

Abstract
Replisomes are dynamic multiprotein machines capable of simultaneously replicating both strands of the DNA duplex. This review focuses on the structure and function of the E. coli replisome, many features of which generalize to other bacteria and eukaryotic cells. For example, the bacterial replisome utilizes clamps and clamp loaders to coordinate the actions required of the trombone model of lagging strand synthesis made famous by Bruce Alberts. All cells contain clamps and clamp loaders and this review summarizes their structure and function. Clamp loaders are pentameric spirals that bind DNA in a structure specific fashion and thread it through the ring shaped clamp. The recent structure of the E. coli β clamp in complex with primed DNA has implications for how multiple polymerases function on sliding clamps and how the primed DNA template is exchanged between them. Recent studies reveal a remarkable fluidity in replisome function that enables it to bypass template lesions on either DNA strand. During these processes the polymerases within the replisome functionally uncouple from one another. Mechanistic processes that underlie these actions may involve DNA looping, similar to the trombone loops that mediate the lagging strand Okazaki fragment synthesis cycle.
Introduction
Bruce Alberts first proposed the “trombone” DNA looping model of lagging strand DNA replication nearly thirty years ago.1 DNA looping on the lagging strand is now well established in the T4 phage replication system,2 and a variety of other replication systems. This review discusses the Escherichia coli system which, like T4, utilizes trombone DNA loops that are mediated by sliding clamps and clamp loading machines. These accessory factors provide high processivity to the polymerase, but also enable the polymerase to hop from one site to another for discontinuous synthesis on the lagging strand.3,4 The use of clamps and clamp loaders for chromosomal replication generalizes to eukaryotes and archae.
This review focuses on the mechanism by which blocks to leading and lagging strands are circumvented and proposes that DNA trombone loops underlie some of these mechanisms.
Encounter of a replication fork with a lesion is typically thought to lead to replication fork collapse. However, discovery of leading strand priming, and of translesion DNA polymerases, provide unexpected routes for lesion bypass without necessarily requiring collapse of a fork.5,6 Observations of discontinuous synthesis on the leading strand, not just the lagging strand, indicates that the leading polymerase is more dynamic than originally thought. This extra plasticity is likely to underlie some of the mechanisms by which replication forks circumvent blocks. First we describe the architecture and function of the E. coli replisome. Then we discuss mechanisms by which the replisome may continue its forward advance after encountering a block on either the leading or lagging strand.
The E. coli replisome
All cellular replisomes contain certain enzymatic activities in common. These include a DNA helicase to unwind the parental duplex, primase which makes short RNA primers to initiate DNA synthesis, and two DNA polymerases for leading and lagging strand synthesis.7 A single-strand DNA (ssDNA) binding protein (SSB) is employed on the lagging strand to protect ssDNA from nucleases and to melt secondary structure. Replicases of cells from all three domains of life utilize circular sliding clamps, proteins that encircle the duplex and tether the polymerase to DNA for high processivity.8 Sliding clamps are assembled onto DNA by a multi-subunit clamp loader machine that opens and closes the clamp around DNA in a reaction driven by ATP hydrolysis.
The architecture of the E. coli replication fork is presented in Fig. 1 and details of the proteins’ subunits are given in Table 1 (reviewed in ref. 9). At the head of the replication fork is the helicase, DnaB. DnaB is a homohexamer that encircles ssDNA on the lagging strand and uses ATP to translocate 5′–3′ along ssDNA to drive strand separation. The replicase, DNA polymerase III (Pol III) holoenzyme, contains one clamp loader which binds two molecules of the heterotrimeric Pol III core for coordinated replication of leading and lagging strands. The connection between the two Pol III cores is made by the two τ subunits within the multi-protein clamp loader. Each τ subunit contains a C-terminal 24 kDa region that is needed to organize the replisome through direct interaction with the Pol III core and DnaB, but is not needed for clamp loading (see Fig. 1). Connection of the leading strand Pol III core to DnaB (through τ) couples the energy of nucleotide incorporation with helicase unwinding action and enables DnaB to unwind DNA at a rate of 500–1000 nucleotides per s, at least 20-fold faster than DnaB acting alone.10
Fig. 1.

Architecture of the E. coli replication fork. The parental duplex DNA is unwound by the homohexameric DnaB (blue). DnaB encircles the lagging strand ssDNA and DnaG primase (purple) interacts with DnaB for activity in synthesis of an RNA primer (orange) on lagging strand ssDNA coated with SSB (white). The Pol III holoenzyme is composed of components as follows: leading and lagging strand Pol III cores (yellow) are held to their respective DNA strands by the β clamp (red), which is loaded onto primed sites by the γ complex clamp loader (green). The γ complex consists of one δ (light green), one δ′ (blue green), one γ and two τ subunits (dark green); the C-terminal extensions of τ bind Pol III core and DnaB. The clamp loader also contains two small subunits, χ and ψ (not shown) that are not required for clamp loading activity. Adapted with permission from ref. 68.
Table 1.
