

Abstract
Differences in gene regulation are thought to play an important role in speciation and adaptation. Comparative genomic studies of gene expression levels have identified a large number of differentially expressed genes among species, and, in a number of cases, also pointed to connections between interspecies differences in gene regulation and differences in ultimate physiological or morphological phenotypes. The mechanisms underlying changes in gene regulation are also being actively studied using comparative genomic approaches. However, the relative importance of different regulatory mechanisms to interspecies differences in gene expression levels is not yet well understood. In particular, it is often difficult to infer causality between apparent differences in regulatory mechanisms and changes in gene expression levels, a challenge that is compounded by the fact that the link between sequence variation and gene regulation is not clear. Indeed, in certain cases, gene regulation can be conserved even when sequences at associated regulatory elements have changed. In this chapter, I examine different genomic approaches to the study of regulatory evolution and the underlying genetic and epigenetic regulatory mechanisms. I try to distinguish between hypothesis-driven and exploratory studies, and argue that the latter class of studies provides valuable information in its own right as well as necessary context for the former. I discuss issues related to study designs and statistical analyses of genomic studies, and review the evidence for natural selection on gene expression levels and associated regulatory mechanisms. Most of the issues that are discussed pertain to the general nature of multivariate genomic data, and thus are often relevant regardless of the technology that is used to collect high-throughput genomic data (for example, microarrays or massively parallel sequencing).
Keywords: Comparative genomics, Gene regulation, Evolution
1. What Can We Learn from Genomic-Scale Comparative Studies of Gene Regulation?
Genomic studies of gene regulatory phenotypes are only rarely hypothesis driven. There are exceptions, for example studies that focus on a difference in phenotypes between populations or species (e.g., 1), and use a genome-wide approach to query regulatory differences that might explain the observed difference in phenotypes. However, most comparative genomic studies of gene regulation are exploratory in nature. Thus, the results of such studies cannot typically be evaluated by the standard metric of considering whether a question was convincingly answered or a hypothesis provided further support. In addition, most genomic studies focus on steady-state gene regulatory phenotypes (such as steady-state gene expression levels or transcription factor binding) and cannot, mainly due to technological limitations, take into account the detailed spatial and temporal dynamics of gene regulation. It is, therefore, important to consider the following question: What can we learn from nonhypothesis-driven comparative genomic explorations of steady-state estimates of gene regulatory phenotypes?
Comparative genomic regulatory studies typically address three general aims. First, they provide a general description of variation in gene expression levels, or variation in regulatory interactions, within and between populations. In itself, such a description is often of no particular interest. However, these descriptions allow investigators to place hypotheses regarding individual genes as well as appreciate observations of differences in regulatory phenotypes between individuals, or across populations and species, in the appropriate context. For example, consider the observation that 20% of the annotated genes in the insulin/IGF-signaling pathway are differentially expressed between human and chimpanzee livers (10). In order to assess the significance of this observation, it needs to be interpreted in the context of overall genome-wide variation in gene regulation between species. In other words, genome-wide data are required to test whether the observation that 20% of genes annotated in the insulin/IGF-signaling pathway are differentially expressed between the two species is indeed unexpected.
The second general aim of comparative genomic investigations of gene regulation is to understand the relative importance of changes in different regulatory mechanisms, and the associated evolutionary pressures, which shape gene regulatory variation within and between species. Functional studies of individual genes are often able to link specific change in regulatory mechanism with a shift in expression levels, which may underlie physiological or morphological phenotypic variation. In some cases, these studies are also able to obtain evidence for the action of natural selection on gene regulation, especially when a strong prior hypothesis exists (for example, in the case of genes related to skin pigmentation and their associated cis regulatory elements (2)). However, while studies of single genes illustrate the connection between regulatory evolution and phenotypic variation, only genome-wide explorations can offer a wide enough perspective to address the more general question of the relative importance of changes in different molecular mechanisms to the evolution of gene regulation. Similarly, genome-wide perspective is required to study the overall impact of natural selection on gene regulatory differences within and between species.
The third aim of comparative genomic studies is to develop specific hypotheses for follow-up functional experiments, which are typically too demanding to be performed on a genome-wide scale. For example, it can be shown, based on genome-wide comparative data, that it is entirely unexpected (by chance) that 20% of the genes annotated in the insulin/IGF-signaling pathway would be differentially expressed between humans and chimpanzees (10). Thus, it may be reasonable to assume that the regulation of this pathway has evolved under directional selection in either humans or chimpanzees (or both). The insulin/IGF-signaling pathway might, therefore, be a promising candidate for subsequent functional studies and analysis. For example, one might choose to proceed by considering interspecies differences in the metabolic phenotypes associated with this pathway.
Beyond these three aims, comparative studies of gene regulation are sometimes motivated by general hypotheses, for example when used as tools to survey possible mechanisms that might explain genetic associations (as in the context of genetic association studies of human diseases (3, 4)). Comparative genomic investigations of regulatory response phenotypes (for example, a response to infection) are another class of studies driven by a general hypothesis.
2. How to Compare Gene Expression Levels Across Species?
Comparative studies of gene expression levels involve related but somewhat different challenges than those involved in studies of the regulatory mechanisms underlying variation in gene expression levels. In what follows, I therefore discuss these classes of studies separately. I begin with a discussion of comparative studies of gene expression levels.
With the advent of massively parallel high-throughput sequencing technologies (“next-generation” sequencing), inter-species comparisons of gene expression levels, while still not straightforward, became more feasible. Prior to the availability of next-generation sequencing technologies, genome-wide comparisons of gene expression levels relied solely on DNA microarrays. Microarrays are still more cost-effective than sequencing for genome-wide transcriptional profiling. Yet, with respect to inter-species comparisons, microarrays fall short. The principal problem is that the collection of gene expression data using microarrays relies on hybridization between the RNA samples being queried and the probes on the arrays. Sequence mismatches between target RNA samples and the microarray probes lead to attenuation of the hybridization intensity, and result in biased estimates of gene expression levels (5). Interspecies comparisons of gene expression levels always involve the hybridization of RNA samples with different sequences. The use of commonly available commercial microarrays, each designed based on the sequence information of only one species (typically, only model organisms and humans), is therefore problematic. Species-specific and multispecies microarrays can be custom designed and used to compare gene expression levels within and between species, without the confounding effects of sequence mismatches on hybridization intensities (e.g., 12). However, the design and manufacturing of such custom arrays is costly, and one can only design arrays for species for which a sequenced genome is available. Moreover, each time another species is added to a comparative study, a new array has to be designed and ordered, and the entire study repeated. Ultimately, due to these considerations, sequencing is generally a more cost-effective choice than microarrays for comparative genomic studies of gene expression levels. Thus, in what follows, I mainly focus on methodological issues related to comparative studies using sequencing.
