic muscles, we performed intramuscular injections of ALK5 AONs in mdx mice, which resulted in a decrease in expression of fibrosis-related genes and upregulation of Myog expression compared to control AON-injected muscles. In summary, our study presents a novel method to target TGF-β signaling cascades with potential beneficial effects for DMD.
Keywords: ALK5, antisense oligonucleotides, Duchenne muscular dystrophy
Introduction
Duchenne muscular dystrophy (DMD) is a lethal and common form of muscular dystrophy affecting ~1 in 5,000 newborn boys worldwide.1,2 DMD is caused by mutations in the DMD gene that disrupt the open reading frame of the transcript. The DMD gene encodes the structural muscle protein dystrophin which is crucial for muscle fiber integrity. In DMD patients, mutations impair dystrophin function, which triggers profound muscle fiber damage and degeneration.3 Currently, no cure exists for DMD, although improved care has extended the lifespan and improved the quality of life of DMD patients.1,4 In addition, antisense oligonucleotide (AON)–mediated exon skipping/correction of mutated DMD transcript is a promising treatment that showed encouraging results in DMD patients in several clinical trials.5,6,7,8 However, pathophysiological processes triggered by the chronic degeneration of muscle fibers and resulting inflammation are an additional hurdle since they elicit fibrosis, impair muscle regeneration, and thus contribute to the progressive loss of muscle fibers and muscle function in DMD patients. Targeting protein signaling cascades involved in fibrosis and regulation of muscle growth/regeneration may therefore provide a supplementary approach to improve muscle quality in DMD patients.9,10
TGF-β signaling is known to play an important role in DMD pathology. The TGF-β family consists of a multitude of structurally related secreted signaling proteins that induce downstream signaling via interaction with type I and type II transmembrane serine/threonine kinase receptors. Up to date, seven type I receptors have been described (ALK1-7) and four type II receptors (TGFBR2, ACVR2A/B, and BMPR2) that confer specificity of the different TGF-β ligands. TGF-β is the prototypical protein of this family, which initiates signaling by forming dimers that bind to the type II receptor TGFBR2. This complex subsequently recruits and activates the type I receptor TGFBR1 (ALK5).11,12 Formation and activation of ligand receptor complexes induces intracellular signaling cascades and transcriptional responses via phosphorylation of receptor-regulated Smad2 and Smad3 proteins (Figure 1a). Importantly, increase of TGF-β signaling has been correlated with the pathophysiology of multiple diseases, including muscle disorders such as DMD.9,10,13 Expression of one specific isoform of TGF-β, TGF-β1, is correlated with the progressive pathology of DMD, since it is upregulated and associated with fibrosis in muscles of DMD patients and mdx mice, a mouse model for DMD.14,15,16,17 Several promising in vivo studies with TGF-β antagonists, such as TGF-β neutralizing antibodies18,19 or losartan,18,19,20 showed that repression of TGF-β signaling reduced fibrosis and enhanced muscle regeneration in mdx mice. However, as yet, no specific inhibitors have advanced to clinical trials involving DMD patients, and moreover, losartan and other small molecule inhibitors that are in clinical trials for other diseases such as cancer are not specific for TGF-β signaling.21 In this study, we therefore adopted a novel AON-mediated strategy to interfere with TGF-β signaling pathways by specifically targeting the type I receptor ALK5. Our aim was to provide proof-of-principle of the efficiency of this knockdown method in vitro. In addition, we used this method to determine the effect of ALK5 knockdown in dystrophic muscle of mdx mice.
Figure 1.
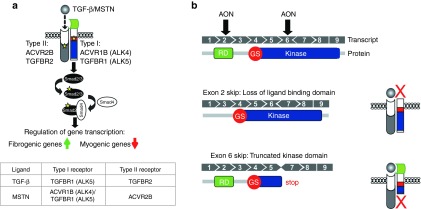
Schematic overview of transforming growth factor-β (TGF-β) signaling cascades and the effect of the designed antisense oligonucleotides (AONs). (a) Overview of the mechanism of TGF-β signaling. TGF-β, MSTN, and activin bind to type II and type I receptors, which are activated and induce downstream Smad2/3-dependent signaling pathways. Yellow stars depict phosphorylation. (b) Overview of ALK5 transcript and the corresponding protein domains, showing the effect of AON-mediated exon skipping. The ALK5 gene consist of nine exons. The different corresponding protein domains are shown below the transcript. Receptor domain (RD, in green), glycine/serine-rich domain (GS, in red) and kinase domain (kinase, in blue). AON-mediated targeting of exon 2 results in transcripts that lack the sequence encoding the ligand-binding domain. AON-mediated targeting of exon 6 disrupts the open reading frame and generates a premature stop triplet (STOP).