Proteins that function at the E. coli replication fork
| Subunit | # | Gene | Monomer mass (kDa) | Function | |
|---|---|---|---|---|---|
| Pol III Core | α | 2 | dnaE | 129.9 | DNA polymerase |
| ε | 2 | dnaQ | 27.5 | 3′–5′ exonuclease | |
| θ | 2 | holE | 8.6 | Binds ε | |
| β Clamp | β2 | 4 | dnaN | 40.6 | Sliding Clamp |
| Clamp loader | γ/τ | 3 | dnaX | 47.5/71.1 | ATPase, Binds Pol & DnaB |
| δ | 1 | holA | 38.7 | Opens the Clamp | |
| δ′ | 1 | holB | 36.9 | Stator | |
| χ | 1 | holC | 16.6 | Binds SSB | |
| ψ | 1 | holD | 15.2 | Binds χand γ | |
| Primase | DnaG | 1 | dnaG | 65.6 | RNA primer synthesis |
| Helicase | DnaB | 6 | dnaB | 52.4 | Unwinds DNA |
| ssDNA binding protein | SSB | 4 | ssb | 18.8 | Binds ssDNA |
The sliding clamp
The β clamp is a homodimer in the shape of a ring.11 Each β protomer consists of three domains that share a common chain folding topology, giving the clamp a six-fold symmetrical appearance. The replication apparatus of eukaryotes and archae also utilize a sliding clamp called PCNA.12 PCNA clamps are homotrimeric 6-domain rings in which each protomer contains two domains.13 Bacterial and eukaryotic clamps have similar inner and outer diameters, and share the same chain folding topology (Fig. 2A). Hence, sliding clamps from each of the three domains of life likely share a common ancestor in evolution. The facinating architecture of these clamps is reviewed elsewhere.14
Fig. 2.

Structures of Pol III holoenzyme components and comparison to its eukaryotic counterparts. (A) Ribbon representations of the E. coli β clamp (left, pdb code 2POL) and human PCNA clamp (right, pdb code 1AXE). Protomers are colored differently. (B) Left: Structure of the E. coli γ3δδ′ circular pentamer (pdb code 1JR3). Location of the gap between δ and δ′, and expected position of the β ring are indicated by arrows. Adapted from ref. 17. Right: Surface representation of yeast RFC-PCNA complex (pdb code 1SXJ). RFC subunits are colored differently and PCNA is shown in grey. DNA is modelled into the central chamber of RFC. Figure reproduced with permission from ref. 24. (C) Left: structure of E. coli α subunit (pdb code 2HQA). Domains are labelled as indicated: PHP (blue), thumb (green), palm (purple), fingers (orange, dark brown, brown, yellow). Right: Model of E. coli α with primed DNA and β subunit. The possible location of ε subunit is indicated by the open circle. Figures reproduced with permission from ref. 35.
The clamp loader
The E. coli clamp loader, referred to as the γ complex, contains 5 subunits required for clamp loading function, three molecules of γ and/or τ (they are interchangable as described below), and one each of δ and δ′; the γ complex also contains two small accessory subunits χ and ψ (reviewed in ref. 9).
The γ and τ subunits are encoded by the same gene, dnaX. The τ subunit (71 kda) is the full length product of dnaX, and γ (47 kda) is a C-terminal truncation product formed by a −1 translational frameshift. The γ subunit contains three domains which are necessary for clamp loading activity, while τ contains an additional C-terminal 24 kda that binds DnaB helicase and Pol III core.15
The clamp loader that organizes a replisome containing two Pol III cores consists of three γ/τ subunits, along with one subunit each of δ, δ′, χ and ψ.9 Only the γ/τ, δ′, and δ subunits are required for clamp loading activity. The χ subunit binds SSB and is held to the clamp loader through its connection to ψ.16 The ψ subunit promotes an ATP-actived conformation that opens the β clamp.18 The structure of the minimal clamp loader reveals a circular pentamer of γ3δδ′.17 Interestingly, the γ/τ, δ and δ′ subunits are all members of the AAA+ family of ATPases. AAA+ proteins (ATPases associated with a variety of functions) generally interact with ATP and remodel other proteins.19,20 In the case of γ complex only the γ/τ subunits bind and hydrolyze ATP; δ and δ′ do not bind ATP. The γ and τ subunits are interchangeable in clamp loader function with δ and δ′.21 For example, a clamp loader consisting of τ3δδ′ is as active as γ3δδ′.21 The two N-terminal domains of all five subunits are homologous to AAA+ proteins, and the third domain forms the tight pentameric contacts. There is a gap between the AAA+ domains of the δ and δ′ subunits, and this gap provides DNA access to the center of the complex (Fig. 2B, left). The β dimer is opened by the δ subunit, and the clamp interactive residues of δ are located on the N-terminal domain, indicating that the β ring binds underneath the complex where it may form connections with all of the subunits.22
Eukaryotes and archae also contain a clamp loading machine consisting of five AAA+ subunits, referred to as RFC (replication factor C).23 The structure of the eukaryotic RFC complex from S. cerevisiae bound to PCNA reveals further details of clamp loader mechanism.24 RFC, like the E. coli γ complex, contains five clamp loading subunits arranged in a circular spiral (Fig. 2B). Each RFC subunit is distinct, but all 5 are members of the AAA+ family, and there is a gap between the AAA+ domains of the RFC1 and RFC5 subunits. The PCNA clamp is located beneath the clamp loader. PCNA is closed in the RFC-PCNA-ATPγS structurewhich is likely due to the use of ATP site mutant RFC subunits, as biochemical studies demonstrate that ATP binding opens PCNA.25–27 An EM reconstruction of an archael RFC-PCNA-ATP complex also reveals an open PCNA clamp.28 The AAA+ domains of RFC are arranged in a spiral and form an inner chamber that approximates the pitch of duplex DNA. Conserved residues that line the inner chamber of the clamp loader bind duplex DNA and position it through the clamp (see Fig. 2B, right).
An outline of the clamp loading mechanism of the E. coli γ complex is illustrated in Fig. 3 (reviewed in ref. 29). ATP binding powers opening of the β clamp; hydrolysis is not required. Molecular simulations indicate that clamps open in a right-handed spiral,30 like a lock-washer, consistent with EM analysis of the archael RFC-PCNA complex and with FRET studies of the T4 clamp.28,31 A right handed open spiral clamp fits nicely with the spiral pitch of the clamp loader subunits. The δ subunit opens the clamp, aligning the gap between δ and δ′ with the gap in the open clamp. Specificity in primer template recognition is achieved by the tight connections of the C-terminal domains of the clamp loader pentamer which block DNA from exiting through the top of the structure, and therefore impose a requirement that one end of DNA exits out of the gap between δ and δ′ in the side of the clamp loader. A primed template can fit into the clamp loader by virtue of the flexible ssDNA template strand which enables the sharp bend necessary to exit through the slot in the side of the clamp loader. In contrast, duplex DNA does not fit into the clamp loader because it lacks the flexibility necessary for this sharp bend. Once primed DNA is properly positioned through the ring, ATP hydrolysis results in ejection of the clamp loader, allowing the clamp to close around DNA (see Fig. 3).