2.1. Multispecies Comparisons of Gene Expression Levels Using RNAseq
Gene expression studies using RNA sequencing (RNAseq) are not free of challenges related to the comparison of expression levels across different species. However, the solutions typically lie in proper analysis of the data rather than in development of new empirical tools (by no means do I intend to argue that all challenges involved in RNAseq data analysis have been solved, only that there are fewer specific difficulties associated with comparative studies when RNAseq is being used instead of microarrays, and most of the remaining difficulties can be solved by proper and cautious analysis). The first set of challenges relate to the requirement of defining the transcriptome. This is necessary because comparisons of estimates of expression levels can only be interpreted in the context of defined transcriptional units (for example, comparison of the expression levels of exons, specific transcripts, or genes). When RNA is being sequenced from a species for which a well-annotated genome is available, RNAseq reads can simply be aligned to the previously defined transcriptional units and expression levels can be estimated based on the number of aligned reads. The problem is that there are only a few well-annotated genomes (such as the human and mouse genomes), and even these are not perfectly annotated (indeed, studies continue to find additional transcriptional units in the human and mouse genomes, such as previously unrecognized exons—typically 5′ to annotated promoters and novel small RNAs (6, 7)).
If one is sequencing RNA from a species for which a sequenced genome is available yet is not well annotated, there are two general alternatives for defining transcriptional units. First, one can rely on the functional annotation of a closely related genome. Consider, for example, a comparative study of gene expression levels among humans, chimpanzees, and rhesus macaques using RNAseq. Sequenced genomes are available for all three species, yet only the human genome is well annotated. Because the three species are closely related, it may seem relatively easy to use the functional annotation of the human genome to define theoretical transcriptional units in the two nonhuman primate genomes. The challenge, however, is to accurately define orthology. If one is conservative (requires exceptionally high sequence similarity) in defining orthology, a large fraction of transcriptional units may be excluded from the analysis. On the other hand, if one defines orthology using relaxed criteria (accepting even weak evidence for homology), falsely classified orthologous regions will often lead to the inclusion of real transcriptional units in human, coupled to spuriously defined transcriptional units in the nonhuman primates. This results in a bias toward estimates of higher expression levels in humans compared to the other two species. Even if a balance is achieved between the desire to include as many transcriptional units as possible and the need to avoid falsely classified orthologous genomic regions, transcriptional units that are specific to the nonhuman primates will never be included in an analysis anchored by annotations based on the human genome. Thus, ultimately this approach will always result in a certain bias. For example, exons that are being used frequently in alternatively spliced transcripts in chimpanzees but not in humans might be excluded from a comparative analysis based on functional annotation of the human genome (Fig. 1).
Fig. 1.
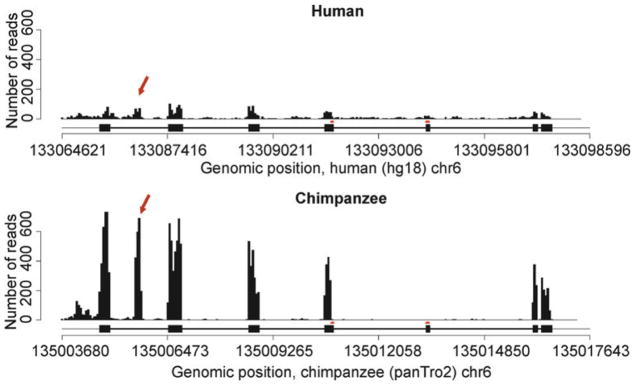
RNAseq data from human and chimpanzee liver samples are plotted along the Vanin-family protein 3 (VNN3) gene region. The human gene structure is provided below each plot and indicates that there are seven annotated exons in this genes (there is no independent annotation of the chimpanzee genome). The arrows indicate a cluster of sequencing reads that does not correspond to any part of the human gene model. A de novo definition of transcriptional units clearly classifies this as an additional exon. Arguably, there is yet another unannotated exon at the 5′ end of the region.
The second alternative is to use the alignment of the RNAseq reads to the available genomes of all studied species in order to define, de novo, the expressed transcriptional units. This is far from a trivial task, as it requires one to distinguish foreground expression levels from the background (such as sequencing reads originating from unspliced introns). At the time this chapter is being written, there are only a handful of algorithms for de novo definition of transcriptional units from aligned sequencing data (e.g., 8), and their effectiveness is still being debated. That said, this is an area of active research, and probably the most promising way to proceed. Comparative gene expression studies that are based on de novo definition of transcriptional units are not affected by biases due to preexisting functional annotations.
When a sequenced genome itself is not available, a third approach is to perform de novo assembly of the transcriptome. This is the most difficult approach because it does not rely on an alignment of the sequencing reads to a known genome. Currently, there is no effective approach for performing de novo assembly of the transcriptome using RNAseq data. Such approaches can in principle rely on successful existing algorithms for de novo assembly of entire genomes (Chap. 5, of volume 1 of this book, ref. 54, where the biggest challenge is typically to identify and resolve repeats. However, de novo assembly of the transcriptome is challenging in a different way because one has to take into account the broad distribution of copy numbers across transcriptional units (namely, the different expression levels). With respect to comparisons of expression levels across species, data processed by using effective de novo assembly of the transcriptome is expected to have the same properties as data processed by de novo definition of the transcriptional units based on aligned RNAseq reads. However, assembly of the transcriptome is an attractive approach because it allows one to perform comparative RNAseq studies on any species, including species for which a sequenced genome is not yet available. That said, with the rapid decrease in sequencing costs and the corresponding increase in sequencing capacity, it might be reasonable to expect that sequencing a new large (e.g., mammalian) genome may not be a prohibitive enterprise in the near future.
For the remainder of the chapter, when issues pertaining to RNAseq studies are discussed, it is assumed that the analysis is being performed using the final dataset of reads that map to a defined set of transcriptional units (regardless of the method used). For simplicity of writing, I will also henceforth refer generally to “genes” as examples of transcriptional units. It should be kept in mind, however, that RNAseq data can be used to study the expression levels of any transcriptional unit, including individual exons, alternatively spliced transcripts, small RNAs, etc.
2.2. General Issues in Design of Comparative Gene Expression Studies
Genome-wide investigations of gene regulation need to take into account a large number of potential confounding sources of variation. These can be technical, such as variation in sample quality and batch effects, or biological, such as variation due to sex, age, and circadian rhythm. Comparative studies of gene expression levels are arguably even more sensitive to confounding effects because of the large number of physical, morphological, and environmental differences between species. Differences in diets, for example which may be unavoidable in a study of multiple species, can affect gene regulation.
One of the main goals of comparative studies of gene expression levels is to understand interspecies genetically regulated differences. However, in many multispecies studies, the environmental and genetic components affecting gene regulation are completely confounded and cannot be distinguished. Similarly, differences in developmental trajectories, organ size, cellular composition, and life histories may all be inherently confounded with genetic effects in a multispecies comparative study.
To some extent, many of these differences can be sidestepped by limiting the investigation to model organisms that can be kept in the lab. In that case, one can often ensure that tissue samples are staged, namely, that samples are being collected from individuals of the same age and sex, who have experienced similar life histories, and that sample collection procedures are identical across individuals, regardless of species. In contrast, studies of non-model species can almost never obtain staged tissues, as in most cases the sample collection is opportunistic in nature (for example, when collecting samples from nonhuman apes that died in accidents, fights, or due to other natural causes).