Results
AON-mediated exon skipping of ALK5
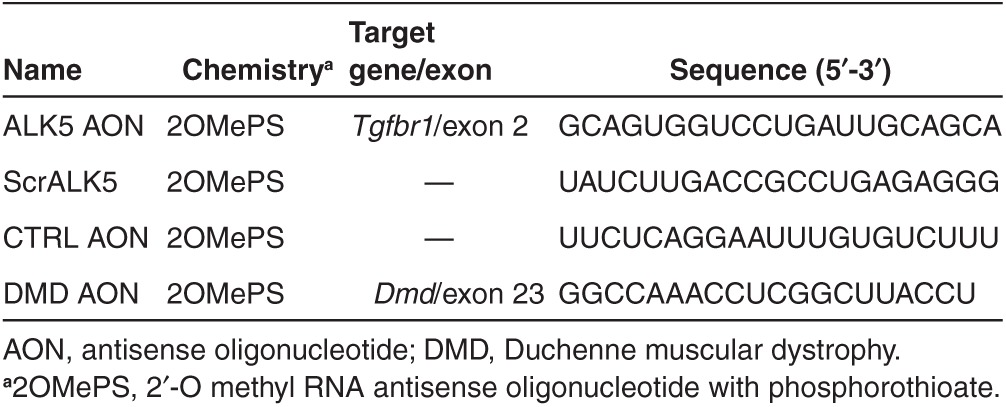
We developed a strategy to selectively inhibit the function of ALK5 receptors in mice based on AON-mediated exon skipping (Figure 1b). Our focus was centered on exon 2 and exon 6, since exclusion of either of these exons in Tgfbr1 (ALK5) transcripts would result in a dysfunctional protein. Exclusion of exon 2 retains the reading frame, but results in an internally truncated protein that lacks the ligand-binding domain (Figure 1b). Exon 6 encodes part of the kinase domain and exclusion of this exon will generate an out-of-frame transcript with a premature stop (Figure 1b). We designed different AONs with phosphorothioate backbones and 2′-O-methyl ribose modifications (2OMePS) for both exon 2 and exon 6 using a previously published protocol for AON design.22 Subsequently, the best AON was selected based on the ability to induce exon skipping after transfection in C2C12 myoblasts. Reverse transcription PCR analysis with primers flanking the targeted exons showed that exon 2 could be effectively excluded during the splicing process in vitro. One AON clearly induced a smaller polymerase chain reaction (PCR) product after transfection in C2C12 myoblasts. Sequence analysis confirmed this fragment lacked exon 2 (Figure 2a,b; Table 1). In addition, quantitative real-time PCR (qPCR) analysis on reverse transcribed cDNA with primers in the targeted exon and outside the targeted exon showed a specific approximately threefold downregulation of ALK5 full length transcript after transfection of the selected ALK5 AON (Figure 2c). Importantly, expression of the related type I receptor ALK4 was not affected by AON-mediated ALK5 knockdown, thereby showing the specificity of the knockdown (Figure 2c). We also evaluated exon-skipping levels in other murine-derived cells expressing ALK5, such as mesenchymal stem cells (C3H10T1/2), endothelial cells, primary and immortalized fibroblasts, and observed comparable exon skipping and knockdown levels (data not shown). Importantly, these experiments showed that the selected AONs can be used to specifically reduce the expression of ALK5. Targeting ALK5 exon 6 was also found to be effective using one AON in vitro, but this AON was later found to be ineffective in vivo and was therefore omitted from this study (data not shown). All further experiments were conducted with the AON targeting exon 2 of ALK5 (from now on referred to as ALK5 AON, Table 1).
Figure 2.

In vitro proof of principle of antisense oligonucleotides (AON)-mediated ALK5 exon skipping. (a) AONs targeting exon 2 of ALK5 (ALK5 AON) were transfected at different concentrations into C2C12 mouse myoblasts. Two days after transfection, the efficacy to induce exon skipping was assessed by reverse transcription PCR ALK5-specific primers in flanking exons (red arrowheads). Nontransfected (NT) cells or cells transfected with control AON (Ctrl AON, nontargeting) served as controls. (b) Sequencing of excised PCR products showed exclusion of the targeted exons ALK5 AON transfection (exon 2 skip) in C2C12 cells. (c) Quantitative real-time PCR of C2C12 samples was performed to compare full-length ALK5 transcript expression using primers in the skipped exon and in the flanking exon. Cells were transfected with 200 nmol/l AON. These data are shown as the average of at least three independent experiments and is shown relative to control AON samples. Error bars represent standard deviations. *P < 0.05; **P < 0.01
Table 1. Antisense oligonucleotides used in this study.