Fig. 3.
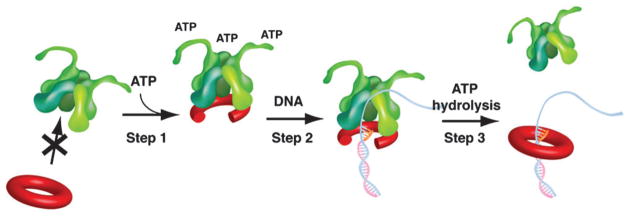
Clamp loading mechanism. Step 1: The clamp loader binds ATP to adopt a conformation that allows it to bind the β clamp and open it. Step 2: The clamp loader binds a primed template which enters into the central chamber of the clamp loader through the gap between δ and δ′ and the open ring. Step 3: ATP hydrolysis ejects the clamp loader from β, allowing the ring to close around DNA.
Pol III core
Pol III core consists of three subunits in 1 : 1 : 1 molar ratio (α, ε, θ).32 The α subunit contains the DNA polymerase activity and binds directly to the β sliding clamp, the ε subunit contains a 3′–5′ proofreading exonuclease activity, and the θ subunit stimulates the activity of ε.9 The α subunit is a member of the C-family of DNA polymerases to which all bacterial replicases belong.33 Until recently the C-family of DNA polymerases lacked a crystal structure representive. However, recent studies have solved the crystal structure of α from both E. coli and Thermus aquaticus.34,35 The structures show a right handed shape common to all DNA polymerases, with palm, fingers and thumb domains. The structures also reveal many unusual features of α subunit, including a fingers domain with four subdomains, plus a novel PHP domain (Fig. 2C). The T. aquaticus PHP domain harbors a cryptic 3′–5′ exonuclease activity.36 The most conserved structural feature among different DNA polymerase families is the catalytic palm domain.37 Suprisingly, the α structures reveal that the palm domain of bacterial α C-family DNA polymerases has the chain folding pattern as found in the X-family DNA polymerases (i.e. eukaryotic DNA polymerase β). Until the structure of bacterial α was solved, the X-family palm chain fold stood alone, and was regarded as an evolutionary offshoot. But the fact that bacterial replicases have similar structures to X-family DNA polymerases suggests that these families may represent an early form of DNA polymerase during evolutionary development.
The lagging strand trombone cycle
The antiparallel structure of DNA and unidirectional action of DNA polymerases imposes the formation of DNA loops on the lagging strand of the replication fork as originally proposed by Bruce Alberts.1 Study of this process in E. coli has shown that the “trombone” cycle is mediated by the clamp loader and sliding clamps, illustrated in Fig. 4. The lagging strand Pol III core travels with the replisome and therefore the lagging ssDNA template is pulled up through the polymerase during synthesis, and the dsDNA product is extruded into the trombone loop. Loop growth is also fueled by continued helicase action and leading strand synthesis which extrudes ssDNA into the loop. The lagging strand polymerase must dissociate upon completing an Okazaki fragment, in order that it can extend an upstream RNA primer for the next Okazaki fragment. The scarce intracellular supply of Pol III (10–20 molecules per cell) and thousands of Okazaki fragments necessitate the rapid and efficient recycling of Pol III.
Fig. 4.

The lagging strand trombone mechanism. (A) The lagging strand DNA loop is produced through the actions of the lagging strand Pol III core-β as it extends an Okazaki fragment, and through continued fork progression to produce ssDNA. DnaG primase synthesizes a new RNA primer. Diagrams B, C and D highlight proteins that act on the lagging strand. (B) The clamp loader loads a new β clamp on the RNA primer. (C) The lagging stand Pol III core finishes a fragment and disengages from its clamp. (D) The lagging Pol III core associates with the new β clamp at an upstream primer to begin extension of the next Okazaki fragment, completing the cycle and starting a new DNA loop. Figure reproduced with permission from ref. 68.
Rapid polymerase recycling conflicts with the picture of a polymerase held tightly to DNA by a ring. This conflict is solved in at least two different ways, both of which involve polymerase separation from its clamp. In one mechanism, termed “collision release”, Pol III core is triggered to disengage from the β clamp upon finishing a DNA fragment, leaving β behind.3,4 The clamp loader repeatedly loads new β clamps at fresh primed sites for the lagging strand Pol III core.38 Under some conditions, Pol III also disengages from its clamp prior to completing an Okazaki fragment.39 This process is refered to as “premature release”. It is also referred to as “signaling release”, as studies in T4 and T7 phage systems indicate that premature release may be signaled by primer formation or assembly of clamps on RNA primers.40,41 In E. coli, the signal for premature release is unclear; it may even be mediated by force exerted on a DNA loop as explained later in this review.