As a result, observations from comparative studies of gene regulation, especially of non-model organisms, should be interpreted with caution. Some patterns are likely robust with respect to the uncontrolled aspects of the study designs, and these can readily be interpreted. For example, it is reasonable to assume that interspecies differences in the environment and life histories experienced by donor individuals will result in perturbation of gene regulation and lead to increased variation in gene expression levels across species. Thus, patterns of similarity (namely, low variation) of gene expression levels between individuals, regardless of species, are probably robust with respect to environmental effects. One can conclude, therefore, with considerable confidence that such patterns are genetically (or epigenetically) controlled (Fig. 2, top panels).
Fig. 2.

Comparative liver gene expression profiles in primates (data from Blekhman et al. 2008). In all panels, the mean (±s.e.m) log gene expression level (y-axis) of six individuals from each species (x-axis) is plotted relative to the human value (which was set to zero). Top panels: Though Blekhman et al. did not obtain staged tissues—the samples were collected opportunistically during postmortem procedures; the expression levels of each of these four genes are remarkably constant across individuals and species (importantly, these four genes are expressed at moderate to high levels, so the observed interindividual low variation is not due to lack of expression). Technical or environmental explanations for these patterns are unlikely. It is, therefore, reasonable to assume that the expression levels of these genes are tightly regulated (indeed, Blekhman and colleagues argue that the regulation of these genes has likely evolved under stabilizing selection in primates). Bottom panels: These genes have similar expression levels in chimpanzees and rhesus macaques, and a significantly different expression level in humans. In these four cases, explanations based on interspecies genetic or environmental differences are completely confounded.
In contrast, the observation of interspecies differences in gene expression levels (Fig. 2, bottom panels) may always be difficult to interpret, as environmental and genetic explanations can be completely confounded. Arguably though, in some cases, the mechanism underlying the observation of a regulatory difference between species is of less importance as long as the difference is indeed between the species rather than between the specific sampled individuals. In that case, care needs to be taken to ensure that a sufficient number of individuals have been sampled to obtain a relatively stable estimate of gene expression levels in the entire species, given specified conditions. Perhaps surprisingly, the number of required individuals to satisfy this criterion can often be quite modest (on the order of a dozen individuals (11); Fig. 3).
Fig. 3.

Examples of strong concordance between expression levels measured using the multispecies arrays from Blekhman et al., 2008, and using the RNAseq data from Blekhman et al., 2009. Six genes are displayed, chosen at random from the data of Blekhman et al., 2008, conditional only on a significant (FDR < 0.05) difference in gene expression level between humans and chimpanzees (expression levels in the rhesus macaques were not considered for the selection process). For each gene, the expression estimate (mean ± s.e.m) from the multispecies array (left ) and normalized expression level (mean ± s.e.m) from the RNAseq data (right ) are shown for each species (H human, C chimpanzee, R rhesus macaque). Each study used different individual samples, yet the patterns are consistent across studies, suggesting that the relative estimates of gene expression levels based on six individuals from each species are mostly stable.
2.3. General Issues in the Analysis of Comparative Gene Expression Data
The challenges involved in the analysis of genome-wide gene expression data are common to nearly all multivariate high-throughput studies, and are not specific to comparative genomics studies. General topics in multivariate analysis are discussed in Chap. 3, Volume 1 (ref. 55) as well as covered in more detail in many dedicated textbooks. Similarly, approaches for modeling gene expression levels based on microarray or sequencing data are discussed elsewhere in detail (e.g., 7, 9, 10). Here, I focus on three particular issues: first, on normalization of gene expression datasets; second, on the relationship between gene length, absolute expression level, and the power to detect differences in gene expression levels, as it pertains to RNAseq data; and third, on the arbitrary nature of the choice of statistical cutoffs.
Normalization
Normalization of gene expression datasets can be performed in a number of ways (e.g., linear shifts, nonlinear extrapolations, median corrections based on smoothing). Microarray studies routinely use a normalization step as part of the low-level analysis of the data. In contrast, most recently published RNAseq studies (including two early studies from my own group) have standardized read count based on transcript length and the total number of sequenced reads in each sample, but have not normalized the sequencing data across samples prior to modeling gene expression levels. In this section, rather than explore particular approaches for normalization, I discuss the reasons for which it is necessary to apply a normalization step to RNAseq data (see refs. 39–41 for details on different normalization approaches).
A normalization step is generally required in genomic studies of gene expression levels to correct for purely technical differences among data from different samples, such as differences in overall RNA quantity and/or quality, sample processing, and batch effects. Arguably, most of these effects can be taken into account in an RNAseq study by correcting gene-specific read counts by the total number of reads sequenced in each sample. Note that this standardization step relies on the assumption of no interacting technical confounding effects, which may or may not be a reasonable assumption. Since I proceed by arguing that normalization is needed, I shall not continue to discuss the validity of this assumption.
A correction based on the total number of sequenced reads, however, cannot account for differences in the distribution of gene expression levels across samples (12, 13). This is a property that we did not need to consider in microarray studies. In contrast to microarrays, where each RNA type hybridizes (we can assume—independently) to a dedicated probe, estimates of gene expression levels using RNAseq are based on the proportion of reads that are sequenced from each gene relative to the total number of sequenced reads in a sample. As the total number of reads sequenced from a given sample is limited, by definition, the range and distribution of gene expression values affect how often genes with a given absolute expression level are being sampled (because the fractions of reads mapped to individual genes must sum to one in each sample). For example, assume that the number of genes expressed in livers and kidneys is identical, but in livers all genes are expressed at low to moderate levels while in kidneys a few genes are expressed at extremely high levels and all other genes at low to moderate levels. In that case, for a given number of RNAseq reads per sample (and when reads are sampled at random), the probability that a lowly expressed gene will be represented is higher in the liver than in the kidney. Normalization of RNAseq data is, therefore, necessary to take these differences into account.
Power to detect differentially expressed genes
Another important property of RNAseq data is that the number of sequence reads that map to a particular gene tends to be roughly proportional to the expression level of the gene multiplied by the gene’s length (14). Thus, long genes tend to be represented by more sequence reads than short genes expressed at the same level. As a result, estimates of expression levels based on RNAseq data, though they are standardized by gene length, tend to be less variable for long genes than for shorter genes (or transcripts, or exons—this property is not specific to a particular class of transcriptional units). The ability to identify differentially expressed genes between samples is, therefore, strongly associated with the length of the transcript. Moreover, when overall sequence coverage is increased, the corresponding increase in the power to detect differences in expression levels across samples is also associated with gene length because the corresponding increase in the number of reads is greater for long than for shorter genes. Microarray data are not susceptible to this complex interaction between gene length and the power to detect differences in expression levels because all probes on the array are typically of the same length.
Since one of the most attractive features of RNAseq is the ability to assay the expression of entire transcriptional units, it may be undesirable to account for this length bias by restricting the analysis to subsections of genes (such as the first n base pairs of 3′ UTRs). The association between gene length and the power to detect expression differences may, therefore, be a constant property of RNAseq studies, and its bias on downstream analyses needs to be considered (15). For example, ranking or testing for functional enrichments (for example, by using gene ontology annotations) among genes that are classified as differentially expressed between species based on RNA-seq data might result in the spurious identification of enriched pathways or functional annotations that include mainly longer genes.