Exon skipping of ALK5 inhibits MSTN/TGF-β signaling in vitro
We next determined whether AON-mediated ALK5 exon skipping interfered with TGF-β signaling in vitro. We transfected ALK5 AONs together with a (CAGA)12-luciferase transcriptional reporter construct, which drives expression of a luciferase reporter gene in a Smad3-dependent manner.23 Transfection with ALK5 AONs inhibited TGF-β signaling in both C2C12 myoblasts and C3H10T1/2 cells, showing the specific effect of AON-mediated knockdown of ALK5 on TGF-β signaling (Figure 3a). Related TGF-β ligand myostatin (MSTN) is known to activate downstream Smad2/3 signaling cascades via type I receptors ACVR1B (ALK4) and ALK5 (Figure 1a).24,25 Therefore, we also determined the effect of AON-mediated ALK5 knockdown on MSTN activity. In contrast to the effect on TGF-β signaling in C2C12 myoblasts, ALK5 AONs did not alter the responsiveness to MSTN in these cultured muscle cells (Figure 3b). However, MSTN-induced signaling was decreased after AON-mediated ALK5 knockdown approximately fourfold in C3H10T1/2 cells and other nonmyogenic cells tested (Figure 3b and data not shown). This is consistent with our recent finding of cell-type–specific utilization of the type I receptor for MSTN signaling using siRNA-mediated knockdown of ALK4 or ALK5 (ref. 24) and confirms the specificity of ALK5 knockdown using AONs.
Figure 3.

ALK5 antisense oligonucleotides (AONs) inhibit MSTN and transforming growth factor-β (TGF-β) signaling. (a) ALK5 AONs (200 nmol/l) specifically repressed TGF-β-induced (CAGA)12-luciferase activity in both C2C12 myoblasts and C3H10T1/2 cells. (b) MSTN-induced Smad3-dependent (CAGA)12-luciferase activity was specifically repressed by ALK5 AONs (200 nmol/l) in C3H10T1/2 mesenchymal stem cells. In contrast, ALK5 AONs did not inhibit MSTN activity in C2C12 myoblasts. Firefly luciferase activity of (CAGA)12-luciferase constructs was corrected with renilla-luciferase activity of cotransfected cytomegalovirus-renilla constructs. The experiments were performed at least three times and each experiment was performed in triplicate. The values were averaged and plotted as fold relative to control AON (Ctrl AON)-transfected samples. Error bars represent standard deviations. *P < 0.05, **P < 0.01.
ALK5 AONs enhance myogenic differentiation of C2C12 myoblasts in vitro
After showing that the AONs exhibited potency in abrogating TGF-β signaling, we investigated the effect of AON-mediated ALK5 knockdown on myoblast differentiation. We induced C2C12 myoblast differentiation by replacing the proliferation medium with low serum differentiation medium. One day after the switch to differentiation medium, cells were transfected with 200 nmol/l ALK5 AONs and the differentiation was monitored for up to 7 days. An increase in myogenic differentiation was observed for ALK5 AON-transfected cells, as measured by immunofluorescent staining of desmin and myosin heavy chain and quantification of the differentiation and fusion indices (Figure 4a,b). Together, these experiments demonstrate that ALK5 AONs enhance myoblast differentiation and that ALK5-mediated signaling cascades are involved in regulation of myogenic differentiation.
Figure 4.

ALK5 antisense oligonucleotides (AONs) enhance myoblast differentiation. (a) Immunofluorescence images of C2C12 cells transfected with control (Ctrl) or ALK5 AONs at different time points after initiation of myogenic differentiation. Immunofluorescent staining is shown for desmin (myogenic cells, red), myosin (differentiated myotubes, green), and 4',6-diamidino-2-phenylindole (nuclear, blue). (b) Fusion and differentiation indices were calculated based on the immunofluorescence images. ALK5 AONs increase fusion and differentiation index in C2C12 cells. Error bars represent standard deviations. **P < 0.01; ns, not significant
Intramuscular injection of ALK5 AONs in mdx mice
Following the encouraging results in vitro, we proceeded to investigate the effect of ALK5 AONs in mdx mice after intramuscular injection. We injected triceps muscles of 5-week-old mdx mice for 4 consecutive days with ALK5 AON and the animals were sacrificed either 4 or 10 days after the final injection. Contralateral muscles were injected with control-scrambled ALK5 AONs (designated as ScrALK5). In addition, the intramuscular injection of ALK5 AONs was combined with 2OMePS AONs targeting exon 23 in Dmd gene (DMD AON). These DMD AONs result in exon skipping of exon 23, restoration of the Dmd open reading frame, and dystrophin expression in mdx mice.26 This combination was chosen to obtain proof-of-principle for a possible combination therapy (improving muscle quality and restoring dystrophin) as well as to provide a positive control for the in vivo administration procedure.