The lagging strand cycle requires stoichiometric use of β clamps, one per Okazaki fragment. Stoichiometric use of β is consistent with its high intracellular concentration (~300 β dimers per cell). However, about 3000 Okazaki fragments are produced in one 30 min cell division cycle. Therefore approximately 10 Okazaki fragments are produced per β clamp, giving about 3 min for each β to recycle. Recycling of β is performed by the δ subunit, a clamp loader subunit that opens β and is present in the cell at a concentration sufficient to perform the clamp recycling task.42
Replication forks can leave DNA lesions behind in ssDNA gaps
Encounter of the replication fork with a DNA lesion may be a process that requires replication fork collapse, followed by a multi-step recombination process to repair the lesion. However, cellular studies show that ssDNA gaps are left behind replicated DNA on both leading and lagging strands in response to DNA damage in bacteria and eukaryotic cells alike.43,44 These observations imply that replication forks have mechanisms to skip over lesions. Several recent biochemical studies reveal mechanisms that may explain how replisomes can bypass lesions without necessarily undergoing replication fork collapse (described below). Lesions that cause replication fork collapse through DNA fragmentation (i.e. nicks) are repaired by the RecBCD dependent double strand break repair pathway. This review presents possible mechanisms that enable replisomes to proceed past lesions, either directly or by skipping over them. Readers that are interested in replication fork collapse and double strand break repair are referred to excellent recent reviews.45,46
Passage of a replication fork over a lesion on the lagging strand is conceptually easier to envision than skipping over a lesion on the leading strand. The reason for this is that the lagging strand is a discontinuous process initiated by multiple priming events, and the lagging strand polymerase rapidly hops from one Okazaki fragment to a new RNA primer. This dynamic process provides a route by which the replisome may skip over a lagging strand lesion (explained below). In contrast, the leading strand is extended in the same direction as fork movement and thus is thought of as a continuous process. It is difficult to envision how a continuous leading strand polymerase can hop over a lesion if there is no new primer on the other side of the lesion for the polymerase to attach to. In this situation the replication fork must collapse (i.e. the replisome must dissociate) to make way for recombination repair of the lesion, followed by reassembly of a new replisome. The kinetic block to reassembly of a replisome is the loading of DnaB helicase onto DNA. This job is performed by the PriA and PriC proteins; mutational analysis of the priA and priC genes supports their role in reassembly of collapsed replication forks in vivo.45,46 However, recent studies reveal that the replisome is surprisingly dynamic, and replication fork collapse may not always be a necessary consequence of replisome encounter with a leading strand block.
These new findings, and how they relate to the fluid processes that allow replication forks to pass lesions on either strand, are the focus of the sections to follow. We begin with the process of skipping over a lagging strand lesion, as it draws on the established model of lagging strand replication with relatively minimal changes. Then we deal with how replication forks skip over a leading strand lesion. In the last section, we will summarize how lesions may be bypassed directly by special translesion DNA polymerases that are designed specifically for this task. Translesion polymerases also utilize the β clamp, and therefore direct lesion bypass requires that DNA polymerases trade places with one another on the DNA sliding clamp.
Skipping over a lesion on the lagging strand
It is interesting to consider that less then ten years ago the commonly held view was that replication of the leading and lagging strands were tightly coupled events, and was considered an important reason that leading and lagging strand polymerases were connected together in a single replisome. Specifically, upon encountering a lagging strand lesion, the stalled lagging strand polymerase would communicate its blocked status to the leading strand enzyme and induce it to stop, thereby halting the replication fork altogether. Only after the lagging strand block was resolved could leading strand synthesis continue. Indeed, coupling such as this has been observed in the bacteriophage T4 and T7 replication systems.47,48
The proposal that leading and lagging strand polymerases are tightly coupled was quite attractive, but cellular studies supported the conflicting view that the two polymerases at replication forks are uncoupled.43,44,49 More recent biochemical studies of the E. coli and T4 replisomes have demonstrated that the actions of the two polymerases uncouple when the lagging strand polymerase encounters a block to forward movement; leading strand synthesis continues at the same rate regardless of the block to lagging strand replication.49–52 In fact, lagging strand synthesis also continues after a block to the leading strand polymerase.53 In each case the fork continues to replicate both strands and simply leaves the block behind in a ssDNA gap. Therefore, the leading and lagging strand Pol III cores are physically coupled, but are functionally uncoupled, and this enables the replisome to skip over lesions.
Uncoupled synthesis allows lagging strand blocks to be circumvented by premature release of the stalled lagging strand polymerase. Furthermore, the released Pol III core simply reassociates with new clamps on upstream RNA primers, allowing the fork to continue but leaving the block behind in a ssDNA gap (illustrated in Fig. 5). Interestingly, Pol III stalled at a lesion, or stalled by nucleotide omission, remains stably attached to its clamp on a simple primed ssDNA substrate, in contrast to premature release observed in replication fork systems that include DnaB helicase and primase.3,4,52,53 Hence, premature release of a stalled Pol III from β is an active process that occurs in the context of a moving replication fork. Premature release of the lagging strand enzyme has been observed in the E. coli replication system in the presence and absence of blocks.39,52 In the absence of blocks, premature release occurs when synthesis of Okazaki fragments is compromised by lowering the concentration of primase or by lowering rNTPs to limit primer formation;39 these conditions result in extraordinarily long Okazaki fragments. Premature release of a blocked lagging strand enzyme is also thought to be preceeded by formation of an abnormally large DNA loop, caused by continued leading strand synthesis.52 It has been suggested that primed sites lacking SSB may trigger release of Pol III from β, provided a replication fork continues until the intracellular supply of SSB is exhausted.52 We propose another possibility in which the force generated by a DNA loop signals premature release of the stalled Pol III (explained below).
Fig. 5.

Model for skipping a lesion on the lagging strand. (A) The lagging strand Pol III core is stalled at a lesion (red stop sign). The helicase-leading strand Pol III core continues forward, producing an abnormally large lagging strand DNA loop. (B) The lagging strand Pol III core disengages prematurely from the clamp, leaving the lesion behind in a ssDNA gap, and transfers to a new β clamp on an upstream RNA primer. The signal for premature release is unknown, but could derive from the force of dragging the large DNA loop along with the replisome.