For that reason, analyses aimed at assessing whether an observation of an enrichment of regulatory differences in a particular pathway or a biological process is unusual need to take into account a background of matching gene lengths or at least a background of matching estimated expression levels. Consider again the observation that 20% of the annotated genes in the insulin/IGF-signaling pathway are differentially expressed between human and chimpanzee livers. In contrast to our simplified discussion above, because of the power-related considerations, it is not appropriate to estimate whether this observation is indeed unexpected by simply considering the overall fraction of differentially expressed genes between the two species. Instead, a proper null expectation should be developed by considering inter-species differences in expression levels in a proper background of genes with similar length as the genes in the insulin/IGF-signaling pathway (15). Alternatively, one can develop a null expectation by sampling at random subsets of n genes—where n is the number of genes in the insulin/IGF-signaling pathway while maintaining a similar distribution of expression levels.
The choice of statistical cutoffs
Genome-wide studies typically use statistical cutoffs to sort genes into different classes, for example to classify genes as differentially expressed between cases and controls. In many contexts, especially when genome-wide studies are used to develop hypotheses for further testing (which typically involve functional experiments that are time consuming and costly), minimizing the number of false positives is nearly the only guiding principle behind the choice of a statistical cutoff. However, comparative studies of gene regulation are often exploratory, and, as such, one of the goals is typically to describe biological processes and pathways that are enriched among different classes of genes, such as those that are differentially expressed between species. The challenge is to provide a description of such patterns that does not rely on the exact choice of the statistical cutoff.
While the choice of cutoffs is nearly always arbitrary, it is often possible to guide it by using prior information regarding related properties of the data. For example, consider “housekeeping” genes (the definition of “housekeeping” genes is controversial, but for the purpose of this discussion, assume that we have an established list of true “housekeeping” genes). A reasonable assumption might be that housekeeping genes will be underrepresented among differentially expressed genes between species. In that case, one approach is to choose a cutoff with which the overall number of genes classified as differentially expressed is maximized while the number of housekeeping genes classified as differentially expressed is minimized. When two or more genomic datasets are combined, the opportunity to leverage information to guide the choice of statistical cutoffs increases. Consider the combination of a transcription factor ChIPseq dataset with genome-wide estimates of gene expression levels following perturbation of the same transcription factor dosage. Two cutoffs need to be chosen: one to classify transcription factor promoter binding events in the ChIPseq data and one to classify differences in gene expression levels following the perturbation of the transcription factor dosage. In choosing these cutoffs, the prior expectation of enrichment in overlap between the two sets of observations can be leveraged. Indeed, true regulatory targets of the transcription factor are expected to be differentially expressed, as well as have the transcription factor bound to their promoters.
Regardless of the type of analysis used or the ability to use prior information to guide the choice of statistical cutoffs, the order of p-values rarely changes. For that reason, an analysis that indicates that the conclusions are robust with respect to a wide range of arbitrary choices always reinforces the study. One way to achieve this is to perform the entire analysis using a range of alternative cutoffs. A more formal way to test specific properties of interest is to use approaches, such as “gene set enrichment analysis” (16), which rely on the order of p-values rather than on specific choices of cutoffs. Using these approaches, one can explore the overall dependence between the choice of cutoff and the examined property of the data (such as an enrichment of differentially expressed genes in a particular pathway).
Strong conclusions can only be based on properties that are demonstrably robust with respect to the choice of statistical cutoffs. For example, the specific number of genes classified as differentially expressed between species obviously depends on the choice of a statistical cutoff. However, the property that the fraction of genes classified as differentially expressed between humans and chimpanzee is smaller than between either humans or chimpanzees, and the more distantly related rhesus macaques, is robust with respect to the specific choice of cutoff (11, 12).
3. What Have We Learned from Comparative Genomic Studies of Gene Expression Levels?
At the time this chapter is being written, comparative studies of gene expression levels are still mostly limited to exploration of variation in gene regulation within and between species. A large number of specific hypotheses have been raised based on the existing studies, but only a few have been followed up. We are still working toward a better understanding of the evolutionary forces that shape gene regulatory phenotypes, and this has still remained the focus of most comparative studies of gene expression levels.
In the first large-scale study to investigate natural variation in gene regulation, Oleksiak et al. (17) compared gene expression levels in heart ventricles from 18 individual postreproductive males from three populations: two of Fundulus heteroclitus (a salt-water fish) and one of its close relative, F. grandis. Despite low migration rates between the two conspecific populations and across the species boundary, fewer than 3% of the 907 genes surveyed were classified as differentially expressed between populations. An order of magnitude more genes were found to be differentially expressed between individuals within populations. In other words, there was little evidence of population structure at the genome-wide expression level. In addition, patterns of variation between populations were inconsistent with the neutral prediction that phenotypic divergence should scale with genetic distance. Instead, gene expression profiles were more similar for the southern F. heteroclitus and F. grandis populations, suggesting that adaptation to different temperatures, rather than genetic drift, drove the differentiation.
Rifkin et al. (18), who studied gene expression variation during Drosophila metamorphosis, took a more explicit quantitative genetic approach to study selection pressures acting on gene regulation. They measured average levels of gene expression in four strains of the cosmopolitan species D. melanogaster and one strain each of D. simulans and D. yakuba at the start of metamorphosis. To identify genes whose regulation evolves under different selective pressures, Rifkin et al. analyzed the gene expression data using a system of related linear models corresponding to the expectations under three different evolutionary scenarios. Using this approach, they could not reject overall low variation for 44% of the expressed genes, could not reject species-specific gene expression patterns for 39% of the genes, and could not reject a model consistent with neutrality for the remaining 17% of genes. They interpreted these results to indicate a dominant signature for stabilizing selection in gene expression evolution with smaller, but important, roles for directional selection and neutral evolution, respectively.
In contrast to Rifkin et al., Lemos and colleagues (19) explicitly tested a null neutral model of gene expression evolution by making two key assumptions about variance in gene expression. First, they used estimates of mutational variance in other quantitative traits as a measure of the mutational variance that might be affecting gene expression. Second, following Lynch (20), they assumed that environmental variance was half the within-population variance—i.e., that broad-sense heritability of gene expression patterns was at most 50%. Using these estimates and based on the neutral model of Lynch and Hill (21), they calculated the minimal and maximal rates of gene expression diversification that would be consistent with neutrality (i.e., evolution without constraint).
Lemos et al. (19) used their approach to perform a meta-analysis of available gene expression datasets from multiple species, and found that the overwhelming majority of genes in all datasets exhibited far less between species variation than expected under a neutral model. They interpreted this pattern to be the result of stabilizing selection acting on within-species gene expression. In fact, Lemos et al. (19) estimated that even if the mutational input to gene expression were two orders of magnitude lower than they had assumed, levels of between-population differentiations in gene expression would still be inconsistent with neutrality. Only in comparisons between mouse lab strains did an appreciable number of genes evolve in a manner consistent with neutrality.