PCR with primers detecting exon skipping showed that ALK5 AONs did not interfere with the exon-skipping potential of DMD AON, resulting in simultaneous exon skipping in both genes (Supplementary Figure S1). All muscles injected with ALK5 AONs demonstrated ALK5 exon 2 skipping, whereas the ScrALK5 AON-injected muscles did not (Supplementary Figure S1). qPCR quantification of triceps samples isolated 10 days after injection showed a significant knockdown of 50% of full-length ALK5 transcripts in the muscles injected with ALK5 AON (Figure 5a). Specific knockdown of full-length ALK5 transcripts was also observed 4 days after injection, although the effect was less pronounced (Supplementary Figure S2a). Target specificity of these AONs was maintained in vivo, because ALK5 AONs did not alter the expression of ALK4 (Figure 5a; Supplementary Figure S2a).
Figure 5.

Effect of ALK5 antisense oligonucleotides (AONs) after intramuscular injections in mdx mice. (a) Quantitative polymerase chain reaction analysis of full-length ALK5 expression in samples from ALK5 or scrambled control AON (ScrALK5 AON)-injected triceps muscles. Muscles were isolated and analyzed 10 days after the last injection. Specific reduction of full-length ALK5 was observed after injection with ALK5 AONs. (b) Treatment with ALK5 AONs increased the expression of Myog (not significant) and decreased expression of Col1a1. ALK5 AONs reduced expression levels of Ctgf (not significant) and Serpine1. (c) Protein lysates were analyzed for Serpine1 and phosphorylated Smad2 by western blot and quantified densitometrically using actin for normalization. Error bars in a, b, and d represent standard deviations. *P < 0.05; **P < 0.01.
Effect of ALK5 AONs on downstream target genes/proteins in mdx mice
To further assess the effect of ALK5 AONs in vivo in mdx mice, Myog expression and expression of genes that are involved in fibrosis (Col1a1, Serpine1, Ctgf) were determined by qPCR. A trend of increased Myog expression was observed upon ALK5 AON injection after 10 days (Figure 5b), but no effect was observed after 4 days (Supplementary Figure S2b). Col1a1 expression level decreased fourfold upon ALK5 knockdown in 10-day samples (Figure 5b). A similar trend, although less pronounced, was observed in 4-day samples (Supplementary Figure S2b). Decreases of Serpine1 (also known as plasminogen activator inhibitor-1 (Pai1)) and connective tissue growth factor (Ctgf) expression, two other genes correlated with fibrosis, were also observed upon Tgfbr1 knockdown after 10 days (Figure 5b) and after 4 days (Serpine1 only (Supplementary Figure S2b)). These changes in Serpine1 gene expression upon ALK5 knockdown were confirmed on protein level by western blot analysis (Figure 5c,d). Western blot analysis also showed a trend of decreased levels of phosphorylated Smad2 upon intramuscular injection with ALK5 AONs, although this was not significant due to the considerable variation between different samples (Figure 5c,d). Histological analysis was performed to assess the percentage of fibrotic/necrotic areas. This revealed no changes between the different groups (Supplementary Figure S3a). We determined the effect of ALK5 AON injections on muscle fiber diameter. No significant differences were observed between scrambled control AON-injected muscles and ALK5-injected muscles (Supplementary Figure S3b).
Discussion
In this study, we present a novel approach to inhibit TGF-β signaling cascades using AON-mediated targeting of type I receptor kinase ALK5 (Tgfbr1). To our knowledge, this is the first study describing the design of a specific ALK5 inhibitor. Whereas other studies described the screening and development of small molecule kinase inhibitors of ALK5, current small molecule ALK5 inhibitors are not specific, since they also inhibit nonrelated protein kinases.27 Additionally, small molecule ALK5 inhibitors also repress related ALK4 and ALK7 function, and therefore, cannot distinguish between the effect on distinct signaling cascades regulated by different TGF-β ligands and type I receptors.27 To specifically inhibit ALK5, we designed and tested 2OMePS AONs that target exon 2 ALK5 transcripts. Sequence-specific ALK5 AONs have the advantage that they specifically modulate ALK5 expression, and therefore, presumably do not induce off-target effects. Consistently, we observed specific knockdown of ALK5 expression, but no change in the expression of the related type I receptor ALK4 after transfection or injection with ALK5 AONs in vitro and in vivo. Although we could not show a knockdown on protein level due to lack of functional antibodies, the selective inhibitory effect of ALK5 AONs on TGF-β activity shown by Smad3-dependent CAGA-luciferase reporter assays was clearly observed. Moreover, myogenic differentiation was enhanced upon transfection with ALK5 AONs. These results suggest that a functional knockdown of ALK5 was achieved after transfection with these AONs.