Uncoupled action at a replication fork with a blocked lagging strand polymerase will lead to an increasingly large lagging strand ssDNA loop due to continued unwinding and forward progression of the leading strand polymerase (see Fig. 5A). As the replisome moves forward, this large loop will be brought along with the replisome and may constitute an ever increasing burden to carry. It seems reasonable to expect that the increasingly large loop may exert a force on replication fork progression due to viscous drag of the growing loop of ssDNA as it is carried along with the moving replisome. The ssDNA loop will also be coated with SSB, which adds several times the weight of ssDNA, thereby increasing the work load yet further. At some point the viscous drag of the loop may match the free energy of association between the blocked lagging strand polymerase and its clamp on DNA. Beyond this point, further loop growth will finally exceed the strength of attachment of Pol III core to β and the polymerase will be pulled away from its clamp, leaving a ssDNA gap (see Fig. 5B). The stalled lagging strand polymerase will now be free to utilize an upstream RNA primed site and resume the generation of new Okazaki fragments. Whether the force generated by a large DNA loop indeed signals premature release of E. coli Pol III, or whether events related to priming provide the signal for premature release will require further studies of this important process.
A leading strand block and the triple Pol III replisome
A block to the continuous leading strand polymerase would appear, at first glance, to be much more difficult to circumvent than a block on the lagging strand. The underlying reason for this is the conception that the leading strand is continuous and thus has no new priming events, and thus no DNA loops to facilitate removal of a stalled polymerase and its replacement on a new upstream primed site. For this reason, most models for the induction of the SOS response to DNA damage imply that the leading strand polymerase stalls at a template lesion while the helicase continues to unwind DNA, thus generating leading strand ssDNA ahead of the stalled leading strand polymerase. The leading strand ssDNA then serves as a substrate for RecA binding which initiates cleavage of LexA repressor and induction of the SOS response that produces numerous proteins which facilitate replisome movement past lesions and recovery of collapsed replication forks.7
Recent studies provide new alternatives to replication fork collapse of a leading strand polymerase stalled at a lesion on DNA. An alternate path is suggested by the remarkable finding that primase can function on the leading strand, and this provides a route for the stalled fork to continue by extending the new primer and leaving the block behind in a ssDNA gap on the leading strand,45 consistent with the in vivo observations in both bacteria and eukaryotes of ssDNA gaps produced after DNA damage.43,44 The ssDNA gaps that are left behind in either leading or lagging stand lesions may trigger the SOS response.
It is interesting to contemplate the mechanism by which the stalled leading strand polymerase departs from its β clamp and transfers to a new clamp at the upstream primer. If the stalled Pol III is stably bound to DNA, it will require a signal to remove it from the β clamp. As proposed above, the signal for premature release of a stalled lagging strand Pol III may be the force generated by viscous drag caused by the large DNA trombone loop attached to the stalled Pol III. Extensive replication fork advancement requires that DnaB helicase be coupled to a moving leading strand polymerase.10 In the absence of a moving leading strand polymerase, DnaB is a poor helicase; it continues slowly and stops after only 0.5–2 kb.10 This action may result in a 0.5–2 kb ssDNA loop between DnaB and the stalled Pol III to which it is connected (i.e. through the τ subunit), and this may provide sufficient force to release Pol III from β.53 However, a larger DNA loop may be required to generate sufficient force to pull a stalled Pol III from β. In this regard it is interesting to note that single molecule experiments reveal that a force of 34 pNs applied to template DNA is sufficient to stop T7 DNA polymerase.54
We would like to suggest a mechanism by which a large DNA loop could be generated on the leading strand. The process makes use of the recent finding of an E. coli Pol III holoenzyme replicase that contains three molecules of Pol III core.21 A triple Pol III replicase is based in studies of the τ and γ subunits and how they assemble with other subunits to form Pol III holoenzyme. Purification of τ and γ from the same cells (i.e. both τ and γ are produced by the same gene, dnaX) yields τ and γ homooligomers, not mixed oligomers, suggesting that the homooligomeric forms of τ and γ are the most stabile state.55 Mixture of γ and τ along with all the other subunits of Pol III holoenzyme results in rapid assembly of a triple Pol III holoenzyme that contains a clamp loader having three τ subunits and no γ.21 The triple Pol III holoenzyme is functional at replication forks in vitro. Hence, the physiological form of Pol III holoenzyme may be a triple polymerase (i.e. only τ and no γ), and the γ complex may be produced to assemble β onto DNA for use by the many other proteins that function with β (i.e. Pols I, II, IV, V, MutS, MutL, ligase, Hda protein).56
In support of a triple Pol III holoenzyme, mutation of the frameshift site of E. coli dnaX, such that it produces only τ, does not impede cell viability suggesting that γ is not required for cell growth.57 This result also implies that a clamp loader with three τ subunits is functional at the E. coli replication fork. In addition, examination of genomic sequences of numerous organisms suggest that many bacteria only produce the full length τ subunit from the dnaX gene, and this has been demonstrated experimentally in Aquifex aeolicus.58 These bacteria lack γ and therefore their replicase will contain three τ subunits and three polymerases.
These several observations suggest that the cellular replicase contains three molecules of the Pol III core. Biochemical in vitro studies suggest that the two Pol III cores in one holoenzyme are sufficent for normal leading and lagging strand synthesis.21 It has been proposed that the third Pol III core may act as a backup polymerase that is used if one of the other two polymerases becomes blocked or inactivated.21 In Fig. 6, we propose a model in which a triple Pol III replisome is uniquely suited to produce a large leading strand DNA loop at a replication fork containing a blocked leading strand enzyme. Upon stalling the leading strand polymerase, primase generates a new RNA primer ahead of the blocked polymerase on the leading strand45 (Fig. 6B). Upon assembly of a clamp on the new RNA primer, the third Pol III within a triple Pol III replicase will bind the new primer and couple with DnaB to form a replisome capable of rapid advance. Forward progression will result in formation of a large leading standDNA loop, and eventually the viscous drag on the growing loop may generate sufficient force to disengage the stalled Pol III from the β clamp, leaving a ssDNA gap behind on the leading strand (see Fig. 6C).