The conclusions of Lemos et al. were supported by several studies that directly measured the mutational input of variation in gene expression levels per generation in a number of model organisms (22–24). Mutational input can be estimated by measuring the variance for a phenotypic trait among a set of initially homogeneous lines maintained with minimally sized populations for many generations. Natural selection is at its weakest under such conditions because genetic drift in such small populations is extremely fast. In an extreme case, when a single, randomly chosen individual propagates each line, the only mutations which can be selected against are those that kill the organism before reproduction or that eliminate fertility altogether. Otherwise, most mutations will be effectively neutral and will quickly either drift to fixation or be lost. As different lines fix different random mutations, the lines drift apart. Variation between lines can then be used to estimate the mutational variance.
These mutation accumulation studies (22–24) provided the first direct estimates of mutational variance in gene expression levels. When comparative gene expression data were analyzed in the context of these estimates (by applying a similar modeling approach to the one used by Lemos et al.) in all systems studied to date, it was concluded that stabilizing selection places severe bounds on gene expression divergence.
3.1. Gene Expression in Apes
Understanding phenotypic evolution in primates is typically more difficult than in model organisms because key experiments often cannot be performed to distinguish between competing hypotheses or to estimate important parameters. Moreover, material is often scarce, leading to largely unknown and uncontrolled environmental variance between samples. These limitations are particularly problematic for dynamic, environmentally sensitive traits, like gene expression.
Perhaps due to the these difficulties, the first few studies that examined the selection pressures that shape gene expression profiles in humans and our extant close evolutionary relatives resulted in somewhat conflicting conclusions (19, 15). However, more recent work on interprimate comparisons of gene expression levels, focusing on patterns of the data that should be robust with respect to the uncontrolled aspects of the study design, indicates that, for most genes, there is little evidence for change in expression levels across primate species. These observations are consistent with widespread stabilizing selection on gene regulation in primates, in agreement with the observations in model organisms (18, 24, 26, 27).
Nonetheless, a subset of genes whose regulation appears to have evolved under positive (directional) selection in the human and chimpanzee lineages was identified. Intriguingly, among this set of genes, there was a significant excess of transcription factors in the human lineage. In addition to the rapid evolution of their expression, genes encoding transcription factors have also been shown to evolve rapidly in the human lineage at the coding sequence level (28). Together, these findings raise the possibility that the function and regulation of transcription factors have been substantially modified in the human lineage, a change that could have propagated to many downstream targets over a short evolutionary time frame. Interestingly, the opposite finding has emerged from studies of closely related Drosophila species, in which the expression levels of transcription factors appear to evolve more slowly than the expression levels of genes encoding other types of proteins (18, 22).
4. How to Compare Regulatory Mechanisms Across Species?
Beyond comparisons of gene expression levels across species, there is a great interest in understanding the underlying regulatory mechanisms. Specifically, we still know little about the relative importance of changes in different regulatory mechanisms to inter-species differences in gene expression levels. Genomic technologies, in particular since the advent of next-generation sequencing techniques, allow us to characterize genome-wide variation in a larger number of genetic and epigenetic regulatory mechanisms and regulatory interactions.
It is important to note at the onset of this discussion that genomic studies can only rarely be used to directly test for causality. Much more often, the inference of causality (for example, between changes in a regulatory mechanism and ultimate differences in gene expression levels) relies on the observation of correlations on a genome-wide scale. Statistical correlation in itself, however, does not provide strong evidence for causality, and, in any case, provides no information for the direction of causality. Instead, most often, inference of causality in comparative studies of gene regulation relies on prior functional knowledge of regulatory mechanisms. For example, enhancer transcription factors are known to bind to promoters of genes, precipitate the assembly of the transcriptional machinery at those promoters, and increase the rate of transcription of the associated genes. Based on this proposed mechanism (which is strongly supported by a large body of independent studies), one may be able to infer causality in a genome-wide study that correlates variation in genome-wide transcription factor binding at promoters and variation in gene expression levels.
4.1. Leveraging Different Sources of Information
Because inference of causality almost always relies on prior information, genome-wide studies of regulatory mechanisms should aspire to build the strongest possible independent “circumstantial case” for a relationship between variation in regulatory interactions and changes in gene expression levels. This can often be done by combining different sources of genome-wide information. For example, consider the task of identifying the direct regulatory targets of a transcription factor. To do so, empirical studies typically use one of the two main approaches: (1) expression profiling following a perturbation of the transcription factor dosage or (2) chromatin immunoprecipitation followed by sequencing (ChIP-seq) using a specific antibody against the transcription factor.
In the first approach, the dosage of the transcription factor is perturbed in cells or in model organisms by a treatment of either overexpression or knockdown (using, for example, siRNA technology (29, 30)) of the transcription factor. Following the treatment, the expression profiles of a large number of genes are studied in order to identify the genes whose regulation has been affected by the perturbation of the transcription factor dosage (29). Typically, a large number of genes—often several thousands—are found to be differentially expressed in such experiments (30, 31). However, it is clear that not all the differentially expressed genes are directly regulated by the transcription factor whose dosage was perturbed. Indeed, a large proportion of the genes are expected to be secondary targets (i.e., regulated by genes that are themselves directly regulated by the transcription factor). In addition, a change in the dosage of a transcription factor often affects the cellular environment in ways that may trigger larger changes in the gene expression profiles, not directly related to the regulatory effects of the perturbed transcription factor (30).
In order to identify the subset of direct transcriptional targets among all the differentially expressed genes, computational predictions of the transcription factor-binding sites are often used. Namely, a gene is considered as a direct regulatory target only if it is differentially expressed following the perturbation of the transcription factor and the binding motif of the transcription factor can be found within the gene’s putative promoter (30, 31). The problem is that computational searches for transcription factor-binding sites are known to have a high error rate (32). In particular, since transcription factor-binding sites are short (6–12-mers), a large number of false positives are expected. In addition, it is unclear how to assign significance to the identification of transcription factor-binding sites based on a single sequence (32).
An alternative approach is to use ChIPseq (33) to directly identify all the sites in the genome to which the transcription factor binds (e.g., refs. 34, 35). In these experiments, sequencing is used to measure the abundance of chromatin that is first precipitated along with the transcription factor of interest. The goal is to identify genomic regions with peaks of aligned sequencing reads, which correspond to regions putatively bound by the transcription factor. When the transcription factor-binding locus is in proximity to a known gene, it is assumed that the gene is being regulated by the transcription factor (35, 36). However, even if the antibody against the transcription factor is highly specific and the number of falsely identified binding events is assumed to be small (37), it is unclear how many binding events reflect a true biological function. Namely, it is unclear how often a transcription factor can bind to genomic regions near genes without participating in the regulation of those genes.
Thus, ChIPseq and dosage perturbation experiments, considered one at a time, suffer from high false-positive rates due to the nonspecificity of the antibody, random binding of the transcription factor in the case of the ChIPseq experiment, or the ripple effect of knocking down a transcription factor in the siRNA experiments. Considered together, however, these approaches enable the reliable identification of genes whose promoter regions are bound to by the transcription factor and whose regulation is affected by the perturbation of the transcription factor dosage. In other words, using this paradigm, one can build a strong circumstantial case for classifying direct regulatory targets of a specific transcription factor.