MSTN is a structurally related protein to TGF-β and is also known to signal via ALK5 and Smad2/3.24,25 However, in contrast to TGF-β, MSTN is a specific inhibitor of muscle growth and is specifically expressed and mainly active in skeletal muscle. Spontaneous mutations in mammals and genetic knockout in mice result in skeletal muscle hypertrophy (reviewed in ref. 28,29). We showed that AON-mediated knockdown of ALK5 inhibits MSTN signaling in C3H10T1/2 cells but not in myoblasts. This suggests that ALK5 AONs specifically target TGF-β and not MSTN signaling in myoblasts, but target both signaling cascades in other cell types. This is in line with a previous study, in which we showed that MSTN activity is regulated by ALK4 an coreceptor Cripto in myoblasts and by ALK5 in other cell types such as fibroblasts.24 Interestingly, recent studies also suggested that MSTN is involved in muscle fibrosis by inhibiting apoptosis and stimulating proliferation of muscle fibroblasts.30,31 Our results therefore suggest that ALK5 knockdown in skeletal muscle will probably result in at least partial cell-type–dependent knockdown of MSTN activity. This is important to note since it has been reported that Mstn inhibition, like TGF-β, has positive effects on muscle function of mdx mice and may therefore be beneficial for improving muscle function in DMD patients.32,33 We determined the effect of ALK5 AONs after intramuscular injections in mdx mice and showed efficient knockdown of ALK5 in vivo. Additionally, we showed that ALK5 AONs repress expression of downstream target genes Col1a1, Ctgf, and Serpine1 and increase Myog expression in dystrophic mdx muscle. Col1a1 is a known downstream target of TGF-β signaling and expression is increased in fibrotic tissue.34 Myog expression is known to be inhibited by MSTN and TGF-β and is a marker for myogenic differentiation and regeneration.19,35 Although we did observe an effect on gene expression and Serpine1/pSmad2 protein levels, no significant effect on the percentage of fibrotic/necrotic areas was observed compared to control-injected muscles and only a slight trend of increased fiber size was shown. This may be due to several reasons. First, we performed these injections in young mdx mice, which do not show pronounced fibrosis and only show twofold increase in collagen expression compared to aged-matched wild-type animals. Additional experiments in old mdx mice, which show more pronounced fibrosis, may therefore be more informative to determine the effect on fibrosis. Second, isolating muscles 10 days after the first injection may not be sufficient to yield a clear effect on fibrosis or muscle mass/fiber size. A recent study indeed showed that intramuscular injections of AONs targeting myostatin only yielded an increase in muscle mass in wild-type mice after 4 weeks.36 More experiments are therefore needed to determine the effect of ALK5 knockdown in mdx mice.
The therapeutic potential of antagonists of TGF-β signaling cascades is well recognized due to the involvement of TGF-β signaling in many pathologies, such as cancer, fibrotic pathologies, and muscle-wasting disorders.13 The approach we have developed may therefore also be applicable for other diseases than DMD. Importantly, in addition to our study, Karkampouna et al.37 showed that AON-mediated knockdown of ALK5 counteracts fibrosis in ex vivo Dupuytren's cultures, suggesting therapeutic potential of ALK5 AONs for Dupuytren's disease. Nonetheless, caution needs to be exercised when inhibiting these pathways, since TGF-β signaling cascades are also involved in numerous physiological processes such as angiogenesis, neurogenesis, and regulation of the immune response. For DMD, systemic treatment with AONs targeting ALK5 would be required since all muscles have to be targeted to improve muscle pathology. However, systemic treatment may result in undesirable side effects due to hindrance of other processes regulated by these signaling cascades. This was underscored by recent clinical trials in DMD patients with ACE-031, a soluble type IIB activin receptor that inhibits MSTN/activin signaling, which resulted in unanticipated side effects such as dilated blood vessels and nosebleeds (clinicaltrials.gov; NCT01099761). These side effects were likely the result of targeting activin signaling rather than MSTN, since MSTN is mostly active in muscle and has no known function in angiogenesis. Importantly, activins are not targeted by our approach and ALK5 knockdown will therefore presumably not result in similar side effects. Furthermore, there are indications that long-term inhibition of TGF-β does not result in adverse side effects.38 Nonetheless, it will be important to determine the effect of systemic injections with ALK5 AONs or to attempt muscle-specific delivery of these AONs.
The exclusion of exons, or exon skipping, induced by 2OMePS AONs is based on steric hindrance by blocking intraexonic sequences used by proteins involved in pre-mRNA splicing. We chose 2OMePS AON chemistry since these AONs have been described to be more stable, display greater sequence specificity, and have favorable pharmacokinetics in vivo compared to other RNA interference methods such as siRNA- and RNase-H-dependent AONs that induce breakdown of the transcript.39,40,41 We clearly show that steric hindrance using these AONs can be used to induce a functional knockdown. Interestingly, our in vivo results show that ALK5 2OMePS AONs can be combined with Dmd 2OMePS AONs in mdx mice, suggesting that a possible cocktail of these AONs may be effective for combined targeting of MSTN/TGF-β signaling and restoration of the DMD transcript. Notably, 2OMePS AONs targeting DMD exon 51 yielded no severe adverse events after local intramuscular injections or systemic injections in DMD patients in three clinical trials.6,8,42 However, replacement of muscle fibers by fibrotic and adipose tissue is a potential problem since AON-mediated restoration of the DMD open reading frame is only effective if enough muscle fibers are preserved in patients. Therefore, combined treatment with MSTNTGF-β antagonists may prove to be advantageous by additionally stimulating muscle regeneration and repressing fibrosis. From a pharmaceutical formulation perspective, combining multiple AONs of the same chemistry would be superior to combining AONs with other TGF-β inhibitors, such as neutralizing antibody18 or small molecule inhibitors.43 Future studies are therefore also aimed at determining the systemic additive or synergistic effects of ALK5 and Dmd AON cocktails on dystrophic muscle function in mdx mice.