Fig. 6.

Use of a triple Pol III replisome to skip over a lesion on the leading stand. (A) A Pol III holoenzyme containing three τ subunits contains three Pol III cores, one of which is “extra”. The leading strand Pol III core is stalled at a lesion, and primase is shown priming the leading strand ahead of the stalled leading Pol III. (B) The “extra” Pol III core associates with β at the new leading strand RNA primer and connects to DnaB, thereby reactivating rapid fork progression. (C) Fork progression results in a growing DNA loop between the moving and stalled leading Pol III cores. (D) The stalled leading Pol III core releases from β, leaving the lesion behind in a ssDNA gap on the leading strand.
Bypass of lesions by translesion polymerase exchange on β
Specialized error-prone translesion DNA polymerases exist that are capable of misincorporating a nucleotide across a template lesion, thereby extending DNA across the lesion.5,59 These DNA polymerases typically lack a 3′-5′ exonuclease, facilitating their advance over a lesion. Thus, another route by which a replisome may traverse a template lesion is by recruiting a translesion DNA polymerase, leaving behind continuous DNA that contains a mutation instead of a ssDNA gap.
E. coli contains two translesion DNA polymerases that are members of the Y-family, Pol IV and Pol V; they are induced by the SOS-response to DNA damage. Pol II is also induced by DNA damage, but is a B-family DNA polymerase and contains a 3′–5′ exonuclease activity. The function of Pol II is unclear, but it appears capable of traversing particular types of DNA lesions. All three damage inducible DNA polymerases of E. coli function with the β clamp, and they contain a consensus β-binding peptide sequence that fits into the protein-binding pocket on the clamp.56 Since each protomer of the β homodimer contains an identical protein-binding pocket, two DNA polymerases could conceivably bind one β clamp simultaneously. Indeed, the structure of Pol IV-β suggests that there may be sufficient room for Pol III to bind β at the same time as Pol IV.59 Biochemical studies have confirmed this prediction and have shown that Pol IV gains control of the primed site when Pol III stalls.60 Additional studies confirm that DNA polymerases can trade places with one another on the clamp.61,62 Thus it may be presumed that all three damage inducible polymerases interact with β and take turns binding the clamp, enabling the template lesion to be sampled by the various translesion polymerases in a process referred to as “replication fork management”.63–65
The crystal structure of β bound to a primed site shows unexpected features of β function and suggests ways that polymerases may trade places on the clamp.66 The structure shows that DNA goes through the β ring at a steep angle, ~22° (Fig. 7A). Residues that bind to duplex DNA are located on surface loops that protrude from β and contribute to the tilt of DNA through the ring. The DNA binding loops of only one β protomer can bind DNA at any given time. Hence, one may expect that the DNA rapidly isomerizes, or “flips”, between the two protomers, and this may facilitate switching of the single primed site among multiple polymerases attached to the same β clamp as illustrated in Fig. 7B. Similar conclusions have been reached from molecular simulation studies of PCNA.67
Fig. 7.

Structure of β on DNA and its implications for TLS polymerase switching on the clamp. (A) Structure of E. coli β bound to DNA (pdb code 3BEP). The two strands of duplex DNA interact with residues R24 and Q149 (front view, left). These residues are located on loops of one protomer (protomer (B) and contribute to the highly tilted orientation of DNA as it passes through the ring (side view, right). Reproduced with permission from ref. 65. (B) Two DNA polymerases can bind one β clamp simultaneously, as indicated in the illustration for Pol III (yellow) and a TLS polymerase (blue). Switching of the primed DNA from one polymerase to the other may be facilitated by the tilted orientation of DNA. Since β is a homodimer, the DNA should isomerise, or flip, from one protomer to the other, enabling the primed site to switch between the two DNA polymerases. Adapted with permission from ref. 65. (C) Two-step polymerase switching may be facilitated by ability of β to bind template ssDNA, holding it at the primed site. Thus if Pol III dissociates after encountering a lesion (left diagram), the protein-binding site of β (white circle) will bind ssDNA, holding β at the 3′ terminus (middle diagram) for association with a TLS polymerase (right diagram).
The ssDNA portion of the primed site binds to the proteinbinding pocket of β.66 This specific interaction of β with a primed template junction may act as a “placeholder” to hold the clamp at the primed site after Pol III dissociates from the clamp at a lesion (see Fig. 7C). Indeed, after premature release of a stalled Pol III from β this interaction may enable β to stay at the primed site where it can be sampled by the various translesion DNA polymerases for lesion bypass.
Concluding remarks
Many recent and exciting studies indicate entirely new ways that the chromosomal replication apparatus may deal with blocks to replication. These studies reveal a remarkable plasticity of the replisome apparatus. The lagging strand polymerase need not hold onto its clamp until an Okazaki fragment is complete, nor must a blocked leading strand polymerase halt replication fork progression. Instead, DNA looping mechanims exist that enable stalled leading and lagging strand polymerases to disengage from their clamps and recycle to new primed sites rather than undergo replication fork collapse. One possible avenue for these events on the leading strand entails use of a triple polymerase replicase. Other mechanisms yet to be discovered may also contribute to replisome function and the ability to cross lesions. These and other exciting questions regarding replisome function await future studies.
Biographies
 Nina Y. Yao received her BSc in Biochemistry from the University of Toronto in Canada, and then obtained her PhD in Biochemistry from the joint graduate program between the Molecular Biology Department at Sloan-KetteringMemorial Institute and Cornell University Medical College in New York City in 1999. Dr Yao is currently a research associate in the laboratory of DNA replication headed by Dr Mike O’Donnell at Rockefeller University, New York City.