4.2. Statistical Challenges in Comparative Studies of Gene Regulation
Most of the statistical challenges involved in genomic studies of gene regulatory mechanisms are related to the multivariate nature of the data. In many ways, therefore, these issues are similar to the ones reviewed above for comparative studies of gene expression levels. For example, effective study designs are still required to test the hypothesis that the variation of regulatory mechanisms between species is significantly larger than the variation between individuals within a species (this seems worth mentioning because a few recent comparative studies of regulatory mechanisms have reported interspecies variation without including independent biological replicates within species).
Similarly, investigations of regulatory mechanisms also rely on mostly arbitrary choices of the statistical cutoffs used to classify the observed patterns. As in most genome-wide studies, regardless of whether the choice of cutoffs is guided to some extent by prior information, the main goal is typically to keep false positives to a minimum. However, comparisons of regulatory mechanisms between species are in that sense more complex because controlling the rate of false negatives is a crucial factor as well. The principal issue is that the data supporting a regulatory mechanism need to be interpreted in the context of each sample (or each species) before variation across samples (or species) can be characterized.
For example, consider a genome-wide comparative study of histone modifications using ChIPseq, namely, a study aimed at characterizing similarities and differences across species in the locations of these epigenetic markers. This may be of interest in order to study the extent to which interspecies variation in gene expression levels can be explained by changes in histone modification profiles. The first step in such a study is to identify all the genomic regions, which are associated with histone modifications, in each species. The characterization of such genomic regions is based on statistical analysis of the data. In the ChIPseq example, the goal is to identify peaks of aligned sequencing reads, which are indicative of enriched chromatin that is associated with histone modifications. In principle, once genomic regions associated with histone modifications are identified in each species independently, a comparison across species can be performed. Here, however, it becomes a bit more challenging.
Typically, one would tend to choose stringent statistical cutoffs to identify peaks of sequencing reads in each species independently, namely, choose such cutoffs that minimize the false positive rate. However, such an approach, while controlling the rate of falsely identified genomic regions associated with histone modification in each species, results in a high rate of spuriously identified differences in this epigenetic regulatory mechanism between species. For example, assume that associations with histone modifications are classified, in each species independently, at an FDR < 0.05 (this would typically refer to the expected proportion of peaks with similarly strong evidence in a negative-control ChIPseq experiment). In that case, an observation of a genomic region associated with histone modifications at an FDR = 0.049 in one species and an FDR = 0.051 in the other species would be considered as evidence for an interspecies difference in histone modifications at this genomic region. Clearly, this would be a problem.
To minimize the number of falsely identified interspecies differences in regulatory mechanisms, one should leverage information from all samples. This can be done using a number of different Bayesian approaches. In its simplest form, such an analysis (although not strictly Bayesian) could use the application of two statistical cutoffs. Considering the example of histone modifications, one can assume that conditional on observing an associated genomic region with high confidence in one species (namely, using a stringent cutoff) the orthologous site in a closely related speciesis also likely to have the modification. Accordingly, one can relax the statistical cutoff for the classification of such secondary observations. Although the choice of statistical cutoffs may still be arbitrary, the distributions of FDR values can be used as a guide, especially with respect to the choice of the second cutoff (Fig. 4). The two cutoff approach uses information across all studied species to increase the power to detect histone modification in any species. This approach is, therefore, conservative with respect to identifying differences across species.
Fig. 4.

Example of how a distribution of FDR values can guide the choice of statistical cutoffs. (a) All ChIPseq peaks with FDR ≤ 20% from a genomic study of histone modification in cell lines from three primate species; the chosen stringent 2% FDR cutoff is indicated with a dashed line. (b) Enrichment peaks with FDR ≤ 20% in each species, which also overlap peaks with FDR ≤ 2% in any of the other species; the chosen relaxed 5% FDR cutoff for a secondary observation is indicated with a dashed line.
5. What Have We Learned from Comparative Studies of Regulatory Mechanisms?
Comparative studies of genetic mechanisms
In contrast to the relative abundance of comparative gene expression data from multiple species, there are far fewer genomic-scale comparative datasets of regulatory mechanisms. At the genetic level, the largest comparative study of regulatory mechanisms to date is that of Schmidt and colleagues (38), who used ChIPseq to compare the genomic locations of binding sites of two transcription factors (CCAAT/ enhancer-binding protein alpha and hepatocyte nuclear factor 4 alpha) in the livers of five vertebrate species (human, mouse, dog, short-tailed opossum, and chicken). Schmidt and colleagues found that most transcription factor-binding locations are species specific, and that orthologous binding locations present in all five species are rare. Quite often, the sequences of orthologous binding loci were identical across species, even when the binding event was inferred to have been lost in one species. On the other hand, in many cases, there was no evidence for conservation at the sequence level even when the location of the transcription factor binding was shared across species.
These observations suggest that interspecies differences in genetic regulation by transcription factors are widespread. However, it should be noted that Schmidt and colleagues did not analyze their data by leveraging information from all species, but rather classified binding events independently in each species. As a result, their analysis was not conservative with respect to classifying differences in binding across species. It is reasonable to assume that to some extent this study overestimated the proportion of differences in binding locations between species.
There are a few other—somewhat smaller in scale—published comparative studies of transcription factor-binding locations across species (39–43). These studies, quite intuitively, suggest that the level of divergence in binding locations largely depends on the specific transcription factor that is being studied (as well as on the evolutionary distance between the species). Most of the comparative ChIPseq studies published to date have not yet been coupled with genome-wide characterization of interspecies gene expression differences. As a result, we still do not have an estimate of the relative importance of changes in transcription factor-binding locations to overall gene expression differences between species. That said, a property that emerges from this collective body of work is that we currently find very little correlation between divergence of inferred transcription factor-binding sites and differences (or similarities) in the observed transcription factor binding. In other words, without additional information, the study of conservation of individual binding sites across species is not very informative with respect to predicting conservation of transcription factor-binding locations.
Comparative studies of epigenetic mechanisms
Parallel surveys of interspecies differences in genetic and epigenetic regulatory mechanisms may provide context that allows us to better appreciate the relationship between differences in transcription factor binding and sequence changes at transcription factor-binding sites. To date, however, genome-wide comparative studies of epigenetic mechanisms have not yet been coupled with other sources of data.
Studies of one class of epigenetic marker, DNA methylation, have suggested that the role of DNA methylation in tissue-specific gene regulation is generally conserved. For example, after identifying tissue-specific differentially methylated regions (T-DMRs (44)) in a number of tissues in mice, Kitamura and colleagues were able to use the methylation status in orthologous human regions to distinguish between the corresponding human tissues (45). In turn, Irizarry and colleagues (46), who studied genome-wide DNA methylation patterns in spleen, liver, and brain tissues from human and mouse, reported that 51% of T-DMRs are shared across both species. However, there also are a large number of potentially functional differences in methylation levels across species. In particular, in primates, Gama-Sosa and colleagues (47) found that relative methylation levels within tissues generally differ between species, with the exception of hypermethylation in the brain and thymus, which were observed regardless of species. In addition, Enard and colleagues (48), who compared methylation profiles of 36 genes in livers, brains, and lymphocytes from humans and chimpanzees, reported significant interspecies methylation level differences in 22 of the 36 genes in at least 1 tissue.