Materials and methods
Ethics statement. All experiments were approved by and performed following the guidelines of the Dier Experimenten Commissie (Animal Experimental Commission) of the Leiden University Medical Center (Permit Numbers: 12032). Effort was put in minimizing the amount of distress caused to the animals as much as possible.
Cell culture and AON transfections. Mouse myoblasts C2C12 (ATCC, Manassas, VA) were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 1% glucose, and 2% glutamax (Invitrogen, Carlsbad, CA), and mesenchymal stem cells C3H10 T1/2 (ATCC) were grown in minimum essential medium α medium with 10% fetal bovine serum (Invitrogen) at 37 °C with 10% and 5% CO2, respectively. The differentiation medium for C2C12 was Dulbecco's modified Eagle's medium with 2% fetal bovine serum, 1% glucose, and 2% glutamax (Invitrogen). AONs with phosphorothioate backbones and 2'-O-methyl ribose modifications were purchased from Eurogentec, Liège, Belgium. The sequences are listed in Table 1. The transfection was done using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions (except in the luciferase and myogenic differentiation assays below). Myogenic differentiation assays for C2C12 were performed and quantified as described before.24 In brief, the differentiation index was calculated as the percentage of myosin-positive cells of all myogenic cells, whereas fusion index is measured as the average number of nuclei in myosin-positive cells.
Luciferase reporter assay. Cells were seeded at a density of 5,000 cells/well on white µclear 96-well plates (Greiner bio-one, Frickenhausen, Germany) until 70% confluent and transiently transfected with 100 ng of (CAGA)12-Luc, 10 ng of pRL-cytomegalovirus, and 200 nmol/l of the indicated AON using Dharmafect Duo (Thermo Scientific, Waltham, MA). Following an overnight serum starvation, cells were stimulated with 1 ng/ml TGF-β (kindly provided by OSI Pharmaceuticals, Melville, NY), 500 ng/ml myostatin (R&D Systems, Minneapolis, MN) in serum-free medium for 8 hours. The cells were lysed using DualGlo Luciferase Assay Kit (Promega, Fitchburg, WI) and the luciferase signals were read in the Multilabel Counter (Perkin Elmer, Waltham, MA). Renilla luciferase signals were used to normalize for the transfection efficiency. Experiments were conducted in triplicates and repeated at least three times. Statistical analysis was performed using Student's t-test, and P values < 0.05 were considered significant.
In vivo AON treatment. All animal experiments were performed after the approval of the Animal Experimental Committee of the Leiden University Medical Center (DEC12032). For the intramuscular injections, the triceps muscles of mdx mice (5–6 weeks old, n = 6) were injected with a mix of 40 µg each of ALK5 AON (Eurogentec) and DMD AON (targeting mouse Dmd exon 23, which harbors the mutation in mdx mice, kindly provided by Prosensa Therapeutics, Leiden, Netherlands). The contralateral triceps, serving as controls, were injected with a mix of ScrALK5 AON and DMD AON at the same dose. The animals were anesthetized with isoflurane and injected at 4 consecutive days, with 24 hours interval between the second and third injections. The injected muscles were sacrificed 4 (n = 12) or 10 days (n = 6) after the last injection by cervical dislocation and tissues were isolated, snap frozen in liquid nitrogen–chilled isopentane and stored in −80 °C until further use. Hematoxilin and Eosin staining and analysis of the percentage of fibrotic/necrotic areas was performed as described before.44 To determine the average fiber size, 300 fibers per section (random fields) were measured using Adobe Photoshop (with ruler tool after setting measurement scale). Fiber size was defined as the greatest distance between the opposite sides of the narrowest aspect of the fiber.