Nina Y. Yao received her BSc in Biochemistry from the University of Toronto in Canada, and then obtained her PhD in Biochemistry from the joint graduate program between the Molecular Biology Department at Sloan-KetteringMemorial Institute and Cornell University Medical College in New York City in 1999. Dr Yao is currently a research associate in the laboratory of DNA replication headed by Dr Mike O’Donnell at Rockefeller University, New York City.
 Mike O’Donnell received his BS in Biochemistry from the University of Portland, Oregon, his PhD in Biochemistry from the University of Michigan, and postdoctoral training on multi-protein replication systems with Drs Arthur Kornberg and I. Robert Lehman, both in the Biochemistry Department at Stanford University. He joined the faculty of Cornell University Medical College in 1986 before moving to Rockefeller in 1996. He is also an investigator in the Howard Hughes Medical Institute and a member of the National Academy of Sciences.
Mike O’Donnell received his BS in Biochemistry from the University of Portland, Oregon, his PhD in Biochemistry from the University of Michigan, and postdoctoral training on multi-protein replication systems with Drs Arthur Kornberg and I. Robert Lehman, both in the Biochemistry Department at Stanford University. He joined the faculty of Cornell University Medical College in 1986 before moving to Rockefeller in 1996. He is also an investigator in the Howard Hughes Medical Institute and a member of the National Academy of Sciences.
Footnotes
This article is part of a Molecular BioSystems special issue dedicated to Professor Bruce Alberts on the occasion of his 70th birthday and in recognition of his important contributions to science and education.
Electronic supplementary information (ESI) available: Links to PDB visualisations in FirstGlance in Jmol. See DOI: 10.1039/b811097b
References
- 1.Sinha NK, Morris CF, Alberts BM. J Biol Chem. 1980;255(9):4290–4293. [PubMed] [Google Scholar]
- 2.Nossal NG, Makhov AM, Chastain PD, 2nd, Jones CE, Griffith JD. J Biol Chem. 2007;282(2):1098–1108. doi: 10.1074/jbc.M606772200. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell ME. J Biol Chem. 1987;262(34):16558–16565. [PubMed] [Google Scholar]
- 4.Stukenberg PT, Turner J, O’Donnell M. Cell. 1994;78(5):877–887. doi: 10.1016/s0092-8674(94)90662-9. [DOI] [PubMed] [Google Scholar]
- 5.Goodman MF. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 6.Heller RC, Marians KJ. Nature. 2006;439(7076):557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 7.Kornberg A, Baker TA. DNA replication. 2. W.H. Freeman; New York: 1992. [Google Scholar]
- 8.Jeruzalmi D, O’Donnell M, Kuriyan J. Curr Opin Struct Biol. 2002;12(2):217–224. doi: 10.1016/s0959-440x(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 9.Johnson A, O’Donnell M. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Dallmann HG, McHenry CS, Marians KJ. Cell. 1996;84(4):643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 11.Kong XP, Onrust R, O’Donnell M, Kuriyan J. Cell. 1992;69(3):425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 12.Stillman B. Mol Cell. 2008;30(3):259–260. doi: 10.1016/j.molcel.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Cell. 1996;87(2):297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 14.Kuriyan J, O’Donnell M. J Mol Biol. 1993;234(4):915–925. doi: 10.1006/jmbi.1993.1644. [DOI] [PubMed] [Google Scholar]
- 15.Gao D, McHenry CS. J Biol Chem. 2001;276(6):4447–4453. doi: 10.1074/jbc.M009827200. [DOI] [PubMed] [Google Scholar]
- 16.Glover BP, McHenry CS. J Biol Chem. 1998;273(36):23476–23484. doi: 10.1074/jbc.273.36.23476. [DOI] [PubMed] [Google Scholar]
- 17.Jeruzalmi D, O’Donnell M, Kuriyan J. Cell. 2001;106(4):429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 18.Anderson SG, Williams CR, O’Donnell M, Bloom LB. J Biol Chem. 2007;282(10):7035–7045. doi: 10.1074/jbc.M610136200. [DOI] [PubMed] [Google Scholar]
- 19.Erzberger JP, Berger JM. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 20.Neuwald AF. Nucleic Acids Res. 2005;33(11):3614–3628. doi: 10.1093/nar/gki674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McInerney P, Johnson A, Katz F, O’Donnell M. Mol Cell. 2007;27(4):527–538. doi: 10.1016/j.molcel.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O’Donnell M, Kuriyan J. Cell. 2001;106(4):417–428. [PubMed] [Google Scholar]
- 23.Tsurimoto T, Stillman B. Mol Cell Biol. 1989;9(2):609–619. doi: 10.1128/mcb.9.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman GD, O’Donnell M, Kuriyan J. Nature. 2004;429(6993):724–730. doi: 10.1038/nature02585. [DOI] [PubMed] [Google Scholar]
- 25.Johnson A, Yao NY, Bowman GD, Kuriyan J, O’Donnell M. J Biol Chem. 2006;281(46):35531–35543. doi: 10.1074/jbc.M606090200. [DOI] [PubMed] [Google Scholar]
- 26.Yao NY, Johnson A, Bowman GD, Kuriyan J, O’Donnell M. J Biol Chem. 2006;281(25):17528–17539. doi: 10.1074/jbc.M601273200. [DOI] [PubMed] [Google Scholar]
- 27.Zhuang Z, Yoder BL, Burgers PM, Benkovic SJ. Proc Natl Acad Sci U S A. 2006;103(8):2546–2551. doi: 10.1073/pnas.0511263103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyata T, Suzuki H, Oyama T, Mayanagi K, Ishino Y, Morikawa K. Proc Natl Acad Sci U S A. 2005;102(39):13795–13800. doi: 10.1073/pnas.0506447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloom LB. Crit Rev Biochem Mol Biol. 2006;41(3):179–208. doi: 10.1080/10409230600648751. [DOI] [PubMed] [Google Scholar]