A somewhat different picture may be emerging from comparative studies of a different class of epigenetic markers, histone modifications. Characterization of several types of histone modifications on human chromosomes 21 and 22, and the syntenic chromosomes in mouse, indicated that the genomic locations of these epigenetic markers at orthologous loci are strongly conserved, even in the absence of sequence conservation (39, 49). Interestingly, the conservation of histone modification patterns was highest in genomic regions proximal to annotated orthologous genes.
With few exceptions, however (e.g., with respect to DNA methylation, ref. 50), genome-wide comparative studies of epigenetic regulatory mechanisms have also not yet explored the extent to which changes in specific regulatory interactions underlie inter-species differences in gene expression levels. As a result, we still cannot assess the relative importance of changes in different genetic and epigenetic regulatory mechanisms to overall regulatory evolution. This status might change rapidly because the main limitation for performing high-throughput investigations of epigenetics markers was technological. Massively parallel sequencing technologies now facilitate comparative epigenetic studies using genome-wide protocols, such as MeDIP and ChIPseq.
6. Summary and Additional Topics
We have gained important insights from comparative genomic studies of gene expression levels. We established that the regulation of most genes evolves under stabilizing selection (51, 52) and described variation in gene expression levels within and between species with sufficient details so that we can now use empirical approaches to identify genes whose regulation likely evolved under directional selection (53). These would be promising candidates for further functional studies. Current efforts are moving beyond the investigation of interspecies variation in gene expression levels to studies of the underlying regulatory mechanisms. In that respect, I did not mention in this chapter many of the types of datasets that are currently being collected, such as measures of chromatin accessibility (using DNase hypersensitive sites, for example), different markers of enhancer elements (such as the cofactors p300 and mediator), maps of nucleosome positions, and expression levels of small regulatory RNA classes. Once we combine different sources of comparative genomic data into a unified model of gene regulation, we should obtain power to truly dissect the genetic and epigenetic architecture of gene regulatory evolution.
7. Exercises
-
You are ready to design a large study to compare gene expression between species using RNAseq. You know that you need to take into account a large number of possible biological and technical effects, but then you also learn that a certain physical environment (such as temperature, humidity, amount of light, etc.) might affect your results. You, therefore, decided to design a pilot experiment to test the effect of this physical environment on the measurements of gene expression level using your platform of choice. Your design should not rely on the availability of “gold standards” (namely, you are not able to obtain samples for which the differences in gene expression are known, neither a priori nor by using additional techniques).
Explain the study design that allows you to test for the effects of the physical environment of choice.
What are the expected results if the physical environment of choice has no effect on the measurement of gene expression levels?
What are the expected results if the effect of the physical environment of choice is random? In that case, how will you take this information into account when you design the larger study?
What are the expected results if the physical environment of choice is nonrandom? In that case, how will you take this information into account when you design the larger study?
-
Design a study that will allow you to compare genome-wide RNA decay rates across species, using RNAseq (using a chemical agent that stops transcription in the cell).
Explain your study design.
As part of the low-level analysis of your data, do you need to perform a normalization step? If so, how would you normalize your data?
Explain, in general terms, how would the data be analyzed to estimate gene-specific RNA decay rates.
References
- 1.Gompel N, Prud’homme B, Wittkopp PJ, Kassner VA, Carroll SB. Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature. 2005;433(7025):481–7. doi: 10.1038/nature03235. [DOI] [PubMed] [Google Scholar]
- 2.Linnen CR, Kingsley EP, Jensen JD, Hoekstra HE. On the origin and spread of an adaptive allele in deer mice. Science. 2009;325(5944):1095–8. doi: 10.1126/science.1175826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drake TA, Schadt EE, Lusis AJ. Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice. Mamm Genome. 2006;17(6):466–79. doi: 10.1007/s00335-005-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, Mouy M, Steinthorsdottir V, Eiriksdottir GH, Bjornsdottir G, Reynisdottir I, Gudbjartsson D, Helgadottir A, Jonasdottir A, Jonasdottir A, Styrkarsdottir U, Gretarsdottir S, Magnusson KP, Stefansson H, Fossdal R, Kristjansson K, Gislason HG, Stefansson T, Leifsson BG, Thorsteinsdottir U, Lamb JR, Gulcher JR, Reitman ML, Kong A, Schadt EE, Stefansson K. Genetics of gene expression and its effect on disease. Nature. 2008;452(7186):423–8. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 5.Gilad Y, Rifkin SA, Bertone P, Gerstein M, White KP. Multi-species microarrays reveal the effect of sequence divergence on gene expression profiles. Genome Res. 2005;15(5):674–80. doi: 10.1101/gr.3335705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 7.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O’Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–60. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 8.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18(9):1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolstad BM, Collin F, Simpson KM, Irizarry RA, Speed TP. Experimental design and low-level analysis of microarray data. Int Rev Neurobiol. 2004;60:25–58. doi: 10.1016/S0074-7742(04)60002-X. [DOI] [PubMed] [Google Scholar]
- 11.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 12.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32(2):261–6. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- 18.Rifkin SA, Kim J, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33(2):138–44. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- 19.Lemos B, Meiklejohn CD, Caceres M, Hartl DL. Rates of divergence in gene expression profiles of primates, mice, and flies: stablizing selection and variability among functional categories. Evolution. 2005;59(1):126–137. [PubMed] [Google Scholar]
- 20.Lynch M. The Rate of Morphological Evolution in Mammals from the Standpoint of the Neutral Expectation. American Naturalist. 1990;136(6):727–741. [Google Scholar]
- 21.Lynch M, Hill WG. Phenotypic Evolution by Neutral Mutation. Evolution. 1986;40(5):915–935. doi: 10.1111/j.1558-5646.1986.tb00561.x. [DOI] [PubMed] [Google Scholar]
- 22.Rifkin SA, Houle D, Kim J, White KP. A mutation accumulation assay reveals extensive capacity for rapid gene expression evolution. Nature. 2005;438(7065):220–3. doi: 10.1038/nature04114. [DOI] [PubMed] [Google Scholar]