RNA isolation, reverse transcription PCR, qPCR. Cells were lysed and RNA was directly isolated using NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instruction. Tissues were first sectioned, collected in 1.4 mm Zirconium Beads prefilled tubes (OPS Diagnostics, Lebanon, NJ), and homogenized using a MagNA Lyser (Roche Diagnostics, Basel, Switzerland) in lysis buffer supplied by the NucleoSpin RNA II kit, followed by RNA isolation. N6-primed cDNA was synthesized from 500 ng RNA using RevertAid H Minus M-Mulv Reverse Transcriptase (MBI Fermentas, Thermo Scientific) according to the manufacturer's instructions. Ten times diluted cDNA was amplified by PCR using AmpliTaq Gold GeneAmp kit (Roche, Basel, Switzerland). A nested PCR was performed for dystrophin as previously published.45 Quantitative PCR was performed in a LightCycler 480 using SensiMix reagents (Bioline, London, UK). The expression levels were analyzed using the LinReg qPCR method46 and normalized to expression values of the housekeeping gene Gapdh, which is expressed at similar levels in muscles of wild-type and mdx mice. Measurements were performed in triplo/biological sample. Primer sequences and detailed PCR conditions are available upon request. Statistical analysis was performed using the Student's t-test.
Protein isolation and western blotting. Protein lysates from the cells were isolated using a previously described method.24 Sectioned muscles collected in the Zirconium beads tubes were homogenized in 500 µl of radioimmunoprecipitation assay homogenizer buffer (50 mmol/l Tris HCl pH 7.4, 150 nmol/l NaCl, 1 mmol/l ethylenediaminetetraacetic acid supplemented with phosphatase and protease inhibitor cocktails (Roche)) and lysed with a MagNA Lyser (Roche). Following two sonification steps, 500 µl of radioimmunoprecipitation assay double detergents buffer (2% deoxycholate, 2% NP40, and 2% Triton X-100 in radioimmunoprecipitation assay homogenizer buffer) was added to the lysates, which were then incubated for 45 minutes at 4 °C with rotation and centrifuged for 10 minutes at 30,000g. The protein concentration of the supernatant was measured using the bicinchoninic acid assay. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blotting were performed using standard protocols. Primary antibodies used were rabbit polyclonal anti-C-terminally phosphorylated Smad2 (Ludwig Institute for Cancer Research, Uppsala, Sweden; 1:10,000) and PAI1/SERPINE1 (Santa Cruz; 1:1,000). Goat anti-rabbit and goat anti-mouse IgG horseradish peroxidase (Santa Cruz, Dallas, TX) were used at 1:5,000 dilution. The detection was performed using SuperSignal West Pico or West Femto Chemiluminescent (Thermo Scientific). Subsequently, blots were stripped and reprobed with mouse monoclonal antibody against β-actin (Sigma, St Louis, MO), dilution 1:5,000 to check for the uniformity of protein loading. Densitometry analysis of protein bands was performed using ImageJ software (National Institutes of Health, Bethesda, MD) according to the method described at the following website: http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/. In short, the average density value of β-actin bands per group was used to normalize the average values per group of pSmad2 and Serpine1. Statistical analysis was performed using the Student's t-test.
SUPPLEMENTARY MATERIAL Figure S1. Detection of exon skipping in AON injected triceps muscles. Figure S2. Effect of ALK5 AONs 4 days after intramuscular injections in mdx mice. Figure S3. Effect of ALK5 AONs on fibrosis and muscle fiber size in mdx muscle.
Acknowledgments
We appreciate the excellent technical help and valuable input from Marianna Kruithof-de Julio, Sofia Karkampouna, Christa Tanganyika-de Winter, Ingrid Verhaart, Wouter Leonhard, Daniela Salvatori, David de Gorter, Frans Prins, Dorien Peters, and Johan den Dunnen (Leiden University Medical Center). We are grateful to Prosensa Therapeutics B.V., Leiden, Netherlands for providing the Duchenne muscular dystrophy antisense oligonucleotides. We thank Mark Einerhand at Vereenigde BV (V.O. Den Haag, Netherlands) for critical reading of the manuscript. Some of the authors are coinventors on several patent applications for antisense sequences, exon-skipping technologies, and therapeutic approach based on modulation of transforming growth factor-β and bone morphogenetic protein expression. The Dutch Duchenne Parent Project, Center for Biomedical Genetics, Netherlands Institute for Regenerative Medicine, and Netherlands Organization for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek) are gratefully acknowledged for financially supporting this study. D.U.K., G.Jv.O., Pt.D., A.A.R., Pt.H., and W.H. declare being employed by Leiden University Medical Center, which has submitted a patent application on ALK5 exon skipping. As coinventors on this patent, they are entitled to a share of royalties should the patent be licensed.
Supplementary material
Detection of exon skipping in AON injected triceps muscles.
Effect of ALK5 AONs 4 days after intramuscular injections in mdx mice.
Effect of ALK5 AONs on fibrosis and muscle fiber size in mdx muscle.