- 30.Kazmirski SL, Zhao Y, Bowman GD, O’Donnell M, Kuriyan J. Proc Natl Acad Sci U S A. 2005;102(39):13801–13806. doi: 10.1073/pnas.0506430102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trakselis MA, Alley SC, Abel-Santos E, Benkovic SJ. Proc Natl Acad Sci U S A. 2001;98(15):8368–8375. doi: 10.1073/pnas.111006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McHenry CS, Crow W. J Biol Chem. 1979;254(5):1748–1753. [PubMed] [Google Scholar]
- 33.Leipe DD, Aravind L, Koonin EV. Nucleic Acids Res. 1999;27(17):3389–3401. doi: 10.1093/nar/27.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey S, Wing RA, Steitz TA. Cell. 2006;126(5):893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Lamers MH, Georgescu RE, Lee SG, O’Donnell M, Kuriyan J. Cell. 2006;126(5):881–892. doi: 10.1016/j.cell.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 36.Stano NM, Chen J, McHenry CS. Nat Struct Mol Biol. 2006;13(5):458–459. doi: 10.1038/nsmb1078. [DOI] [PubMed] [Google Scholar]
- 37.Brautigam CA, Steitz TA. Curr Opin Struct Biol. 1998;8(1):54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 38.Yuzhakov A, Turner J, O’Donnell M. Cell. 1996;86(6):877–886. doi: 10.1016/s0092-8674(00)80163-4. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Marians KJ. J Biol Chem. 2000;275(44):34757–34765. doi: 10.1074/jbc.M006556200. [DOI] [PubMed] [Google Scholar]
- 40.Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. Nature. 2006;439(7076):621–624. doi: 10.1038/nature04317. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Nelson SW, Benkovic SJ. Mol Cell. 2006;21(2):153–164. doi: 10.1016/j.molcel.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 42.Leu FP, Hingorani MM, Turner J, O’Donnell M. J Biol Chem. 2000;275(44):34609–34618. doi: 10.1074/jbc.M005495200. [DOI] [PubMed] [Google Scholar]
- 43.Lopes M, Foiani M, Sogo JM. Mol Cell. 2006;21(1):15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 44.Wang TC. Bioessays. 2005;27(6):633–636. doi: 10.1002/bies.20233. [DOI] [PubMed] [Google Scholar]
- 45.Heller RC, Marians KJ. Nat Rev Mol Cell Biol. 2006;7(12):932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 46.Marians KJ. Nat Struct Mol Biol. 2008;15(2):125–127. doi: 10.1038/nsmb0208-125. [DOI] [PubMed] [Google Scholar]
- 47.Lee J, Chastain PD, 2nd, Griffith JD, Richardson CC. J Mol Biol. 2002;316(1):19–34. doi: 10.1006/jmbi.2001.5325. [DOI] [PubMed] [Google Scholar]
- 48.Salinas F, Benkovic SJ. Proc Natl Acad Sci U S A. 2000;97(13):7196–7201. doi: 10.1073/pnas.97.13.7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pages V, Fuchs RP. Science. 2003;300(5623):1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 50.Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H. Genes Cells. 2003;8(5):437–449. doi: 10.1046/j.1365-2443.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 51.Kadyrov FA, Drake JW. Nucleic Acids Res. 2002;30(20):4387–4397. doi: 10.1093/nar/gkf576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McInerney P, O’Donnell M. J Biol Chem. 2004;279(20):21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- 53.McInerney P, O’Donnell M. J Biol Chem. 2007;282(35):25903–25916. doi: 10.1074/jbc.M703777200. [DOI] [PubMed] [Google Scholar]
- 54.Wuite GL, Smith SB, Young M, Keller D, Bustamante C. Nature. 2000;404(6773):103–106. doi: 10.1038/35003614. [DOI] [PubMed] [Google Scholar]
- 55.Maki S, Kornberg A. J Biol Chem. 1988;263(14):6555–6560. [PubMed] [Google Scholar]
- 56.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. Proc Natl Acad Sci U S A. 2001;98(20):11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blinkova A, Hervas C, Stukenberg PT, Onrust R, O’Donnell ME, Walker JR. J Bacteriol. 1993;175(18):6018–6027. doi: 10.1128/jb.175.18.6018-6027.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bruck I, Yuzhakov A, Yurieva O, Jeruzalmi D, Skangalis M, Kuriyan J, O’Donnell M. J Biol Chem. 2002;277(19):17334–17348. doi: 10.1074/jbc.M110198200. [DOI] [PubMed] [Google Scholar]
- 59.Bunting KA, Roe SM, Pearl LH. EMBO J. 2003;22(21):5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M. Mol Cell. 2005;19(6):805–815. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Johnson DE, Takahashi M, Hamdan SM, Lee SJ, Richardson CC. Proc Natl Acad Sci U S A. 2007;104(13):5312–5317. doi: 10.1073/pnas.0701062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J, Zhuang Z, Roccasecca RM, Trakselis MA, Benkovic SJ. Proc Natl Acad Sci U S A. 2004;101(22):8289–8294. doi: 10.1073/pnas.0402625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maul RW, Ponticelli SK, Duzen JM, Sutton MD. Mol Microbiol. 2007;65(3):811–827. doi: 10.1111/j.1365-2958.2007.05828.x. [DOI] [PubMed] [Google Scholar]
- 64.Sutton MD, Duzen JM. DNA Repair. 2006;5(3):312–323. doi: 10.1016/j.dnarep.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 65.Sutton MD, Walker GC. Proc Natl Acad Sci U S A. 2001;98(15):8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O’Donnell M. Cell. 2008;132(1):43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ivanov I, Chapados BR, McCammon JA, Tainer JA. Nucleic Acids Res. 2006;34(20):6023–6033. doi: 10.1093/nar/gkl744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langston LD, O’Donnell M. Mol Cell. 2006;23:155–160. doi: 10.1016/j.molcel.2006.05.034. [DOI] [PubMed] [Google Scholar]