- 23.Keightley PD, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter ML. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 2009;19(7):1195–201. doi: 10.1101/gr.091231.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denver DR, Morris K, Streelman JT, Kim SK, Lynch M, Thomas WK. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat Genet. 2005;37(5):544–8. doi: 10.1038/ng1554. [DOI] [PubMed] [Google Scholar]
- 25.Khaitovich P, Weiss G, Lachmann M, Hellmann I, Enard W, Muetzel B, Wirkner U, Ansorge W, Paabo S. A neutral model of transcriptome evolution. PLoS Biol. 2004;2(5):E132. doi: 10.1371/journal.pbio.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14(20):2551–69. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 27.Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317(5834):118–21. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- 28.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Todd Hubisz M, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez R, Civello D, Adams MD, Cargill M, Clark AG. Natural Selection on Protein Coding Genes in the Human Genome. Nature. 2005;437(7062):1153–7. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 29.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447(7144):550–5. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 30.Murphy CT. The search for DAF-16/ FOXO transcriptional targets: Approaches and discoveries. Experimental Gerontology. 2006 doi: 10.1016/j.exger.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 31.Chavez V, Mohri-Shiomi A, Maadani A, Vega LA, Garsin DA. Oxidative Stress Enzymes Are Required for DAF-16-Mediated Immunity Due to Generation of Reactive Oxygen Species by Caenorhabditis elegans. Genetics. 2007;176(3):1567–77. doi: 10.1534/genetics.107.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vavouri T, Elgar G. Prediction of cis-regulatory elements using binding site matrices–the successes, the failures and the reasons for both. Curr Opin Genet Dev. 2005;15(4):395–402. doi: 10.1016/j.gde.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Negre N, Lavrov S, Hennetin J, Bellis M, Cavalli G. Mapping the distribution of chromatin proteins by ChIP on chip. Methods Enzymol. 2006;410:316–41. doi: 10.1016/S0076-6879(06)10015-4. [DOI] [PubMed] [Google Scholar]
- 34.Sandmann T, Jakobsen JS, Furlong EE. ChIP-on-chip protocol for genome-wide analysis of transcription factor binding in Drosophila melanogaster embryos. Nat Protoc. 2006;1(6):2839–55. doi: 10.1038/nprot.2006.383. [DOI] [PubMed] [Google Scholar]
- 35.Ceribelli M, Alcalay M, Vigano MA, Mantovani R. Repression of new p53 targets revealed by ChIP on chip experiments. Cell Cycle. 2006;5(10):1102–10. doi: 10.4161/cc.5.10.2777. [DOI] [PubMed] [Google Scholar]
- 36.Lin Z, Reierstad S, Huang CC, Bulun SE. Novel estrogen receptor-alpha binding sites and estradiol target genes identified by chromatin immunoprecipitation cloning in breast cancer. Cancer Res. 2007;67(10):5017–24. doi: 10.1158/0008-5472.CAN-06-3696. [DOI] [PubMed] [Google Scholar]
- 37.Qi Y, Rolfe A, MacIsaac KD, Gerber GK, Pokholok D, Zeitlinger J, Danford T, Dowell RD, Fraenkel E, Jaakkola TS, Young RA, Gifford DK. High-resolution computational models of genome binding events. Nat Biotechnol. 2006;24(8):963–70. doi: 10.1038/nbt1233. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, Talianidis I, Flicek P, Odom DT. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328(5981):1036–40. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VL, Fisher EM, Tavare S, Odom DT. Species-specific transcription in mice carrying human chromosome 21. Science. 2008;322(5900):434–8. doi: 10.1126/science.1160930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, Macisaac KD, Rolfe PA, Conboy CM, Gifford DK, Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet. 2007;39(6):730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Candia P, Blekhman R, Chabot AE, Oshlack A, Gilad Y. A combination of genomic approaches reveals the role of FOXO1a in regulating an oxidative stress response pathway. PLoS ONE. 2008;3(2):e1670. doi: 10.1371/journal.pone.0001670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradley RK, Li XY, Trapnell C, Davidson S, Pachter L, Chu HC, Tonkin LA, Biggin MD, Eisen MB. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol. 2010;8(3):e1000343. doi: 10.1371/journal.pbio.1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wittkopp PJ. Variable transcription factor binding: a mechanism of evolutionary change. PLoS Biol. 2010;8(3):e1000342. doi: 10.1371/journal.pbio.1000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, Tomazou EM, Backdahl L, Johnson N, Herberth M, Howe KL, Jackson DK, Miretti MM, Fiegler H, Marioni JC, Birney E, Hubbard TJP, Carter NP, Tavaré S, Beck S. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) Genome Research. 2008;18(9):1518–29. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makino S, Adachi M, Ago Y, Akiyama K, Baba M, Egashira Y, Fujimura M, Fukuda T, Furusho K, Iikura Y, Inoue H, Ito K, Iwamoto I, Kabe J, Kamikawa Y, Kawakami Y, Kihara N, Kitamura S, Kudo K, Mano K, Matsui T, Mikawa H, Miyagi S, Miyamoto T, Morita Y, Nagasaka Y, Nakagawa T, Nakajima S, Nakazawa T, Nishima S, Ohta K, Okubo T, Sakakibara H, Sano Y, Shinomiya K, Takagi K, Takahashi K, Tamura G, Tomioka H, Yoyoshima K, Tsukioka K, Ueda N, Yamakido M, Hosoi S, Sagara H. Definition, diagnosis, disease types, and classification of asthma. Int Arch Allergy Immunol. 2005;136(Suppl 1):3–4. doi: 10.1159/000085901. [DOI] [PubMed] [Google Scholar]
- 46.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 2009;41(2):178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gama-Sosa MA, Midgett RM, Slagel VA, Githens S, Kuo KC, Gehrke CW, Ehrlich M. Tissue-specific differences in DNA methylation in various mammals. Biochimica et Biophysica Acta. 1983;740:212–219. doi: 10.1016/0167-4781(83)90079-9. [DOI] [PubMed] [Google Scholar]
- 48.Enard W, Fassbender A, Model F, Adorjan P, Paabo S, Olek A. Differences in DNA methylation patterns between humans and chimpanzees. Current Biology. 2004;14(4):R148–R149. [PubMed] [Google Scholar]
- 49.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Farcas R, Schneider E, Frauenknecht K, Kondova I, Bontrop R, Bohl J, Navarro B, Metzler M, Zischler H, Zechner U, Daser A, Haaf T. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol Biol Evol. 2009;26(6):1379–89. doi: 10.1093/molbev/msp046. [DOI] [PubMed] [Google Scholar]
- 51.Fay JC, Wittkopp PJ. Evaluating the role of natural selection in the evolution of gene regulation. Heredity. 2008;100(2):191–9. doi: 10.1038/sj.hdy.6801000. [DOI] [PubMed] [Google Scholar]
- 52.Whitehead A, Crawford DL. Neutral and adaptive variation in gene expression. Proc Natl Acad Sci U S A. 2006;103(14):5425–30. doi: 10.1073/pnas.0507648103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilad Y, Oshlack A, Rifkin SA. Natural selection on gene expression. Trends Genet. 2006;22(8):456–61. doi: 10.1016/j.tig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Lee H, Tang H. Next generation sequencing technology and fragment assembly algorithms. In: Anisimova M, editor. Evolutionary Genomics: Statistical and Computational Methods. Methods in Molecular Biology. Springer Science+Business Media; New York: 2012. [Google Scholar]
- 55.Beerenwinkel N, Siebourg J. Probability, statistics and computational science. In: Anisimova M, editor. Evolutionary Genomics: Statistical and Computational Methods. Methods in Molecular Biology. Springer Science+Business Media; New York: 2012. [Google Scholar]