References
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. BMJ. 1998;317:991–995. doi: 10.1136/bmj.317.7164.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–189. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- Kemaladewi DU, Dijke P, van Ommen GJ, Hoogaars WM. TGF-beta signaling in Duchenne muscular dystrophy. Future Neurology. 2012;7:209–224. [Google Scholar]
- Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Muñoz-Cánoves P. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol. 2011;96:167–201. doi: 10.1016/B978-0-12-385940-2.00007-3. [DOI] [PubMed] [Google Scholar]
- Cárcamo J, Weis FM, Ventura F, Wieser R, Wrana JL, Attisano L, et al. Type I receptors specify growth-inhibitory and transcriptional responses to transforming growth factor beta and activin. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzén P, ten Dijke P, Ichijo H, Yamashita H, Schulz P, Heldin CH, et al. Cloning of a TGF beta type I receptor that forms a heteromeric complex with the TGF beta type II receptor. Cell. 1993;75:681–692. doi: 10.1016/0092-8674(93)90489-d. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, et al. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest. 1995;96:1137–1144. doi: 10.1172/JCI118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–834. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest. 2009;119:1583–1594. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–1752. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA, et al. Inhibiting TGF-β activity improves respiratory function in mdx mice. Am J Pathol. 2011;178:2611–2621. doi: 10.1016/j.ajpath.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney CF, Sali A, Guerron AD, Iantorno M, Yu Q, Gordish-Dressman H, et al. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011;16:87–95. doi: 10.1177/1074248410381757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, van Vliet L, Hirschi M, Janson AA, Heemskerk H, de Winter CL, et al. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol Ther. 2009;17:548–553. doi: 10.1038/mt.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemaladewi DU, de Gorter DJ, Aartsma-Rus A, van Ommen GJ, ten Dijke P, ‘t Hoen PA, et al. Cell-type specific regulation of myostatin signaling. FASEB J. 2012;26:1462–1472. doi: 10.1096/fj.11-191189. [DOI] [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P, et al. Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther. 2010;18:1210–1217. doi: 10.1038/mt.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell Signal. 2011;23:1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Amthor H, Hoogaars WM. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr Gene Ther. 2012;12:245–259. doi: 10.2174/156652312800840577. [DOI] [PubMed] [Google Scholar]
- Lee SJ. Extracellular Regulation of Myostatin: A Molecular Rheostat for Muscle Mass. Immunol Endocr Metab Agents Med Chem. 2010;10:183–194. doi: 10.2174/187152210793663748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo Li Z, Zhang J, Wagner KR. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci. 2012;125 Pt 17:3957–3965. doi: 10.1242/jcs.090365. [DOI] [PubMed] [Google Scholar]
- Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283:19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- Lindahl GE, Chambers RC, Papakrivopoulou J, Dawson SJ, Jacobsen MC, Bishop JE, et al. Activation of fibroblast procollagen alpha 1(I) transcription by mechanical strain is transforming growth factor-beta-dependent and involves increased binding of CCAAT-binding factor (CBF/NF-Y) at the proximal promoter. J Biol Chem. 2002;277:6153–6161. doi: 10.1074/jbc.M108966200. [DOI] [PubMed] [Google Scholar]
- Ríos R, Carneiro I, Arce VM, Devesa J. Myostatin is an inhibitor of myogenic differentiation. Am J Physiol Cell Physiol. 2002;282:C993–C999. doi: 10.1152/ajpcell.00372.2001. [DOI] [PubMed] [Google Scholar]
- Malerba A, Kang JK, McClorey G, Saleh AF, Popplewell L, Gait MJ, et al. Dual Myostatin and Dystrophin Exon Skipping by Morpholino Nucleic Acid Oligomers Conjugated to a Cell-penetrating Peptide Is a Promising Therapeutic Strategy for the Treatment of Duchenne Muscular Dystrophy. Mol Ther Nucleic Acids. 2012;1:e62. doi: 10.1038/mtna.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkampouna S, Kruithof BP, Kloen P, Obdeijn MC, van der Laan AM, Tanke HJ, et al. Novel Ex Vivo Culture Method for the Study of Dupuytren's Disease: Effects of TGFβ Type 1 Receptor Modulation by Antisense Oligonucleotides. Mol Ther Nucleic Acids. 2014;3:e142. doi: 10.1038/mtna.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan KM, Voit T, Rosales XQ, Servais L, Kraus JE, Wardell C, et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: Results of a double-blind randomized clinical trial. Neuromuscul Disord. 2014;24:16–24. doi: 10.1016/j.nmd.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- van Putten M, de Winter C, van Roon-Mom W, van Ommen GJ, ‘t Hoen PA, Aartsma-Rus A. A 3 months mild functional test regime does not affect disease parameters in young mdx mice. Neuromuscul Disord. 2010;20:273–280. doi: 10.1016/j.nmd.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Kemaladewi DU, Hoogaars WM, van Heiningen SH, Terlouw S, de Gorter DJ, den Dunnen JT, et al. Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med Genomics. 2011;4:36. doi: 10.1186/1755-8794-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detection of exon skipping in AON injected triceps muscles.
Effect of ALK5 AONs 4 days after intramuscular injections in mdx mice.
Effect of ALK5 AONs on fibrosis and muscle fiber size in mdx muscle.